
AML under the Scope: Current Strategies and Treatment Involving FLT3 Inhibitors and Venetoclax-Based Regimens

Abstract
:1. Introduction
2. Results
2.1. BCL-2 Inhibitors
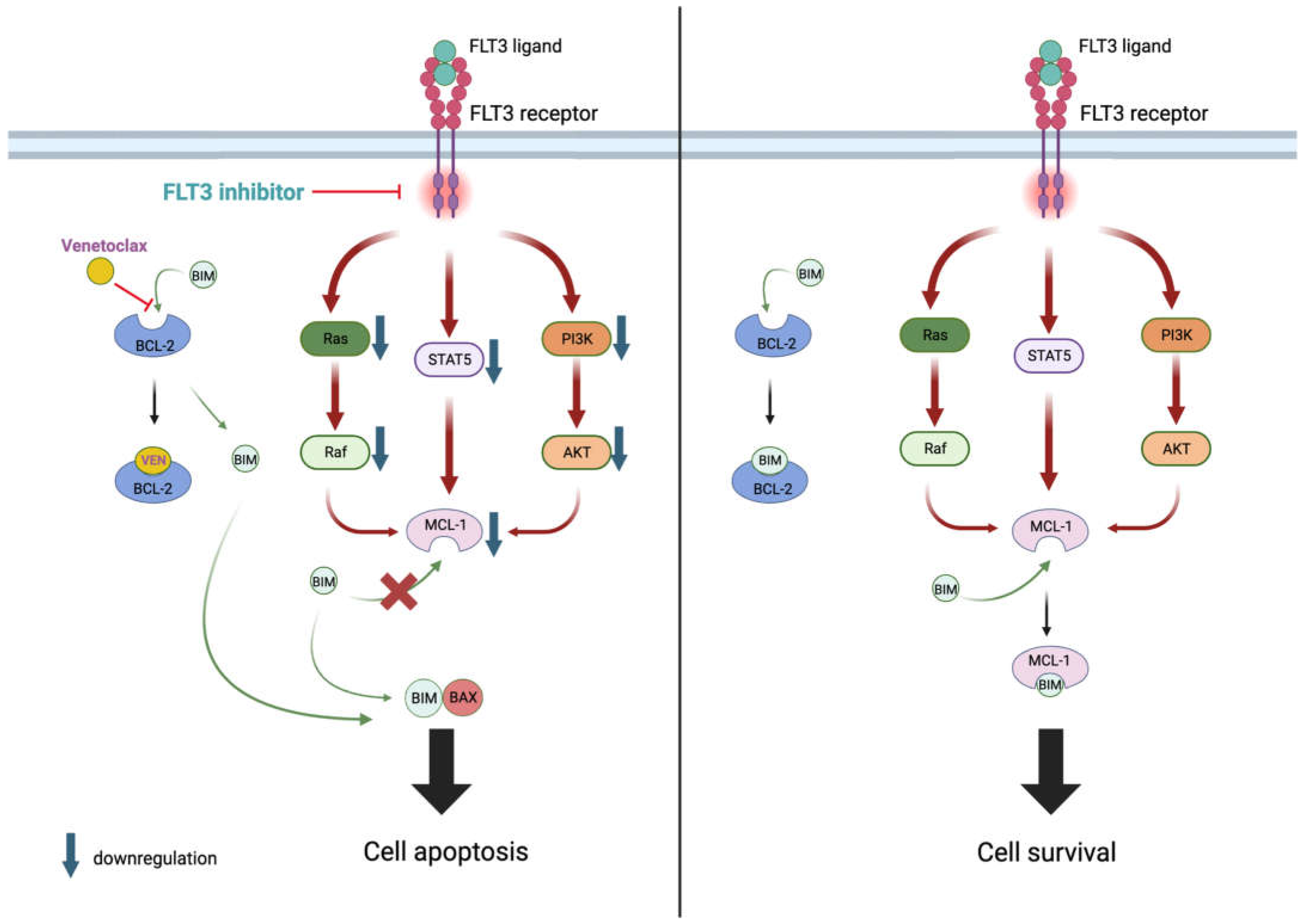
2.1.1. Mechanism of Action of Venetoclax
2.1.2. Clinical Data for Venetoclax
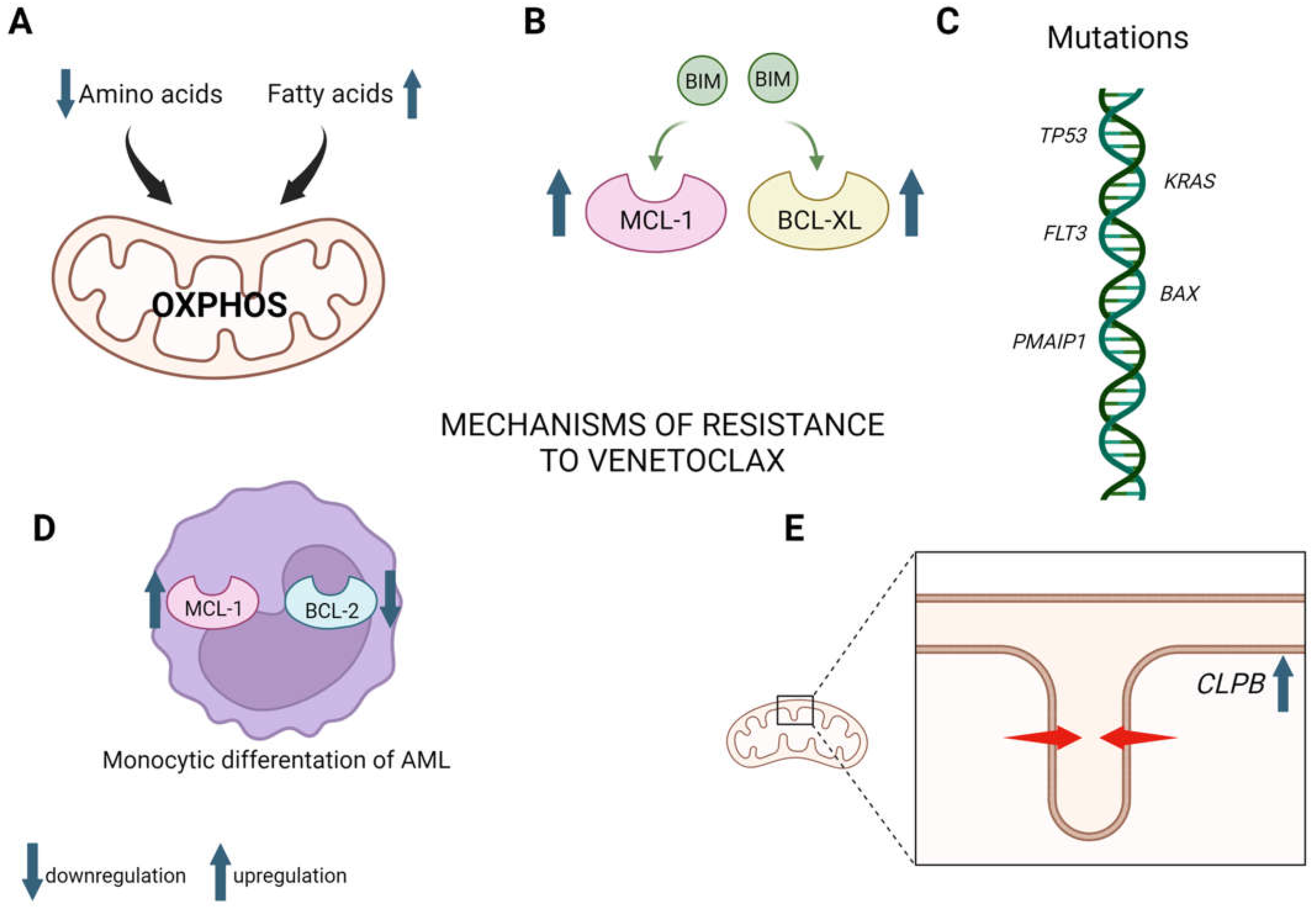
2.1.3. Mechanisms of Venetoclax Resistance
2.2. FLT3
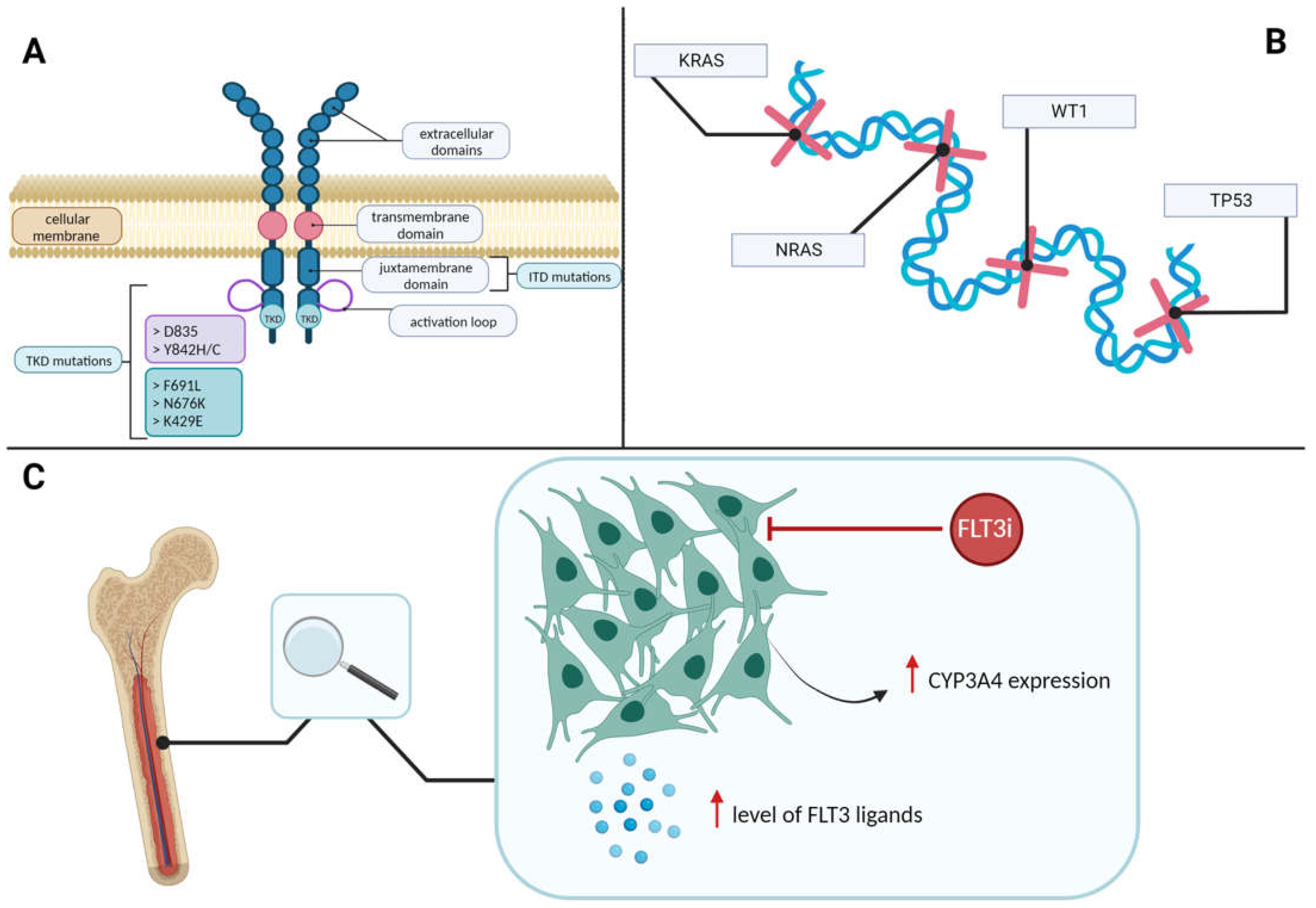
FLT3 Mutations
2.3. FLT3 Inhibitors
2.4. First-Generation FLT3 Inhibitors
2.4.1. Midostaurin
2.4.2. Sorafenib
2.5. Second-Generation FLT3 Inhibitors
2.5.1. Gilteritinib
2.5.2. Quizartinib
2.6. Mechanisms of FLT3 Inhibitors Resistance
2.7. Predictive Factors of Venetoclax and FLT3 Inhibitors
2.8. BCL-2 and FLT3 Inhibitors
3. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vakiti, A.; Mewawalla, P. Acute Myeloid Leukemia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- SEER Cancer Stat Facts: Acute Myeloid Leukemia. Available online: https://seer.cancer.gov/statfacts/html/amyl.html (accessed on 1 August 2023).
- Yi, M.; Li, A.; Zhou, L.; Chu, Q.; Song, Y.; Wu, K. The Global Burden and Attributable Risk Factor Analysis of Acute Myeloid Leukemia in 195 Countries and Territories from 1990 to 2017: Estimates Based on the Global Burden of Disease Study 2017. J. Hematol. Oncol. 2020, 13, 72. [Google Scholar] [CrossRef]
- Chen, X.; Pan, J.; Wang, S.; Hong, S.; Hong, S.; He, S. The Epidemiological Trend of Acute Myeloid Leukemia in Childhood: A Population-Based Analysis. J. Cancer 2019, 10, 4824–4835. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, D.; Antoniou, E.; Waack, K. Pediatric Acute Myeloid Leukemia—Past, Present, and Future. J. Clin. Med. 2022, 11, 504. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 Family Proteins: Changing Partners in the Dance towards Death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef]
- Moldoveanu, T. Apoptotic Mitochondrial Poration by a Growing List of Pore-forming BCL-2 Family Proteins. BioEssays 2023, 45, 2200221. [Google Scholar] [CrossRef]
- Pfeffer, C.; Singh, A. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef]
- Valentin, R.; Grabow, S.; Davids, M.S. The Rise of Apoptosis: Targeting Apoptosis in Hematologic Malignancies. Blood 2018, 132, 1248–1264. [Google Scholar] [CrossRef]
- Konopleva, M.; Letai, A. BCL-2 Inhibition in AML: An Unexpected Bonus? Blood 2018, 132, 1007–1012. [Google Scholar] [CrossRef]
- Totiger, T.M.; Ghoshal, A.; Zabroski, J.; Sondhi, A.; Bucha, S.; Jahn, J.; Feng, Y.; Taylor, J. Targeted Therapy Development in Acute Myeloid Leukemia. Biomedicines 2023, 11, 641. [Google Scholar] [CrossRef]
- Mohamad Anuar, N.N.; Nor Hisam, N.S.; Liew, S.L.; Ugusman, A. Clinical Review: Navitoclax as a Pro-Apoptotic and Anti-Fibrotic Agent. Front. Pharmacol. 2020, 11, 564108. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity While Sparing Platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Fairbrother, W.J.; Leverson, J.D.; Souers, A.J. From Basic Apoptosis Discoveries to Advanced Selective BCL-2 Family Inhibitors. Nat. Rev. Drug Discov. 2017, 16, 273–284. [Google Scholar] [CrossRef]
- Deng, J.; Carlson, N.; Takeyama, K.; Dal Cin, P.; Shipp, M.; Letai, A. BH3 Profiling Identifies Three Distinct Classes of Apoptotic Blocks to Predict Response to ABT-737 and Conventional Chemotherapeutic Agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef]
- Chonghaile, T.N.; Sarosiek, K.A.; Vo, T.-T.; Ryan, J.A.; Tammareddi, A.; Moore, V.D.G.; Deng, J.; Anderson, K.C.; Richardson, P.; Tai, Y.-T.; et al. Pretreatment Mitochondrial Priming Correlates with Clinical Response to Cytotoxic Chemotherapy. Science 2011, 334, 1129–1133. [Google Scholar] [CrossRef]
- FDA Grants Regular Approval to Venetoclax in Combination for Untreated Acute Myeloid Leukemia. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-venetoclax-combination-untreated-acute-myeloid-leukemia (accessed on 1 August 2023).
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax Combined with Decitabine or Azacitidine in Treatment-Naive, Elderly Patients with Acute Myeloid Leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Jonas, B.A.; Wei, A.H.; Recher, C.; DiNardo, C.D.; Jang, J.; Pratz, K.; Panayiotidis, P.; Montesinos, P.; Yeh, S.; Ivanov, V.; et al. Timing of Response with Venetoclax Combination Treatment in Patients with Newly Diagnosed Acute Myeloid Leukemia. Am. J. Hematol. 2022, 97, E299. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Hoff, F.W.; Patel, P.A.; Belli, A.J.; Hansen, E.; Foss, H.; Schulte, M.; Wang, C.-K.; Madanat, Y.F. Real-World Outcomes of Frontline Venetoclax-Based Therapy in Older Adults with Acute Myeloid Leukemia: An Analysis Utilizing EHR Data. Leuk. Lymphoma 2023, 64, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Thirman, M.J.; Pratz, K.W.; Garcia, J.S.; Recher, C.; Pullarkat, V.; Kantarjian, H.M.; DiNardo, C.D.; Dail, M.; Duan, Y.; et al. Impact of FLT3 Mutation on Outcomes after Venetoclax and Azacitidine for Patients with Treatment-Naïve Acute Myeloid Leukemia. Clin. Cancer Res. 2022, 28, 2744–2752. [Google Scholar] [CrossRef] [PubMed]
- Murphy, T.; Yee, K.W.L. Cytarabine and Daunorubicin for the Treatment of Acute Myeloid Leukemia. Expert Opin. Pharmacother. 2017, 18, 1765–1780. [Google Scholar] [CrossRef] [PubMed]
- Lachowiez, C.A.; Reville, P.K.; Kantarjian, H.; Jabbour, E.; Borthakur, G.; Daver, N.; Loghavi, S.; Furudate, K.; Xiao, L.; Pierce, S.; et al. Venetoclax Combined with Induction Chemotherapy in Patients with Newly Diagnosed Acute Myeloid Leukaemia: A Post-Hoc, Propensity Score-Matched, Cohort Study. Lancet Haematol. 2022, 9, e350–e360. [Google Scholar] [CrossRef]
- Wang, H.; Mao, L.; Yang, M.; Qian, P.; Lu, H.; Tong, H.; Xie, W.; Zhou, D.; Huang, X.; Wang, Y.; et al. Venetoclax plus 3 + 7 Daunorubicin and Cytarabine Chemotherapy as First-Line Treatment for Adults with Acute Myeloid Leukaemia: A Multicentre, Single-Arm, Phase 2 Trial. Lancet Haematol. 2022, 9, e415–e424. [Google Scholar] [CrossRef]
- Suo, X.; Zheng, F.; Wang, D.; Zhao, L.; Liu, J.; Li, L.; Zhang, Z.; Zhang, C.; Li, Y.; Yang, S.; et al. Venetoclax Combined with Daunorubicin and Cytarabine (2 + 6) as Induction Treatment in Adults with Newly Diagnosed Acute Myeloid Leukemia: A Phase 2, Multicenter, Single-Arm Trial. Exp. Hematol. Oncol. 2023, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Lachowiez, C.A.; Atluri, H.; DiNardo, C.D. Advancing the Standard: Venetoclax Combined with Intensive Induction and Consolidation Therapy for Acute Myeloid Leukemia. Ther. Adv. Hematol. 2022, 13, 204062072210939. [Google Scholar] [CrossRef] [PubMed]
- Lachowiez, C.; DiNardo, C.D.; Takahashi, K.; Loghavi, S.; Xiao, L.-C.; Kadia, T.M.; Daver, N.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax Combined with FLAG-IDA Induction and Consolidation in Newly Diagnosed Acute Myeloid Leukemia. Blood 2021, 138 (Suppl. S1), 701. [Google Scholar] [CrossRef]
- Wolach, O.; Frisch, A.; Shargian, L.; Yeshurun, M.; Apel, A.; Vainstein, V.; Moshe, Y.; Shimony, S.; Amit, O.; Bar-On, Y.; et al. Venetoclax in Combination with FLAG-IDA-Based Protocol for Patients with Acute Myeloid Leukemia: A Real-World Analysis. Ann. Hematol. 2022, 101, 1719–1726. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, B.; Deng, L.; Qiao, Y.; Jian, J. The Efficacy and Adverse Events of Venetoclax in Combination with Hypomethylating Agents Treatment for Patients with Acute Myeloid Leukemia and Myelodysplastic Syndrome: A Systematic Review and Meta-Analysis. Hematology 2020, 25, 414–423. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Xiao, L.; Kadia, T.; Daver, N.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax Combined With FLAG-IDA Induction and Consolidation in Newly Diagnosed and Relapsed or Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2021, 39, 2768–2778. [Google Scholar] [CrossRef]
- Karol, S.E.; Alexander, T.B.; Budhraja, A.; Pounds, S.B.; Canavera, K.; Wang, L.; Wolf, J.; Klco, J.M.; Mead, P.E.; Das Gupta, S.; et al. Venetoclax in Combination with Cytarabine with or without Idarubicin in Children with Relapsed or Refractory Acute Myeloid Leukaemia: A Phase 1, Dose-Escalation Study. Lancet Oncol. 2020, 21, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Trabal, A.; Gibson, A.; He, J.; McCall, D.; Roth, M.; Nuñez, C.; Garcia, M.; Buzbee, M.; Toepfer, L.; Bidikian, A.; et al. Venetoclax for Acute Myeloid Leukemia in Pediatric Patients: A Texas Medical Center Experience. Cancers 2023, 15, 1983. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide Metabolism Mediates Resistance to Venetoclax in Relapsed Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2020, 27, 748–764.e4. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty Acid Metabolism Underlies Venetoclax Resistance in Acute Myeloid Leukemia Stem Cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Yu, L.; Yang, L. Mechanisms of Venetoclax Resistance and Solutions. Front. Oncol. 2022, 12, 1005659. [Google Scholar] [CrossRef]
- Saliba, A.N.; John, A.J.; Kaufmann, S.H. Resistance to Venetoclax and Hypomethylating Agents in Acute Myeloid Leukemia. Cancer Drug Resist. 2020, 4, 125–142. [Google Scholar] [CrossRef]
- Xu, Y.; Ye, H. Progress in Understanding the Mechanisms of Resistance to BCL-2 Inhibitors. Exp. Hematol. Oncol. 2022, 11, 31. [Google Scholar] [CrossRef]
- Waclawiczek, A.; Leppä, A.-M.; Renders, S.; Stumpf, K.; Reyneri, C.; Betz, B.; Janssen, M.; Shahswar, R.; Donato, E.; Karpova, D.; et al. Combinatorial BCL2 Family Expression in Acute Myeloid Leukemia Stem Cells Predicts Clinical Response to Azacitidine/Venetoclax. Cancer Discov. 2023, 13, 1408–1427. [Google Scholar] [CrossRef]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef]
- Lin, K.H.; Winter, P.S.; Xie, A.; Roth, C.; Martz, C.A.; Stein, E.M.; Anderson, G.R.; Tingley, J.P.; Wood, K.C. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci. Rep. 2016, 6, 27696. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, K.; Maiti, A.; Konopleva, M. Targeting FLT3 Mutation in Acute Myeloid Leukemia: Current Strategies and Future Directions. Cancers 2023, 15, 2312. [Google Scholar] [CrossRef]
- Paul, M.K.; Mukhopadhyay, A.K. Tyrosine Kinase—Role and Significance in Cancer. Int. J. Med. Sci. 2004, 1, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R. The Molecular Mechanisms behind Activation of FLT3 in Acute Myeloid Leukemia and Resistance to Therapy by Selective Inhibitors. Biochim. Biophys. Acta BBA Rev. Cancer 2022, 1877, 188666. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.-S.; Kim, H.-J. FLT3 Mutations in Acute Myeloid Leukemia: A Review Focusing on Clinically Applicable Drugs. Blood Res. 2022, 57 (Suppl. S1), S32–S36. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.C.; Agarwal, S.; Ahmad, H.; Amin, K.; Bewersdorf, J.P.; Zeidan, A.M. A Review of FLT3 Inhibitors in Acute Myeloid Leukemia. Blood Rev. 2022, 52, 100905. [Google Scholar] [CrossRef] [PubMed]
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The Genetics of Myelodysplastic Syndrome: From Clonal Haematopoiesis to Secondary Leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Daver, N.; Perl, A.E.; Maly, J.; Levis, M.; Ritchie, E.; Litzow, M.; McCloskey, J.; Smith, C.C.; Schiller, G.; Bradley, T.; et al. Venetoclax Plus Gilteritinib for FLT3-Mutated Relapsed/Refractory Acute Myeloid Leukemia. J. Clin. Oncol. 2022, 40, 4048–4059. [Google Scholar] [CrossRef]
- Daver, N.; Venugopal, S.; Ravandi, F. FLT3 Mutated Acute Myeloid Leukemia: 2021 Treatment Algorithm. Blood Cancer J. 2021, 11, 104. [Google Scholar] [CrossRef]
- Schneider, F.; Hoster, E.; Unterhalt, M.; Schneider, S.; Dufour, A.; Benthaus, T.; Mellert, G.; Zellmeier, E.; Kakadia, P.M.; Bohlander, S.K.; et al. The FLT3ITD MRNA Level Has a High Prognostic Impact in NPM1 Mutated, but Not in NPM1 Unmutated, AML with a Normal Karyotype. Blood 2012, 119, 4383–4386. [Google Scholar] [CrossRef] [PubMed]
- Cismowski, M.J. Tyrosine Kinase Inhibitors. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. ISBN 978-0-08-055232-3. [Google Scholar]
- Fathi, A.T.; Le, L.; Hasserjian, R.P.; Sadrzadeh, H.; Levis, M.; Chen, Y.-B. FLT3 Inhibitor-Induced Neutrophilic Dermatosis. Blood 2013, 122, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Ghelli Luserna Di Rorà, A.; Jandoubi, M.; Martinelli, G.; Simonetti, G. Targeting Proliferation Signals and the Cell Cycle Machinery in Acute Leukemias: Novel Molecules on the Horizon. Molecules 2023, 28, 1224. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 Mutations in AML: Review of Current Knowledge and Evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, A.S.; Yilmaz, M.; Kanagal-Shamanna, R.; Loghavi, S.; Kadia, T.M.; DiNardo, C.D.; Borthakur, G.; Konopleva, M.; Pierce, S.A.; Wang, S.A.; et al. Patterns of Resistance Differ in Patients with Acute Myeloid Leukemia Treated with Type I versus Type II FLT3 Inhibitors. Blood Cancer Discov. 2021, 2, 125–134. [Google Scholar] [CrossRef]
- Arai, Y.; Chi, S.; Minami, Y.; Yanada, M. FLT3-Targeted Treatment for Acute Myeloid Leukemia. Int. J. Hematol. 2022, 116, 351–363. [Google Scholar] [CrossRef]
- Perrone, S.; Ottone, T.; Zhdanovskaya, N.; Molica, M. How Acute Myeloid Leukemia (AML) Escapes from FMS-Related Tyrosine Kinase 3 (FLT3) Inhibitors? Still an Overrated Complication? Cancer Drug Resist. 2023, 6, 223–238. [Google Scholar] [CrossRef]
- Abbas, H.A.; Alfayez, M.; Kadia, T.; Ravandi-Kashani, F.; Daver, N. Midostaurin in Acute Myeloid Leukemia: An Evidence-Based Review And Patient Selection. Cancer Manag. Res. 2019, 11, 8817–8828. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Lazarus, H.M.; Cooper, B.W. Midostaurin: A Novel Therapeutic Agent for Patients with FLT3-Mutated Acute Myeloid Leukemia and Systemic Mastocytosis. Ther. Adv. Hematol. 2017, 8, 245–261. [Google Scholar] [CrossRef]
- Virchis, A.; Ganeshaguru, K.; Hart, S.; Jones, D.; Fletcher, L.; Wright, F.; Wickremasinghe, R.; Man, A.; Csermak, K.; Meyer, T.; et al. A Novel Treatment Approach for Low Grade Lymphoproliferative Disorders Using PKC412 (CGP41251), an Inhibitor of Protein Kinase C. Hematol. J. 2002, 3, 131–136. [Google Scholar] [CrossRef]
- Millward, M.J.; House, C.; Bowtell, D.; Webster, L.; Olver, I.N.; Gore, M.; Copeman, M.; Lynch, K.; Yap, A.; Wang, Y.; et al. The Multikinase Inhibitor Midostaurin (PKC412A) Lacks Activity in Metastatic Melanoma: A Phase IIA Clinical and Biologic Study. Br. J. Cancer 2006, 95, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Eder, J.P.; Garcia-Carbonero, R.; Clark, J.W.; Supko, J.G.; Puchalski, T.A.; Ryan, D.P.; Deluca, P.; Wozniak, A.; Campbell, A.; Rothermel, J.; et al. A Phase I Trial of Daily Oral 4′-N-Benzoyl-Staurosporine in Combination with Protracted Continuous Infusion 5-Fluorouracil in Patients with Advanced Solid Malignancies. Investig. New Drugs 2004, 22, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Levis, M. Midostaurin Approved for FLT3-Mutated AML. Blood 2017, 129, 3403–3406. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Mandrekar, S.J.; Huebner, L.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Döhner, K.; et al. Midostaurin Reduces Relapse in FLT3-Mutant Acute Myeloid Leukemia: The Alliance CALGB 10603/RATIFY Trial. Leukemia 2021, 35, 2539–2551. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weber, D.; Krzykalla, J.; Fiedler, W.; Wulf, G.; Salih, H.; Lübbert, M.; Kühn, M.W.M.; Schroeder, T.; Salwender, H.; et al. Midostaurin plus Intensive Chemotherapy for Younger and Older Patients with AML and FLT3 Internal Tandem Duplications. Blood Adv. 2022, 6, 5345–5355. [Google Scholar] [CrossRef] [PubMed]
- Solana-Altabella, A.; Ballesta-López, O.; Megías-Vericat, J.E.; Martínez-Cuadrón, D.; Montesinos, P. Emerging FLT3 Inhibitors for the Treatment of Acute Myeloid Leukemia. Expert Opin. Emerg. Drugs 2022, 27, 1–18. [Google Scholar] [CrossRef]
- Midostaurin. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Gong, L.; Giacomini, M.M.; Giacomini, C.; Maitland, M.L.; Altman, R.B.; Klein, T.E. PharmGKB Summary: Sorafenib Pathways. Pharmacogenetics Genom. 2017, 27, 240–246. [Google Scholar] [CrossRef]
- Xuan, L.; Wang, Y.; Huang, F.; Fan, Z.; Xu, Y.; Sun, J.; Xu, N.; Deng, L.; Li, X.; Liang, X.; et al. Sorafenib Maintenance in Patients with FLT3-ITD Acute Myeloid Leukaemia Undergoing Allogeneic Haematopoietic Stem-Cell Transplantation: An Open-Label, Multicentre, Randomised Phase 3 Trial. Lancet Oncol. 2020, 21, 1201–1212. [Google Scholar] [CrossRef]
- Burchert, A.; Bug, G.; Fritz, L.V.; Finke, J.; Stelljes, M.; Röllig, C.; Wollmer, E.; Wäsch, R.; Bornhäuser, M.; Berg, T.; et al. Sorafenib Maintenance After Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myeloid Leukemia With FLT3 –Internal Tandem Duplication Mutation (SORMAIN). J. Clin. Oncol. 2020, 38, 2993–3002. [Google Scholar] [CrossRef]
- Pollard, J.A.; Alonzo, T.A.; Gerbing, R.; Brown, P.; Fox, E.; Choi, J.; Fisher, B.; Hirsch, B.; Kahwash, S.; Getz, K.; et al. Sorafenib in Combination With Standard Chemotherapy for Children With High Allelic Ratio FLT3 /ITD+ Acute Myeloid Leukemia: A Report From the Children’s Oncology Group Protocol AAML1031. J. Clin. Oncol. 2022, 40, 2023–2035. [Google Scholar] [CrossRef]
- Molica, M.; Perrone, S.; Rossi, M. Gilteritinib: The Story of a Proceeding Success into Hard-to-Treat FLT3-Mutated AML Patients. J. Clin. Med. 2023, 12, 3647. [Google Scholar] [CrossRef]
- Ezelarab, H.A.A.; Ali, T.F.S.; Abbas, S.H.; Hassan, H.A.; Beshr, E.A.M. Indole-Based FLT3 Inhibitors and Related Scaffolds as Potential Therapeutic Agents for Acute Myeloid Leukemia. BMC Chem. 2023, 17, 73. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Larson, R.A.; Podoltsev, N.A.; Strickland, S.; Wang, E.S.; Atallah, E.; Schiller, G.J.; Martinelli, G.; Neubauer, A.; Sierra, J.; et al. Follow-up of Patients with R/R FLT3- Mutation–Positive AML Treated with Gilteritinib in the Phase 3 ADMIRAL Trial. Blood 2022, 139, 3366–3375. [Google Scholar] [CrossRef] [PubMed]
- Perl, A.E.; Hosono, N.; Montesinos, P.; Podoltsev, N.; Martinelli, G.; Panoskaltsis, N.; Recher, C.; Smith, C.C.; Levis, M.J.; Strickland, S.; et al. Clinical Outcomes in Patients with Relapsed/Refractory FLT3-Mutated Acute Myeloid Leukemia Treated with Gilteritinib Who Received Prior Midostaurin or Sorafenib. Blood Cancer J. 2022, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- Numan, Y.; Abdel Rahman, Z.; Grenet, J.; Boisclair, S.; Bewersdorf, J.P.; Collins, C.; Barth, D.; Fraga, M.; Bixby, D.L.; Zeidan, A.M.; et al. Gilteritinib Clinical Activity in Relapsed/Refractory FLT3 Mutated ACUTE MYELOID LEUKEMIA Previously Treated with FLT3 Inhibitors. Am. J. Hematol. 2022, 97, 322–328. [Google Scholar] [CrossRef]
- Wang, E.S.; Montesinos, P.; Minden, M.D.; Lee, J.-H.; Heuser, M.; Naoe, T.; Chou, W.-C.; Laribi, K.; Esteve, J.; Altman, J.K.; et al. Phase 3 Trial of Gilteritinib plus Azacitidine vs Azacitidine for Newly Diagnosed FLT3 Mut + AML Ineligible for Intensive Chemotherapy. Blood 2022, 140, 1845–1857. [Google Scholar] [CrossRef]
- Cortes, J.E.; Kantarjian, H.; Foran, J.M.; Ghirdaladze, D.; Zodelava, M.; Borthakur, G.; Gammon, G.; Trone, D.; Armstrong, R.C.; James, J.; et al. Phase I Study of Quizartinib Administered Daily to Patients With Relapsed or Refractory Acute Myeloid Leukemia Irrespective of FMS-Like Tyrosine Kinase 3–Internal Tandem Duplication Status. J. Clin. Oncol. 2013, 31, 3681–3687. [Google Scholar] [CrossRef]
- Erba, H.P.; Montesinos, P.; Kim, H.-J.; Patkowska, E.; Vrhovac, R.; Žák, P.; Wang, P.-N.; Mitov, T.; Hanyok, J.; Kamel, Y.M.; et al. Quizartinib plus Chemotherapy in Newly Diagnosed Patients with FLT3-Internal-Tandem-Duplication-Positive Acute Myeloid Leukaemia (QuANTUM-First): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2023, 401, 1571–1583. [Google Scholar] [CrossRef]
- FDA Approves Quizartinib for Newly Diagnosed Acute Myeloid Leukemia. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-quizartinib-newly-diagnosed-acute-myeloid-leukemia (accessed on 1 August 2023).
- Zhou, F.; Ge, Z.; Chen, B. Quizartinib (AC220): A Promising Option for Acute Myeloid Leukemia. Drug Des. Dev. Ther. 2019, 13, 1117–1125. [Google Scholar] [CrossRef]
- Cortes, J.E.; Khaled, S.; Martinelli, G.; Perl, A.E.; Ganguly, S.; Russell, N.; Krämer, A.; Dombret, H.; Hogge, D.; Jonas, B.A.; et al. Quizartinib versus Salvage Chemotherapy in Relapsed or Refractory FLT3-ITD Acute Myeloid Leukaemia (QuANTUM-R): A Multicentre, Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 984–997. [Google Scholar] [CrossRef]
- Cortes, J.; Perl, A.E.; Döhner, H.; Kantarjian, H.; Martinelli, G.; Kovacsovics, T.; Rousselot, P.; Steffen, B.; Dombret, H.; Estey, E.; et al. Quizartinib, an FLT3 Inhibitor, as Monotherapy in Patients with Relapsed or Refractory Acute Myeloid Leukaemia: An Open-Label, Multicentre, Single-Arm, Phase 2 Trial. Lancet Oncol. 2018, 19, 889–903. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Nguyen, B.; Li, L.; Brown, P.; Levis, M.; Leahy, D.; Small, D. Mutations of FLT3/ITD Confer Resistance to Multiple Tyrosine Kinase Inhibitors. Leukemia 2013, 27, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, M.; Minami, Y.; Kuzume, A.; Chi, S. Mechanisms Underlying Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Biomedicines 2020, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Heidel, F.; Solem, F.K.; Breitenbuecher, F.; Lipka, D.B.; Kasper, S.; Thiede, M.H.; Brandts, C.; Serve, H.; Roesel, J.; Giles, F.; et al. Clinical Resistance to the Kinase Inhibitor PKC412 in Acute Myeloid Leukemia by Mutation of Asn-676 in the FLT3 Tyrosine Kinase Domain. Blood 2006, 107, 293–300. [Google Scholar] [CrossRef]
- Zhang, H.; Savage, S.; Schultz, A.R.; Bottomly, D.; White, L.; Segerdell, E.; Wilmot, B.; McWeeney, S.K.; Eide, C.A.; Nechiporuk, T.; et al. Clinical Resistance to Crenolanib in Acute Myeloid Leukemia Due to Diverse Molecular Mechanisms. Nat. Commun. 2019, 10, 244. [Google Scholar] [CrossRef]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.-S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous Resistance to Quizartinib in Acute Myeloid Leukemia Revealed by Single-Cell Analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Smith, C.C.; Lin, K.; Stecula, A.; Sali, A.; Shah, N.P. FLT3 D835 Mutations Confer Differential Resistance to Type II FLT3 Inhibitors. Leukemia 2015, 29, 2390–2392. [Google Scholar] [CrossRef]
- Schmalbrock, L.K.; Dolnik, A.; Cocciardi, S.; Sträng, E.; Theis, F.; Jahn, N.; Panina, E.; Blätte, T.J.; Herzig, J.; Skambraks, S.; et al. Clonal Evolution of Acute Myeloid Leukemia with FLT3-ITD Mutation under Treatment with Midostaurin. Blood 2021, 137, 3093–3104. [Google Scholar] [CrossRef]
- Lindblad, O.; Cordero, E.; Puissant, A.; Macaulay, L.; Ramos, A.; Kabir, N.N.; Sun, J.; Vallon-Christersson, J.; Haraldsson, K.; Hemann, M.T.; et al. Aberrant Activation of the PI3K/mTOR Pathway Promotes Resistance to Sorafenib in AML. Oncogene 2016, 35, 5119–5131. [Google Scholar] [CrossRef]
- Rummelt, C.; Gorantla, S.P.; Meggendorfer, M.; Charlet, A.; Endres, C.; Döhner, K.; Heidel, F.H.; Fischer, T.; Haferlach, T.; Duyster, J.; et al. Activating JAK-Mutations Confer Resistance to FLT3 Kinase Inhibitors in FLT3-ITD Positive AML in Vitro and in Vivo. Leukemia 2021, 35, 2017–2029. [Google Scholar] [CrossRef]
- Patel, R.K.; Weir, M.C.; Shen, K.; Snyder, D.; Cooper, V.S.; Smithgall, T.E. Expression of Myeloid Src-Family Kinases Is Associated with Poor Prognosis in AML and Influences Flt3-ITD Kinase Inhibitor Acquired Resistance. PLoS ONE 2019, 14, e0225887. [Google Scholar] [CrossRef]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-Regulates MCL-1 to Promote Survival of Stem Cells in Acute Myeloid Leukemia via FLT3-ITD–Specific STAT5 Activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef]
- O’Reilly, E.; Dhami, S.P.S.; Baev, D.V.; Ortutay, C.; Halpin-McCormick, A.; Morrell, R.; Santocanale, C.; Samali, A.; Quinn, J.; O’Dwyer, M.E.; et al. Repression of Mcl-1 Expression by the CDC7/CDK9 Inhibitor PHA-767491 Overcomes Bone Marrow Stroma-Mediated Drug Resistance in AML. Sci. Rep. 2018, 8, 15752. [Google Scholar] [CrossRef] [PubMed]
- Kohl, T.M.; Hellinger, C.; Ahmed, F.; Buske, C.; Hiddemann, W.; Bohlander, S.K.; Spiekermann, K. BH3 Mimetic ABT-737 Neutralizes Resistance to FLT3 Inhibitor Treatment Mediated by FLT3-Independent Expression of BCL2 in Primary AML Blasts. Leukemia 2007, 21, 1763–1772. [Google Scholar] [CrossRef]
- Hunter, H.M.; Pallis, M.; Seedhouse, C.H.; Grundy, M.; Gray, C.; Russell, N.H. The Expression of P-glycoprotein in AML Cells with FLT3 Internal Tandem Duplications Is Associated with Reduced Apoptosis in Response to FLT3 Inhibitors. Br. J. Haematol. 2004, 127, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yang, X.; Knapper, S.; White, P.; Smith, B.D.; Galkin, S.; Small, D.; Burnett, A.; Levis, M. FLT3 Ligand Impedes the Efficacy of FLT3 Inhibitors in Vitro and in Vivo. Blood 2011, 117, 3286–3293. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-T.; Hernandez, D.; Alonso, S.; Gao, M.; Su, M.; Ghiaur, G.; Levis, M.J.; Jones, R.J. Role of CYP3A4 in Bone Marrow Microenvironment–Mediated Protection of FLT3/ITD AML from Tyrosine Kinase Inhibitors. Blood Adv. 2019, 3, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Kivioja, J.; Malani, D.; Kumar, A.; Kontro, M.; Parsons, A.; Kallioniemi, O.; Heckman, C.A. FLT3-ITD Allelic Ratio and HLF Expression Predict FLT3 Inhibitor Efficacy in Adult AML. Sci. Rep. 2021, 11, 23565. [Google Scholar] [CrossRef]
- Malani, D.; Kumar, A.; Brück, O.; Kontro, M.; Yadav, B.; Hellesøy, M.; Kuusanmäki, H.; Dufva, O.; Kankainen, M.; Eldfors, S.; et al. Implementing a Functional Precision Medicine Tumor Board for Acute Myeloid Leukemia. Cancer Discov. 2022, 12, 388–401. [Google Scholar] [CrossRef]
- Waldeck, S.; Rassner, M.; Keye, P.; Follo, M.; Herchenbach, D.; Endres, C.; Charlet, A.; Andrieux, G.; Salzer, U.; Boerries, M.; et al. CCL5 Mediates Target-kinase Independent Resistance to FLT3 Inhibitors in FLT3-ITD-positive AML. Mol. Oncol. 2020, 14, 779–794. [Google Scholar] [CrossRef]
- Tecik, M.; Adan, A. Therapeutic Targeting of FLT3 in Acute Myeloid Leukemia: Current Status and Novel Approaches. OncoTargets Ther. 2022, 15, 1449–1478. [Google Scholar] [CrossRef] [PubMed]
- Kontro, M.; Kumar, A.; Majumder, M.M.; Eldfors, S.; Parsons, A.; Pemovska, T.; Saarela, J.; Yadav, B.; Malani, D.; Fløisand, Y.; et al. HOX Gene Expression Predicts Response to BCL-2 Inhibition in Acute Myeloid Leukemia. Leukemia 2017, 31, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Karjalainen, R.; Liu, M.; Kumar, A.; He, L.; Malani, D.; Parsons, A.; Kontro, M.; Kallioniemi, O.; Porkka, K.; Heckman, C.A. Elevated Expression of S100A8 and S100A9 Correlates with Resistance to the BCL-2 Inhibitor Venetoclax in AML. Leukemia 2019, 33, 2548–2553. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Tiong, I.S.; Quaglieri, A.; MacRaild, S.; Loghavi, S.; Brown, F.C.; Thijssen, R.; Pomilio, G.; Ivey, A.; Salmon, J.M.; et al. Molecular Patterns of Response and Treatment Failure after Frontline Venetoclax Combinations in Older Patients with AML. Blood 2020, 135, 791–803. [Google Scholar] [CrossRef]
- Bhatt, S.; Pioso, M.S.; Olesinski, E.A.; Yilma, B.; Ryan, J.A.; Mashaka, T.; Leutz, B.; Adamia, S.; Zhu, H.; Kuang, Y.; et al. Reduced Mitochondrial Apoptotic Priming Drives Resistance to BH3 Mimetics in Acute Myeloid Leukemia. Cancer Cell 2020, 38, 872–890.e6. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Konopleva, M.Y. A Venetoclax Bench-to-Bedside Story. Nat. Cancer 2021, 2, 3–5. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, J.; Ma, J.; Xie, C.; Edwards, H.; Wang, G.; Caldwell, J.T.; Xiang, S.; Zhang, X.; Chu, R.; et al. Binding of Released Bim to Mcl-1 Is a Mechanism of Intrinsic Resistance to ABT-199 Which Can Be Overcome by Combination with Daunorubicin or Cytarabine in AML Cells. Clin. Cancer Res. 2016, 22, 4440–4451. [Google Scholar] [CrossRef]
- Ong, F.; Kim, K.; Konopleva, M.Y. Venetoclax Resistance: Mechanistic Insights and Future Strategies. Cancer Drug Resist. 2022, 5, 380–400. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, S.; Qiao, X.; Knight, T.; Edwards, H.; Polin, L.; Kushner, J.; Dzinic, S.H.; White, K.; Wang, G.; et al. Inhibition of Bcl-2 Synergistically Enhances the Antileukemic Activity of Midostaurin and Gilteritinib in Preclinical Models of FLT3-Mutated Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 6815–6826. [Google Scholar] [CrossRef]
- Zhu, R.; Li, L.; Nguyen, B.; Seo, J.; Wu, M.; Seale, T.; Levis, M.; Duffield, A.; Hu, Y.; Small, D. FLT3 Tyrosine Kinase Inhibitors Synergize with BCL-2 Inhibition to Eliminate FLT3/ITD Acute Leukemia Cells through BIM Activation. Signal Transduct. Target. Ther. 2021, 6, 186. [Google Scholar] [CrossRef]
- Mali, R.S.; Zhang, Q.; De Filippis, R.A.; Cavazos, A.; Kuruvilla, V.M.; Raman, J.; Mody, V.; Choo, E.F.; Dail, M.; Shah, N.P.; et al. Venetoclax Combines Synergistically with FLT3 Inhibition to Effectively Target Leukemic Cells in FLT3-ITD+ Acute Myeloid Leukemia Models. Haematologica 2020, 106, 1034–1046. [Google Scholar] [CrossRef]
- Janssen, M.; Schmidt, C.; Bruch, P.-M.; Blank, M.F.; Rohde, C.; Waclawiczek, A.; Heid, D.; Renders, S.; Göllner, S.; Vierbaum, L.; et al. Venetoclax Synergizes with Gilteritinib in FLT3 Wild-Type High-Risk Acute Myeloid Leukemia by Suppressing MCL-1. Blood 2022, 140, 2594–2610. [Google Scholar] [CrossRef]
- Fang, D.D.; Zhu, H.; Tang, Q.; Wang, G.; Min, P.; Wang, Q.; Li, N.; Yang, D.; Zhai, Y. FLT3 Inhibition by Olverembatinib (HQP1351) Downregulates MCL-1 and Synergizes with BCL-2 Inhibitor Lisaftoclax (APG-2575) in Preclinical Models of FLT3-ITD Mutant Acute Myeloid Leukemia. Transl. Oncol. 2022, 15, 101244. [Google Scholar] [CrossRef]
- Aldoss, I.; Zhang, J.; Mei, M.; Al Malki, M.M.; Arslan, S.; Ngo, D.; Aribi, A.; Ali, H.; Sandhu, K.; Salhotra, A.; et al. Venetoclax and Hypomethylating Agents in FLT3-mutated Acute Myeloid Leukemia. Am. J. Hematol. 2020, 95, 1193–1199. [Google Scholar] [CrossRef]
- Morsia, E.; McCullough, K.; Joshi, M.; Cook, J.; Alkhateeb, H.B.; Al-Kali, A.; Begna, K.; Elliott, M.; Hogan, W.; Litzow, M.; et al. Venetoclax and Hypomethylating Agents in Acute Myeloid Leukemia: Mayo Clinic Series on 86 Patients. Am. J. Hematol. 2020, 95, 1511–1521. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Maiti, A.; DiNardo, C.D.; Wang, S.A.; Jorgensen, J.; Kadia, T.M.; Daver, N.G.; Short, N.J.; Yilmaz, M.; Pemmaraju, N.; Borthakur, G.; et al. Prognostic Value of Measurable Residual Disease after Venetoclax and Decitabine in Acute Myeloid Leukemia. Blood Adv. 2021, 5, 1876–1883. [Google Scholar] [CrossRef]
- Maiti, A.; DiNardo, C.D.; Daver, N.G.; Rausch, C.R.; Ravandi, F.; Kadia, T.M.; Pemmaraju, N.; Borthakur, G.; Bose, P.; Issa, G.C.; et al. Triplet Therapy with Venetoclax, FLT3 Inhibitor and Decitabine for FLT3-Mutated Acute Myeloid Leukemia. Blood Cancer J. 2021, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Kantarjian, H.; Short, N.J.; Reville, P.; Konopleva, M.; Kadia, T.; DiNardo, C.; Borthakur, G.; Pemmaraju, N.; Maiti, A.; et al. Hypomethylating Agent and Venetoclax with FLT3 Inhibitor “Triplet” Therapy in Older/Unfit Patients with FLT3 Mutated AML. Blood Cancer J. 2022, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Muftuoglu, M.; Kantarjian, H.M.; Dinardo, C.D.; Kadia, T.M.; Konopleva, M.; Borthakur, G.; Pemmaraju, N.; Short, N.J.; Alvarado Valero, Y.; et al. Quizartinib (QUIZ) with Decitabine (DAC) and Venetoclax (VEN) Is Active in Patients (Pts) with FLT3-ITD Mutated Acute Myeloid Leukemia (AML): A Phase I/II Clinical Trial. J. Clin. Oncol. 2022, 40 (Suppl. S16), 7036. [Google Scholar] [CrossRef]
- Short, N.; DiNardo, C.D.; Daver, N.; Macaron, W.; Yilmaz, M.; Borthakur, G.; Montalban-Bravo, G.; Garcia-Manero, G.; Issa, G.C.; Sasaki, K.; et al. Updated Results from a Phase I/II Study of the Triplet Combination of Azacitidine, Venetoclax and Gilteritinib for Patients with FLT3 -Mutated Acute Myeloid Leukemia. Blood 2022, 140 (Suppl. S1), 2007–2009. [Google Scholar] [CrossRef]
- Chua, C.C.; Anstee, N.S.; Enjeti, A.K.; Hiwase, D.; Marlton, P.; Bajel, A.; Tan, S.Y.; Morris, E.S.; Ma, C.-K.-K.; Grove, C.; et al. High Deliverability of a Midostaurin Triplet Regimen Incorporating Venetoclax and Low Dose Cytarabine in Non-Adverse Cytogenetic Risk Acute Myeloid Leukaemia: A Sub-Analysis of the Australasian Leukaemia Lymphoma Group (ALLG) Intervene Study. Blood 2022, 140 (Suppl. S1), 3362–3364. [Google Scholar] [CrossRef]
- Bergua-Burgues, J.M.; Rodríguez-Veiga, R.; Cano, I.; Vall-llovera, F.; García-Guiñon, A.; Gómez-Estruch, J.; Colorado, M.; Casas-Avilés, I.; Esteve-Reyner, J.; Verdugo, M.V.; et al. P512: Preliminary results of ven-a-qui study: A phase 1-2 trial to assess the safety and efficacy of the combination of azacitidine or low-dose cytarabine with venetoclax and quizartinib in newly diagnosed. HemaSphere 2022, 6, 411–412. [Google Scholar] [CrossRef]
- Short, N.; Macaron, W.; Dinardo, C.; Daver, N.; Yilmaz, M.; Borthakur, G.; Montalban-Bravo, G.; Garcia-Manero, G.; Issa, G.; Sasaki, K.; et al. P485: Azacitidine, venetoclax and gilteritinib for patients with newly diagnosed flt3-mutated acute myeloid leukemia: A subgroup analysis from a phase ii study. HemaSphere 2023, 7, e535260f. [Google Scholar] [CrossRef]
- Abuasab, T.; Kantarjian, H.; Garcia-Manero, G.; Montalban-Bravo, G.; Alvarado, Y.; Yilmaz, M.; Pemmaraju, N.; Chien, K.S.; Mohamed, S.F.; Daver, N.; et al. Phase II Study of Cladribine, Idarubicin, Cytarabine (CLIA) Plus Gilteritinib in Patients with FLT3 Mutated Acute Myeloid Leukemia (AML). Blood 2021, 138 (Suppl. S1), 2330. [Google Scholar] [CrossRef]
- Jin, H.; Zhang, Y.; Yu, S.; Du, X.; Xu, N.; Shao, R.; Lin, D.; Chen, Y.; Xiao, J.; Sun, Z.; et al. Venetoclax Combined with Azacitidine and Homoharringtonine in Relapsed/Refractory AML: A Multicenter, Phase 2 Trial. J. Hematol. Oncol. 2023, 16, 42. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Authors | Treatment Regimen | Phase of a Trial | Number of Participants | Median Age | Outcomes | Survivability |
|---|---|---|---|---|---|---|
| Abhishek Maiti et al. [125] | DEC + VEN + FLT3i | II | ND AML—12 | 70 | CRc—92% | 2-year OS—80% |
| R/R AML—13 | 50 | CRc—62% | mOS—6.8 months | |||
| Musa Yilmaz et al. [126] | LIC + VEN + FLT3i | retrospective | ND AML—27 | 69 | CR/CRi—93% | mOS—NR with a median 12-month follow-up |
| Musa Yilmaz et al. [127] | DEC + VEN + QUIZ | I/II | ND AML—5 | 69 | CR/CRi—100% | mOS—14.5 months |
| R/R AML—23 | - | CR/CRi—78% | mOS—7.6 months | |||
| Nicholas J. Short et al. [128] | AZA + VEN + GILT | I/II | ND AML—21 | 68 | ORR—100% | 6-month OS—95%, estimated 1-year OS—80% |
| R/R AML—19 | 68 | ORR—74% | mOS—5.8 months, 1-year OS—27% | |||
| Chong Chyn Chua et al. [129] | LDAC + VEN + MIDO | Ib/II | ND AML—18 | 77 | ORR—77.8% | mOS—NR, with a median 18-months follow-up |
| Juan Miguel Bergua-Bergues et al. [130] | AZA + VEN + QUIZ or LDAC + VEN + QUIZ | I/II | ND AML—45 | 76.5 | ORR—54% | unknown |
| Nicholas Short et al. [131] | AZA + VEN + GILT | II | ND AML—30 | 71 | CR/CRi—96% | Estimated 1-year OS—86% |
| Tareq Abuasab et al. [132] | CLIA + VEN + GILT | II | ND AML—8 | 55 | CR—88% | mOS—22.4 months |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milnerowicz, S.; Maszewska, J.; Skowera, P.; Stelmach, M.; Lejman, M. AML under the Scope: Current Strategies and Treatment Involving FLT3 Inhibitors and Venetoclax-Based Regimens. Int. J. Mol. Sci. 2023, 24, 15849. https://doi.org/10.3390/ijms242115849
Milnerowicz S, Maszewska J, Skowera P, Stelmach M, Lejman M. AML under the Scope: Current Strategies and Treatment Involving FLT3 Inhibitors and Venetoclax-Based Regimens. International Journal of Molecular Sciences. 2023; 24(21):15849. https://doi.org/10.3390/ijms242115849
Chicago/Turabian StyleMilnerowicz, Szymon, Julia Maszewska, Paulina Skowera, Magdalena Stelmach, and Monika Lejman. 2023. "AML under the Scope: Current Strategies and Treatment Involving FLT3 Inhibitors and Venetoclax-Based Regimens" International Journal of Molecular Sciences 24, no. 21: 15849. https://doi.org/10.3390/ijms242115849