
Leigh Syndrome: Spectrum of Molecular Defects and Clinical Features in Russia
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
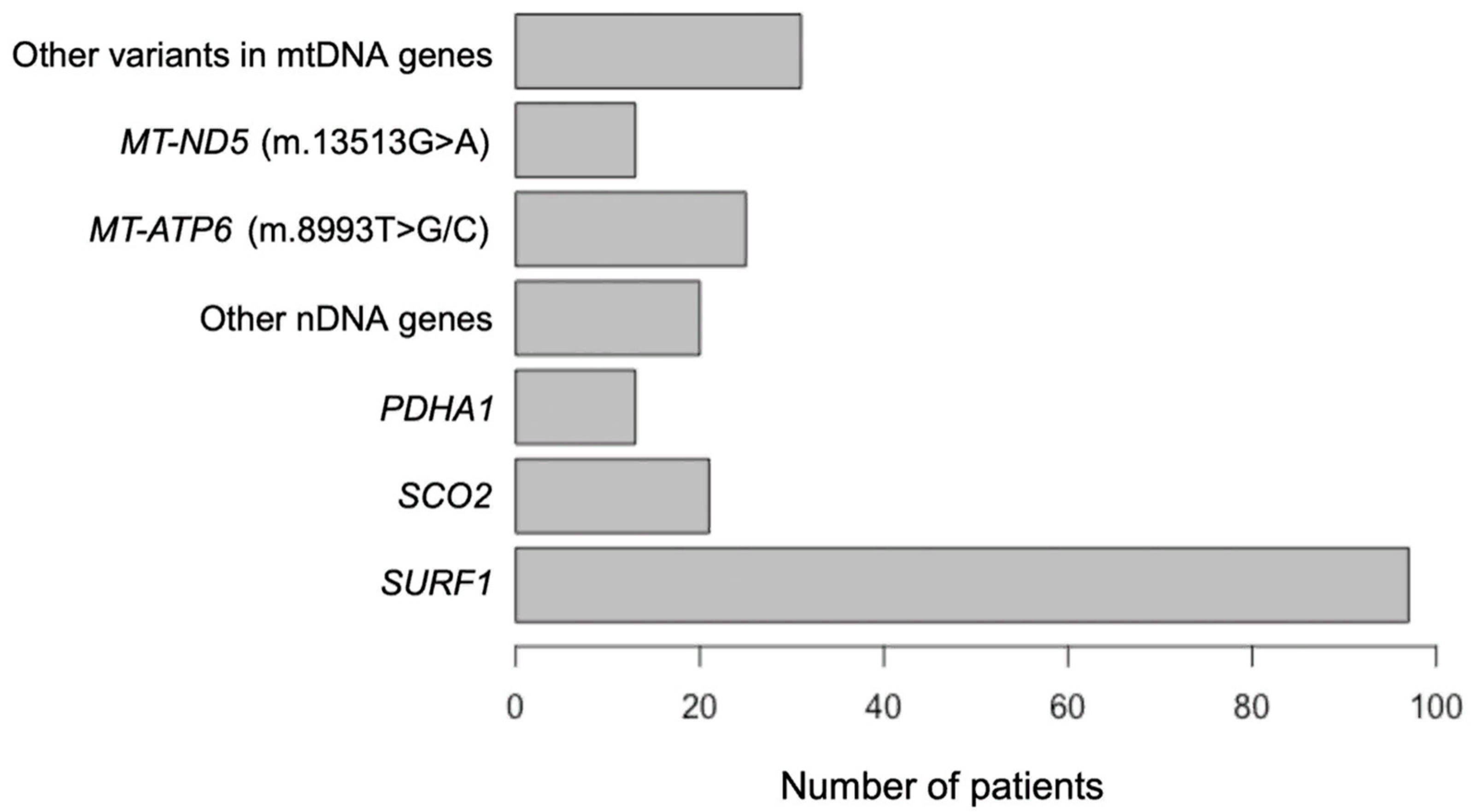
2.1. Demography
2.2. Molecular, Clinical, Radiological and Biochemical Findings in Patients with Nuclear Genes’ Variants
2.2.1. The SURF1 Gene
2.2.2. The SCO2 Gene
2.2.3. The PDHA1 Gene
2.3. Molecular, Clinical, Radiological and Biochemical Findings in Patients with mtDNA Genes’ Variatns
2.4. Clinical Cases
3. Discussion
4. Materials and Methods
4.1. Editorial Policies and Ethical Considerations
4.2. Patients
4.3. DNA and RNA Extraction, Analysis and Sanger Sequencing
4.4. Metabolite Analyses
4.5. WES and WGS
4.6. Minigene Assay
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial Diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Schlieben, L.D.; Prokisch, H. The Dimensions of Primary Mitochondrial Disorders. Front. Cell Dev. Biol. 2020, 8, 600079. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.A.P.F.; Teixeira, S.R.; Martin-Saavedra, J.S.; Guimarães Gonçalves, F.; Lo Russo, F.; Muraresku, C.; McCormick, E.M.; Falk, M.J.; Zolkipli-Cunningham, Z.; Ganetzky, R.; et al. Pediatric Leigh Syndrome: Neuroimaging Features and Genetic Correlations. Ann. Neurol. 2020, 88, 218–232. [Google Scholar] [CrossRef]
- Leigh, D. Subacute necrotizing encephalomyelopathy in an infant. J. Neurol. Neurosurg. Psychiatry 1951, 14, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.H.; Mayatepek, E.; Morava, E.; Distelmaier, F. A Guide to Diagnosis and Treatment of Leigh Syndrome. J. Neurol. Neurosurg. Psychiatry 2014, 85, 257–265. [Google Scholar] [CrossRef]
- Gerards, M.; Sallevelt, S.C.E.H.; Smeets, H.J.M. Leigh Syndrome: Resolving the Clinical and Genetic Heterogeneity Paves the Way for Treatment Options. Mol. Genet. Metab. 2016, 117, 300–312. [Google Scholar] [CrossRef]
- Finsterer, J. Leigh and Leigh-Like Syndrome in Children and Adults. Pediatr. Neurol. 2008, 39, 223–235. [Google Scholar] [CrossRef]
- Schubert Baldo, M.; Vilarinho, L. Molecular Basis of Leigh Syndrome: A Current Look. Orphanet J. Rare Dis. 2020, 15, 31. [Google Scholar] [CrossRef] [Green Version]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh Syndrome: One Disorder, more than 75 Monogenic Causes: Leigh Syndrome. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef]
- Stenton, S.L.; Kremer, L.S.; Kopajtich, R.; Ludwig, C.; Prokisch, H. The Diagnosis of Inborn Errors of Metabolism by an Integrative “Multi-omics” Approach: A Perspective Encompassing Genomics, Transcriptomics, and Proteomics. J. Inherit. Metab. Dis. 2020, 43, 25–35. [Google Scholar] [CrossRef]
- Guillen Sacoto, M.J.; Tchasovnikarova, I.A.; Torti, E.; Forster, C.; Andrew, E.H.; Anselm, I.; Baranano, K.W.; Briere, L.C.; Cohen, J.S.; Craigen, W.J.; et al. De Novo Variants in the ATPase Module of MORC2 Cause a Neurodevelopmental Disorder with Growth Retardation and Variable Craniofacial Dysmorphism. Am. J. Hum. Genet. 2020, 107, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Naess, K.; Freyer, C.; Bruhn, H.; Wibom, R.; Malm, G.; Nennesmo, I.; von Döbeln, U.; Larsson, N.-G. MtDNA Mutations Are a Common Cause of Severe Disease Phenotypes in Children with Leigh Syndrome. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 484–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sue, C.M.; Karadimas, C.; Checcarelli, N.; Tanji, K.; Papadopoulou, L.C.; Pallotti, F.; Guo, F.L.; Shanske, S.; Hirano, M.; De Vivo, D.C.; et al. Differential Features of Patients with Mutations in Two COX Assembly Genes, SURF-1 and SCO2. Ann. Neurol. 2000, 47, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, M.; Horvath, R.; Horn, N.; Auer, D.P.; Macmillan, C.; Peters, J.; Gerbitz, K.-D.; Kraegeloh-Mann, I.; Muntau, A.; Karcagi, V.; et al. Homozygosity (E140K) in SCO2 Causes Delayed Infantile Onset of Cardiomyopathy and Neuropathy. Neurology 2001, 57, 1440–1446. [Google Scholar] [CrossRef]
- Pronicka, E.; Piekutowska-Abramczuk, D.; Szymańska-Dębińska, T.; Bielecka, L.; Kowalski, P.; Łuczak, S.; Karkucińska-Więckowska, A.; Migdał, M.; Kubalska, J.; Zimowski, J.; et al. The Natural History of SCO2 Deficiency in 36 Polish Children Confirmed the Genotype–Phenotype Correlation. Mitochondrion 2013, 13, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Monlleo-Neila, L.; del Toro, M.; Bornstein, B.; Garcia-Arumi, E.; Sarrias, A.; Roig-Quilis, M.; Munell, F. Leigh Syndrome and the Mitochondrial m.13513G>A Mutation: Expanding the Clinical Spectrum. J. Child Neurol. 2013, 28, 1531–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreeva, N.A.; Murakhovskaya, Y.K.; Tsygankova, P.G.; Krilova, T.D.; Sheremet, N.L. Leber’s Hereditary Optic Neuropathy Clinical Features in Patients with Mitochondrial DNA m.13513G>A Candidate Mutation. Vestn. Oftalmol. 2022, 138, 208. [Google Scholar] [CrossRef]
- Wedatilake, Y.; Brown, R.M.; McFarland, R.; Yaplito-Lee, J.; Morris, A.A.M.; Champion, M.; Jardine, P.E.; Clarke, A.; Thorburn, D.R.; Taylor, R.W.; et al. SURF1 Deficiency: A Multi-Centre Natural History Study. Orphanet J. Rare Dis. 2013, 8, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Østergaard, E.; Bradinova, I.; Ravn, S.H.; Hansen, F.J.; Simeonov, E.; Christensen, E.; Wibrand, F.; Schwartz, M. Hypertrichosis in Patients WithSURF1 Mutations. Am. J. Med. Genet. 2005, 138, 384–388. [Google Scholar] [CrossRef]
- Ganesan, K.; Desai, S.; Udwadia-Hegde, A.; Ursekar, M. Mitochondrial Leukodystrophy: An Unusual Manifestation of Leigh’s Disease: A Report of Three Cases and Review of the Literature. Neuroradiol. J. 2007, 20, 271–277. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Lewis, B.; Mock, D.M.; Said, H.M.; Tarailo-Graovac, M.; Mattman, A.; van Karnebeek, C.D.; Thorburn, D.R.; Rodenburg, R.J.; Christodoulou, J. Leigh-Like Syndrome Due to Homoplasmic m.8993T>G Variant with Hypocitrullinemia and Unusual Biochemical Features Suggestive of Multiple Carboxylase Deficiency (MCD). JIMD Rep. 2017, 33, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassone, E.; Rahman, S. Complex I Deficiency: Clinical Features, Biochemistry and Molecular Genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakare, A.B.; Lesnefsky, E.J.; Iyer, S. Leigh Syndrome: A Tale of Two Genomes. Front. Physiol. 2021, 12, 693734. [Google Scholar] [CrossRef] [PubMed]
- Pronicki, M.; Matyja, E.; Piekutowska-Abramczuk, D.; Szymańska-Dębińska, T.; Karkucińska-Więckowska, A.; Karczmarewicz, E.; Grajkowska, W.; Kmieć, T.; Popowska, E.; Sykut-Cegielska, J. Light and Electron Microscopy Characteristics of the Muscle of Patients with SURF1 Gene Mutations Associated with Leigh Disease. J. Clin. Pathol. 2008, 61, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Sofou, K.; De Coo, I.F.M.; Isohanni, P.; Ostergaard, E.; Naess, K.; De Meirleir, L.; Tzoulis, C.; Uusimaa, J.; De Angst, I.B.; Lönnqvist, T.; et al. A Multicenter Study on Leigh Syndrome: Disease Course and Predictors of Survival. Orphanet J. Rare Dis. 2014, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Liu, Z.; Liu, Y.; Wu, M.; Fang, F.; Deng, X.; Liu, Z.; Song, L.; Murayama, K.; Zhang, C.; et al. Novel ECHS1 Mutations in Leigh Syndrome Identified by Whole-Exome Sequencing in Five Chinese Families: Case Report. BMC Med. Genet. 2020, 21, 149. [Google Scholar] [CrossRef]
- Oláhová, M.; Hardy, S.A.; Hall, J.; Yarham, J.W.; Haack, T.B.; Wilson, W.C.; Alston, C.L.; He, L.; Aznauryan, E.; Brown, R.M.; et al. LRPPRC Mutations Cause Early-Onset Multisystem Mitochondrial Disease Outside of the French-Canadian Population. Brain 2015, 138, 3503–3519. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.-C.; El-Hattab, A.W.; Wang, J.; Li, F.-Y.; Weng, S.-W.; Craigen, W.J.; Wong, L.-J.C. SURF1-Associated Leigh Syndrome: A Case Series and Novel Mutations. Hum. Mutat. 2012, 33, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Piekutowska-Abramczuk, D.; Magner, M.; Popowska, E.; Pronicki, M.; Karczmarewicz, E.; Sykut-Cegielska, J.; Kmiec, T.; Jurkiewicz, E.; Szymanska-Debinska, T.; Bielecka, L.; et al. SURF1 Missense Mutations Promote a Mild Leigh Phenotype. Clin. Genet. 2009, 76, 195–204. [Google Scholar] [CrossRef]
- Yüksel, A.; Seven, M.; Cetincelik, Ü.; Yeşil, G.; Köksal, V. Facial Dysmorphism in Leigh Syndrome With SURF-1 Mutation and COX Deficiency. Pediatr. Neurol. 2006, 34, 486–489. [Google Scholar] [CrossRef]
- Coenen, M.J.H.; Smeitink, J.A.M.; Farhoud, M.H.; Nijtmans, L.G.J.; Rodenburg, R.; Janssen, A.; van Kaauwen, E.P.M.; Trijbels, F.J.M.; van den Heuvel, L.P. The First Patient Diagnosed with Cytochrome c Oxidase Deficient Leigh Syndrome: Progress Report. J. Inherit. Metab. Dis. 2006, 29, 212–213. [Google Scholar] [CrossRef]
- Lee, J.S.; Yoo, T.; Lee, M.; Lee, Y.; Jeon, E.; Kim, S.Y.; Lim, B.C.; Kim, K.J.; Choi, M.; Chae, J. Genetic Heterogeneity in Leigh Syndrome: Highlighting Treatable and Novel Genetic Causes. Clin. Genet. 2020, 97, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Kurkina, M.V.; Mihaylova, S.V.; Baydakova, G.V.; Saifullina, E.V.; Korostelev, S.A.; Pyankov, D.V.; Kanivets, I.V.; Yunin, M.A.; Pechatnikova, N.L.; Zakharova, E.Y. Molecular and Biochemical Study of Glutaric Aciduria Type 1 in 49 Russian Families: Nine Novel Mutations in the GCDH Gene. Metab. Brain Dis. 2020, 35, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symptoms | SURF1 | SCO2 | PDHA1 | m.8993T>G/C | m.13513G>A | Other mtDNA |
|---|---|---|---|---|---|---|
| Onset (median age, range; months) | 13.0 (3–44) | 3.0 (0–11) | 4.0 (0–18) | 4.5 (0.5–24) | 15.0 (0–132) | 14.5 (3–60) |
| Delay or regress of psychomotor development | 90.6% (29/32) | 50.0% (8/16) | 60.0% (6/10) | 72.7% (8/11) | 70.0% (7/10) | 87.5% (14/16) |
| Muscle hypotonia/weakness | 90.6% (29/32) | 75.0% (12/16) | 90.0% (9/10) | 81.8% (9/11) | 50.0% (5/10) | 62.5% (10/16) |
| Ataxia | 40.6% (13/32) | 0.0% (0/16) | 70.0% (7/10) | 18.2% (2/11) | 30.0% (3/10) | 37.5% (6/16) |
| Epileptic seizures and/or convulsions | 6.3% (2/32) | 31.3% (5/16) | 20.0% (2/10) | 36.4% (4/11) | 30.0% (3/10) | 31.3% (5/16) |
| Pyramidal symptoms | 9.4% (3/32) | 25.0% (4/16) | 20.0% (2/10) | 36.4% (4/11) | 80.0% (8/10) | 50.0% (8/16) |
| Dystonia | 18.8% (6/32) | 43.8% (7/16) | 10.0% (1/10) | 27.3% (3/11) | 30.0% (3/10) | 56.3% (9/16) |
| Myoclonus and choreiform hyperkinesis | 18.8% (6/32) | 6.3% (1/16) | 10.0% (1/10) | 36.4% (4/11) | 0.0% (0/10) | 31.3% (5/16) |
| Hypertrichosis | 65.6% (21/32) | 0.0% (0/16) | 0.0% (0/10) | 0.0% (0/11) | 20.0% (2/10) | 6.3% (1/16) |
| Nystagmus | 46.9% (15/32) | 25.0% (4/16) | 10.0% (1/10) | 18.2% (2/11) | 30.0% (3/10) | 25.0% (4/16) |
| Strabismus | 31.3% (10/32) | 25.0% (4/16) | 20.0% (2/10) | 18.2% (2/11) | 60.0% (6/10) | 37.5% (6/16) |
| Ptosis | 12.5% (4/32) | 31.3% (5/16) | 40.0% (4/10) | 9.1% (1/11) | 30.0% (3/10) | 6.3% (1/16) |
| Optic nerve atrophy | 18.9% (6/32) | 6.3% (1/16) | 10.0% (1/10) | 9.1% (1/11) | 20.0% (2/10) | 12.5% (2/16) |
| Feeding difficulties | 25.0% (8/32) | 18.8% (3/16) | 20.0% (2/10) | 9.1% (1/11) | 10.0% (1/10) | 12.5% (2/16) |
| Dysmorphic features | 18.8% (6/32) | 25.0% (4/16) | 20.0% (2/10) | 0.0% (0/11) | 40.0% (4/10) | 0.0% (0/16) |
| Liver damage | 21.9% (7/32) | 0.0% (0/16) | 20.0% (2/10) | 18.2% (2/11) | 40.0% (4/10) | 6.3% (1/16) |
| Cardiological symptoms | 21.9% (7/32) | 68.8% (11/16) | 30.0% (3/10) | 18.2% (2/11) | 70.0% (7/10) | 18.8% (3/16) |
| Hematological changes | 6.3% (2/32) | 37.5% (6/16) | 10.0% (1/10) | 18.2% (2/11) | 20.0% (2/10) | 0.0% (0/16) |
| Respiratory problems | 9.4% (3/32) | 93.8% (15/16) | 10.0% (1/10) | 63.6% (7/11) | 10.0% (1/10) | 12.5% (2/16) |
| Lesions on brain MRI: | ||||||
| Basal ganglia | 77.3% (17/22) | 30.0% (5/15) | 66.7% (6/9) | 77.8% (7/9) | 66.7% (6/9) | 85.7% (12/14) |
| Brain stem | 86.4% (19/22) | 13.3% (2/15) | 11.1% (1/9) | 44.5% (4/9) | 33.3% (3/9) | 35.7% (5/14) |
| Thalamus | 18.2% (4/22) | 0.0% (0/15) | 11.1% (1/9) | 11.1% (1/9) | 22.2% (2/9) | 28.6% (4/14) |
| Cerebellar | 40.9% (9/22) | 0.0% (0/15) | 11.1% (1/9) | 11.1% (1/9) | 0.0% (0/9) | 0.0% (0/14) |
| Spinal cord | 36.4% (8/22) | 0.0% (0/15) | 0.0% (0/9) | 0.0% (0/9) | 0.0% (0/9) | 0.0% (0/14) |
| Leukodystrophy | 18.2% (4/22) | 6.7% (1/15) | 0.0% (0/9) | 11.1% (1/9) | 11.1% (1/9) | 7.1% (1/14) |
| Lactate level (median concentration, range; mM/L) | 3.9 (2.2–8.5) | 4.9 (3.3–9.1) | 6.1 (3.0–14.0) | 5.5 (1.2–16.0) | 5.5 (3.6–10.5) | 4.4 (4.1–7.0) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kistol, D.; Tsygankova, P.; Krylova, T.; Bychkov, I.; Itkis, Y.; Nikolaeva, E.; Mikhailova, S.; Sumina, M.; Pechatnikova, N.; Kurbatov, S.; et al. Leigh Syndrome: Spectrum of Molecular Defects and Clinical Features in Russia. Int. J. Mol. Sci. 2023, 24, 1597. https://doi.org/10.3390/ijms24021597
Kistol D, Tsygankova P, Krylova T, Bychkov I, Itkis Y, Nikolaeva E, Mikhailova S, Sumina M, Pechatnikova N, Kurbatov S, et al. Leigh Syndrome: Spectrum of Molecular Defects and Clinical Features in Russia. International Journal of Molecular Sciences. 2023; 24(2):1597. https://doi.org/10.3390/ijms24021597
Chicago/Turabian StyleKistol, Denis, Polina Tsygankova, Tatiana Krylova, Igor Bychkov, Yulia Itkis, Ekaterina Nikolaeva, Svetlana Mikhailova, Maria Sumina, Natalia Pechatnikova, Sergey Kurbatov, and et al. 2023. "Leigh Syndrome: Spectrum of Molecular Defects and Clinical Features in Russia" International Journal of Molecular Sciences 24, no. 2: 1597. https://doi.org/10.3390/ijms24021597