

Nipbl Haploinsufficiency Leads to Delayed Outflow Tract Septation and Aortic Valve Thickening

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
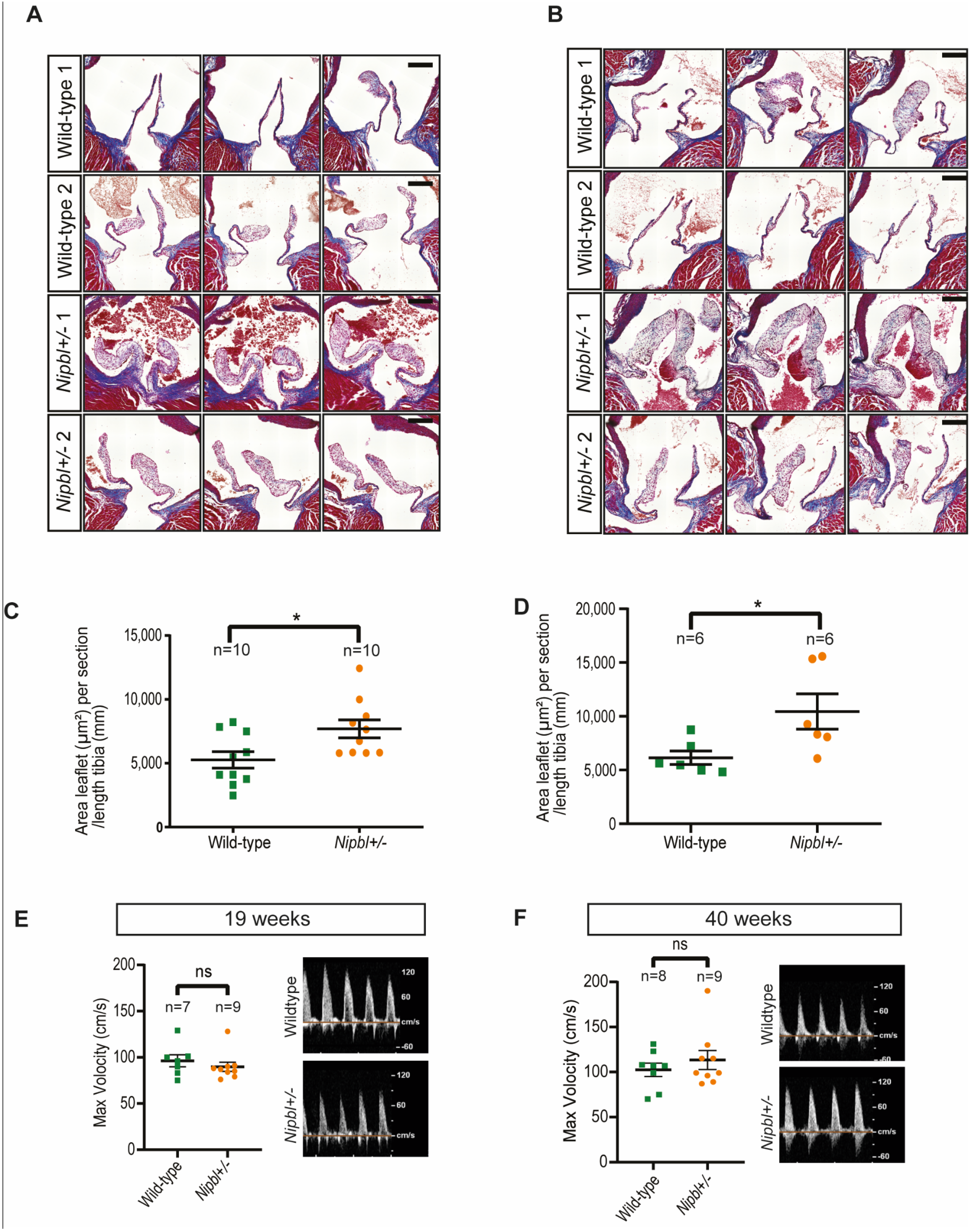
2.1. Aortic Valve Thickening in Nipbl+/- Mice
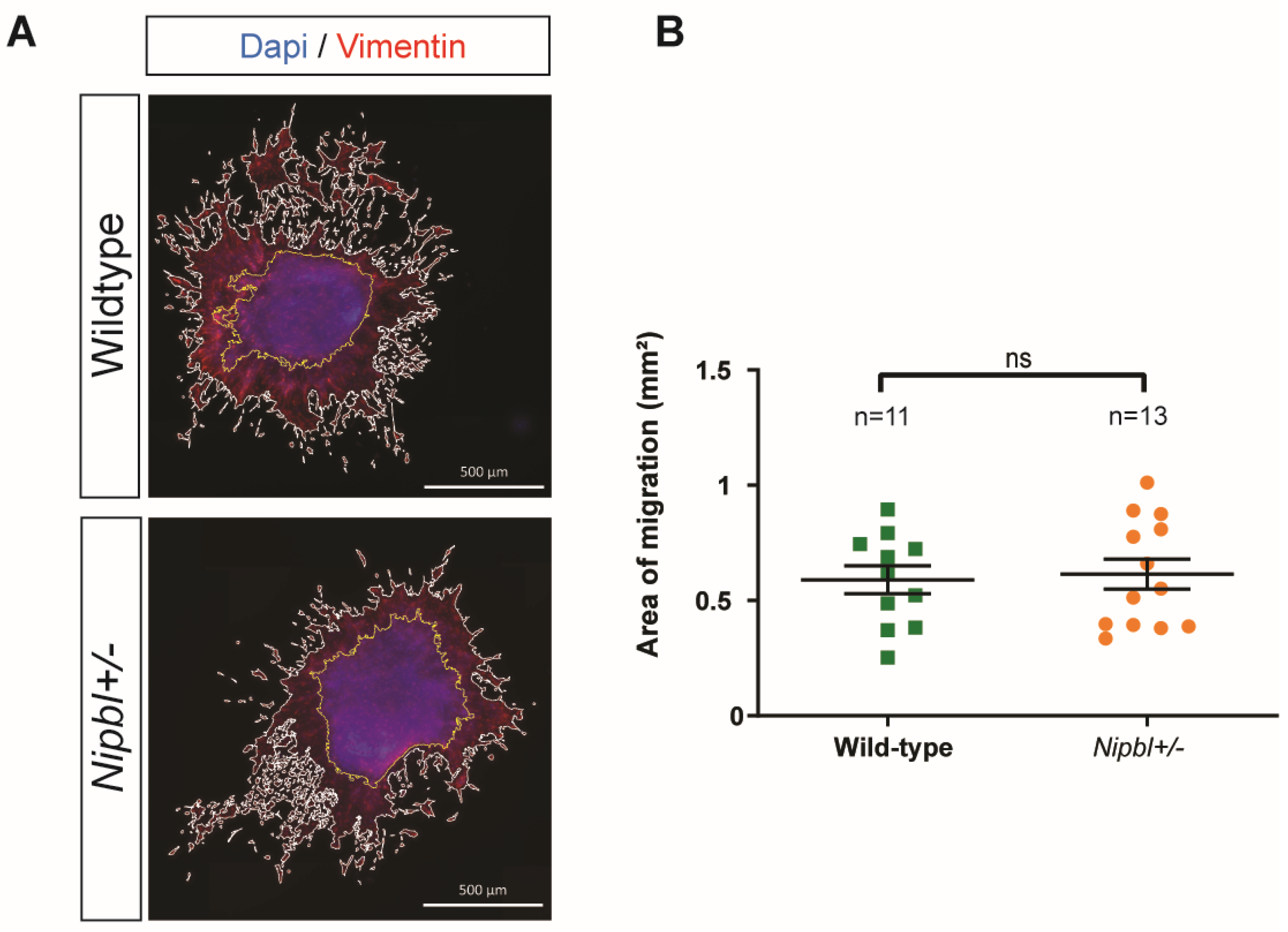
2.2. Normal Endothelial-to-Mesenchymal Transition in Nipbl+/- Outflow Tracts
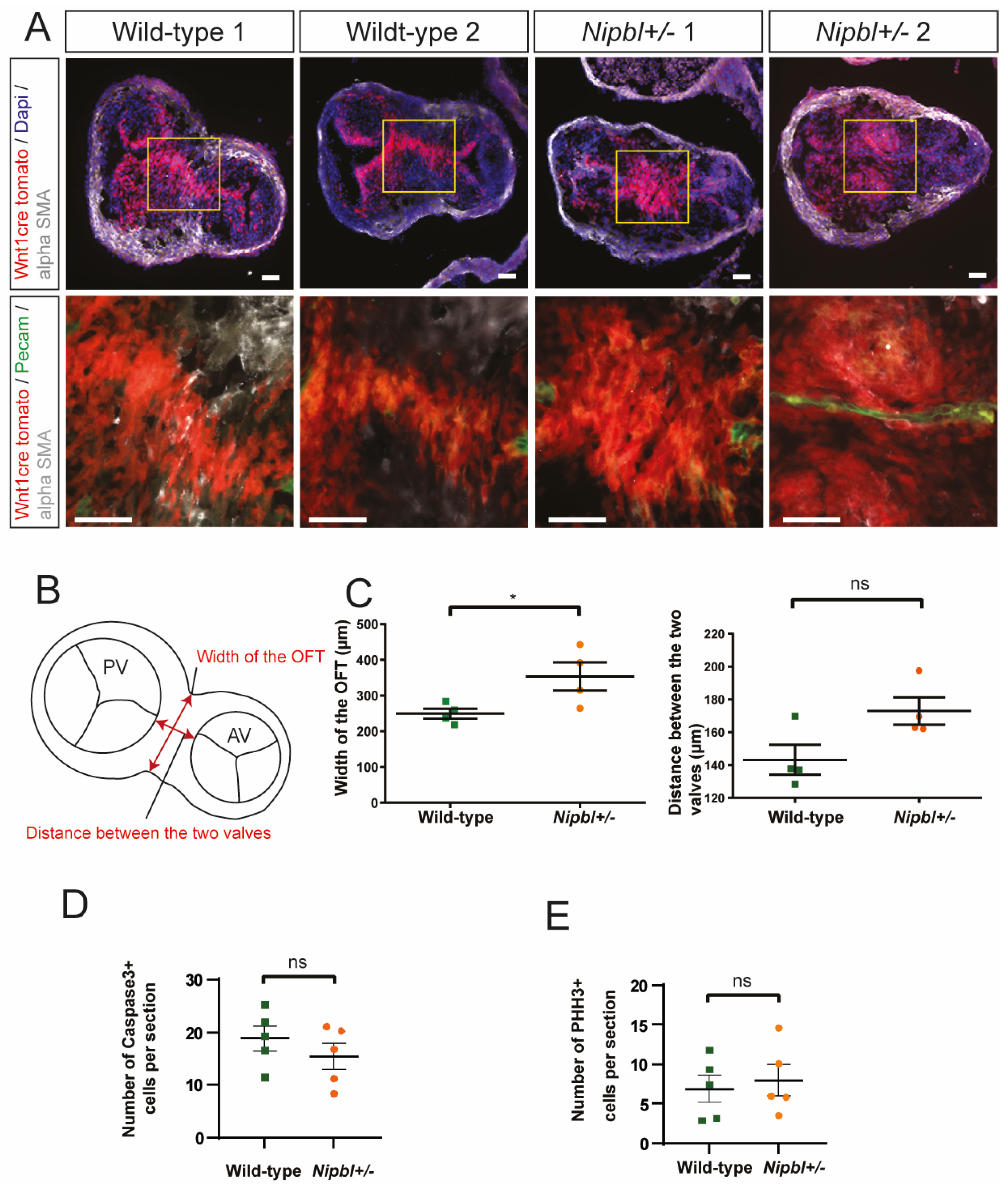
2.3. Cardiac Neural Crest Cell Condensation Is Delayed in Nipbl+/- OFTs
3. Discussion
4. Materials and Methods
- Primers:
- Nipbl:
- Fwd: GCCGATTCGCCCAGAGTTT
- Rev: CCTGAAGTTCTGGAATGGTGT
- HPRT:
- Fwd CTG GTG AAA AGG ACC TCT CG
- Rev TGG CAA CAT CAA CAG GAC TC
- Antibodies
| Antigen | Antibody | Dilution |
| Vimentin | Abcam, ab139878 | 1/100 |
| Pecam | BD pharmigen, 550274 | 1/100 |
| αSMA | Abcam, ab5694 | 1/200 |
| Phospho-Histone H3 | CST, 3377T | 1/500 |
| Cleaved Caspase-3 | CST, 9661T | 1/500 |
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nkomo, V.T.; Gardin, J.M.; Skelton, T.N.; Gottdiener, J.S.; Scott, C.G.; Enriquez-Sarano, M. Burden of valvular heart diseases: A population-based study. Lancet 2006, 368, 1005–1011. [Google Scholar] [CrossRef]
- Baumgartner, H.; Falk, V.; Bax, J.J.; De Bonis, M.; Hamm, C.; Holm, P.J.; Iung, B.; Lancellotti, P.; Lansac, E.; Rodriguez Muñoz, D.; et al. 2017 ESC/EACTS Guidelines for the management of valvular heart disease. Eur. Heart J. 2017, 38, 2739–2791. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.G.; Dvir, D. Transcatheter aortic valve replacement for bioprosthetic aortic valve failure: The valve-in-valve procedure. Circulation 2013, 127, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Javier Delmo, E.M.; Hetzer, R. Mitral valve surgery in infants and children. Transl. Pediatr. 2020, 9, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.L.; Gale, T.F.; Stewart, D.E. Neural crest cells contribute to normal aorticopulmonary septation. Science 1983, 220, 1059–1061. [Google Scholar] [CrossRef]
- Armstrong, E.J.; Bischoff, J. Heart valve development: Endothelial cell signaling and differentiation. Circ. Res. 2004, 95, 459–470. [Google Scholar] [CrossRef]
- de Lange, F.J.; Moorman, A.F.M.; Anderson, R.H.; Männer, J.; Soufan, A.T.; de Gier-de Vries, C.; Schneider, M.D.; Webb, S.; van den Hoff, M.J.B.; Christoffels, V.M. Lineage and morphogenetic analysis of the cardiac valves. Circ. Res. 2004, 95, 645–654. [Google Scholar] [CrossRef]
- Kagey, M.H.; Newman, J.J.; Bilodeau, S.; Zhan, Y.; Orlando, D.A.; van Berkum, N.L.; Ebmeier, C.C.; Goossens, J.; Rahl, P.B.; Levine, S.S.; et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature 2010, 467, 430–435. [Google Scholar] [CrossRef]
- Moore-Morris, T.; van Vliet, P.P.; Andelfinger, G.; Puceat, M. Role of Epigenetics in Cardiac Development and Congenital Diseases. Physiol. Rev. 2018, 98, 2453–2475. [Google Scholar] [CrossRef]
- Kawauchi, S.; Calof, A.L.; Santos, R.; Lopez-Burks, M.E.; Young, C.M.; Hoang, M.P.; Chua, A.; Lao, T.; Lechner, M.S.; Daniel, J.A.; et al. Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/-) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet. 2009, 5, e1000650. [Google Scholar] [CrossRef]
- Garcia, P.; Fernandez-Hernandez, R.; Cuadrado, A.; Coca, I.; Gomez, A.; Maqueda, M.; Latorre-Pellicer, A.; Puisac, B.; Ramos, F.J.; Sandoval, J.; et al. Disruption of NIPBL/Scc2 in Cornelia de Lange Syndrome provokes cohesin genome-wide redistribution with an impact in the transcriptome. Nat. Commun. 2021, 12, 4551. [Google Scholar] [CrossRef]
- Mills, J.A.; Herrera, P.S.; Kaur, M.; Leo, L.; McEldrew, D.; Tintos-Hernandez, J.A.; Rajagopalan, R.; Gagne, A.; Zhang, Z.; Ortiz-Gonzalez, X.R.; et al. Nipbl+/- haploinsufficiency reveals a constellation of transcriptome disruptions in the pluripotent and cardiac states. Sci. Rep. 2018, 8, 1056. [Google Scholar] [CrossRef] [PubMed]
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.-M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, E.T.; Wang, T.-J.; Lisgo, S.; Bamshad, M.J.; Strachan, T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 2004, 36, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Krantz, I.D.; McCallum, J.; DeScipio, C.; Kaur, M.; Gillis, L.A.; Yaeger, D.; Jukofsky, L.; Wasserman, N.; Bottani, A.; Morris, C.A.; et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004, 36, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Chatfield, K.C.; Schrier, S.A.; Li, J.; Clark, D.; Kaur, M.; Kline, A.D.; Deardorff, M.A.; Jackson, L.S.; Goldmuntz, E.; Krantz, I.D. Congenital heart disease in Cornelia de Lange syndrome: Phenotype and genotype analysis. Am. J. Med. Genet. A 2012, 158A, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Barisic, I.; Tokic, V.; Loane, M.; Bianchi, F.; Calzolari, E.; Garne, E.; Wellesley, D.; Dolk, H.; EUROCAT Working Group. Descriptive epidemiology of Cornelia de Lange syndrome in Europe. Am. J. Med. Genet. A 2008, 146A, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Kawauchi, S.; Jacobs, R.E.; Lopez-Burks, M.E.; Choi, H.; Wikenheiser, J.; Hallgrimsson, B.; Jamniczky, H.A.; Fraser, S.E.; Lander, A.D.; et al. Conditional Creation and Rescue of Nipbl-Deficiency in Mice Reveals Multiple Determinants of Risk for Congenital Heart Defects. PLoS Biol. 2016, 14, e2000197. [Google Scholar] [CrossRef]
- Schuster, K.; Leeke, B.; Meier, M.; Wang, Y.; Newman, T.; Burgess, S.; Horsfield, J.A. A neural crest origin for cohesinopathy heart defects. Hum. Mol. Genet. 2015, 24, 7005–7016. [Google Scholar] [CrossRef]
- Smith, T.G.; Laval, S.; Chen, F.; Rock, M.J.; Strachan, T.; Peters, H. Neural crest cell-specific inactivation of Nipbl or Mau2 during mouse development results in a late onset of craniofacial defects. Genesis 2014, 52, 687–694. [Google Scholar] [CrossRef]
- Cosmi, J.E.; Kort, S.; Tunick, P.A.; Rosenzweig, B.P.; Freedberg, R.S.; Katz, E.S.; Applebaum, R.M.; Kronzon, I. The risk of the development of aortic stenosis in patients with “benign” aortic valve thickening. Arch. Intern. Med. 2002, 162, 2345–2347. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, L.M.; Markwald, R.R. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ. Res. 1995, 77, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Choudhry, A.; Berlan, M.; Singal, A.; Siwik, E.; Mohr, S.; Fisher, S.A. Developmental remodeling and shortening of the cardiac outflow tract involves myocyte programmed cell death. Development 1998, 125, 3809–3820. [Google Scholar] [CrossRef] [PubMed]
- Selicorni, A.; Colli, A.M.; Passarini, A.; Milani, D.; Cereda, A.; Cerutti, M.; Maitz, S.; Alloni, V.; Salvini, L.; Galli, M.A.; et al. Analysis of congenital heart defects in 87 consecutive patients with Brachmann-de Lange syndrome. Am. J. Med. Genet. A 2009, 149A, 1268–1272. [Google Scholar] [CrossRef]
- Kline, A.D.; Krantz, I.D.; Deardorff, M.A.; Shirahige, K.; Dorsett, D.; Gerton, J.L.; Wu, M.; Mehta, D.; Mills, J.A.; Carrico, C.S.; et al. Cornelia de Lange syndrome and molecular implications of the cohesin complex: Abstracts from the 7th biennial scientific and educational symposium 2016. Am. J. Med. Genet. A 2017, 173, 1172–1185. [Google Scholar] [CrossRef]
- van den Hoff, M.J.; Moorman, A.F.; Ruijter, J.M.; Lamers, W.H.; Bennington, R.W.; Markwald, R.R.; Wessels, A. Myocardialization of the cardiac outflow tract. Dev. Biol. 1999, 212, 477–490. [Google Scholar] [CrossRef]
- de la Pompa, J.L.; Epstein, J.A. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev. Cell 2012, 22, 244–254. [Google Scholar] [CrossRef]
- Zuin, J.; Franke, V.; van Ijcken, W.F.J.; van der Sloot, A.; Krantz, I.D.; van der Reijden, M.I.J.A.; Nakato, R.; Lenhard, B.; Wendt, K.S. A cohesin-independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet. 2014, 10, e1004153. [Google Scholar] [CrossRef]
- El-Brolosy, M.A.; Kontarakis, Z.; Rossi, A.; Kuenne, C.; Günther, S.; Fukuda, N.; Kikhi, K.; Boezio, G.L.M.; Takacs, C.M.; Lai, S.-L.; et al. Genetic compensation triggered by mutant mRNA degradation. Nature 2019, 568, 193–197. [Google Scholar] [CrossRef]
- LaHaye, S.; Majumdar, U.; Yasuhara, J.; Koenig, S.N.; Matos-Nieves, A.; Kumar, R.; Garg, V. Developmental origins for semilunar valve stenosis identified in mice harboring congenital heart disease-associated GATA4 mutation. Dis. Model. Mech. 2019, 12, dmm036764. [Google Scholar] [CrossRef]
- Jain, R.; Engleka, K.A.; Rentschler, S.L.; Manderfield, L.J.; Li, L.; Yuan, L.; Epstein, J.A. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J. Clin. Investig. 2011, 121, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Plein, A.; Calmont, A.; Fantin, A.; Denti, L.; Anderson, N.A.; Scambler, P.J.; Ruhrberg, C. Neural crest-derived SEMA3C activates endothelial NRP1 for cardiac outflow tract septation. J. Clin. Investig. 2015, 125, 2661–2676. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Selever, J.; Wang, D.; Lu, M.-F.; Moses, K.A.; Schwartz, R.J.; Martin, J.F. Bmp4 signaling is required for outflow-tract septation and branchial-arch artery remodeling. Proc. Natl. Acad. Sci. USA 2004, 101, 4489–4494. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Rowitch, D.H.; Soriano, P.; McMahon, A.P.; Sucov, H.M. Fate of the mammalian cardiac neural crest. Development 2000, 127, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Madisen, L.; Zwingman, T.A.; Sunkin, S.M.; Oh, S.W.; Zariwala, H.A.; Gu, H.; Ng, L.L.; Palmiter, R.D.; Hawrylycz, M.J.; Jones, A.R.; et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 2010, 13, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Papoutsi, T.; Odelin, G.; Moore-Morris, T.; Pucéat, M.; de la Pompa, J.L.; Robert, B.; Zaffran, S. Msx1CreERT2 knock-In allele: A useful tool to target embryonic and adult cardiac valves. Genesis 2015, 53, 337–345. [Google Scholar] [CrossRef]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boulet, F.; Odelin, G.; Harrington, A.; Moore-Morris, T. Nipbl Haploinsufficiency Leads to Delayed Outflow Tract Septation and Aortic Valve Thickening. Int. J. Mol. Sci. 2023, 24, 15564. https://doi.org/10.3390/ijms242115564
Boulet F, Odelin G, Harrington A, Moore-Morris T. Nipbl Haploinsufficiency Leads to Delayed Outflow Tract Septation and Aortic Valve Thickening. International Journal of Molecular Sciences. 2023; 24(21):15564. https://doi.org/10.3390/ijms242115564
Chicago/Turabian StyleBoulet, Fanny, Gaelle Odelin, Alenca Harrington, and Thomas Moore-Morris. 2023. "Nipbl Haploinsufficiency Leads to Delayed Outflow Tract Septation and Aortic Valve Thickening" International Journal of Molecular Sciences 24, no. 21: 15564. https://doi.org/10.3390/ijms242115564