
Urokinase System in Pathogenesis of Pulmonary Fibrosis: A Hidden Threat of COVID-19
 , , , , , , , ,
, , , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
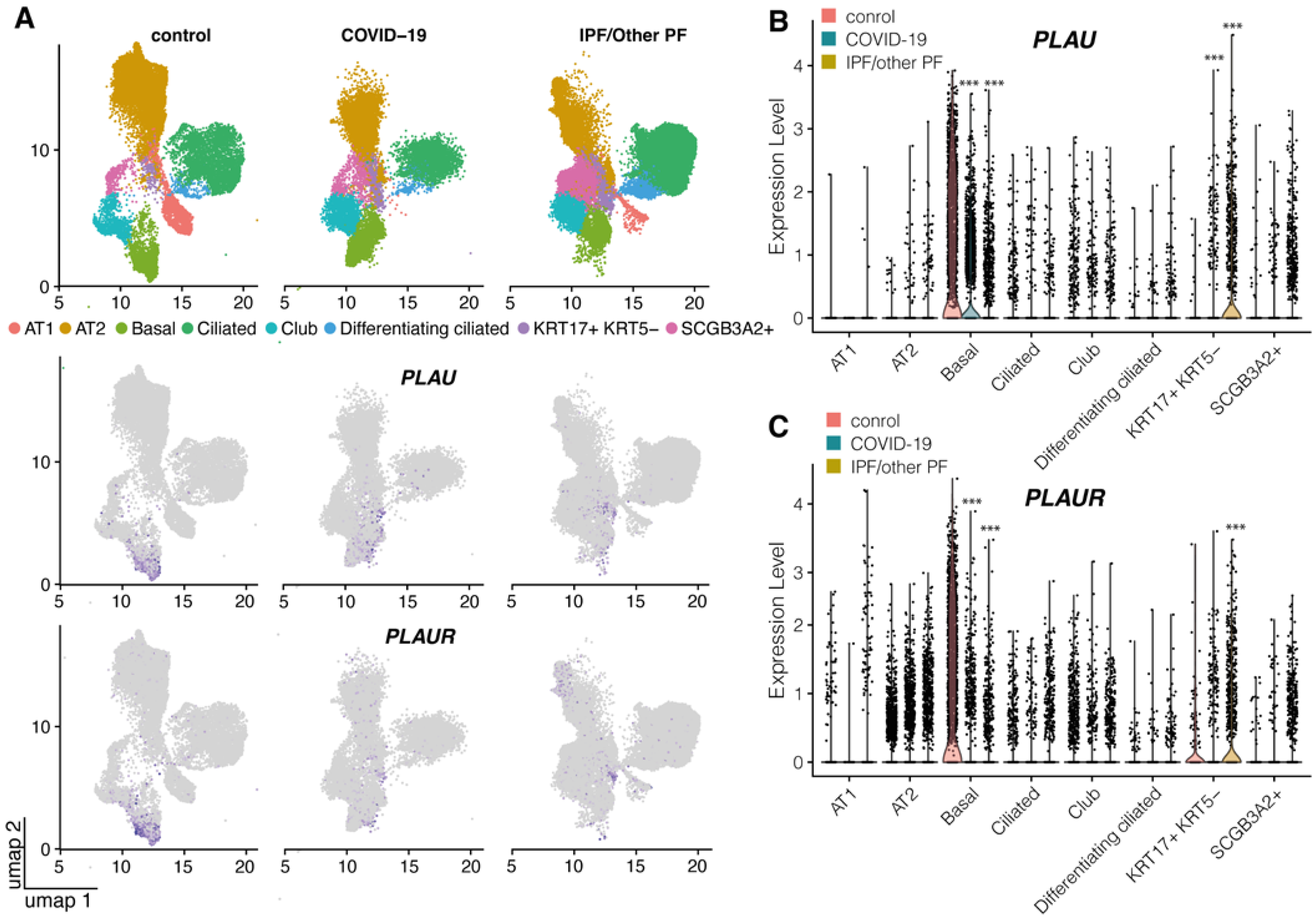
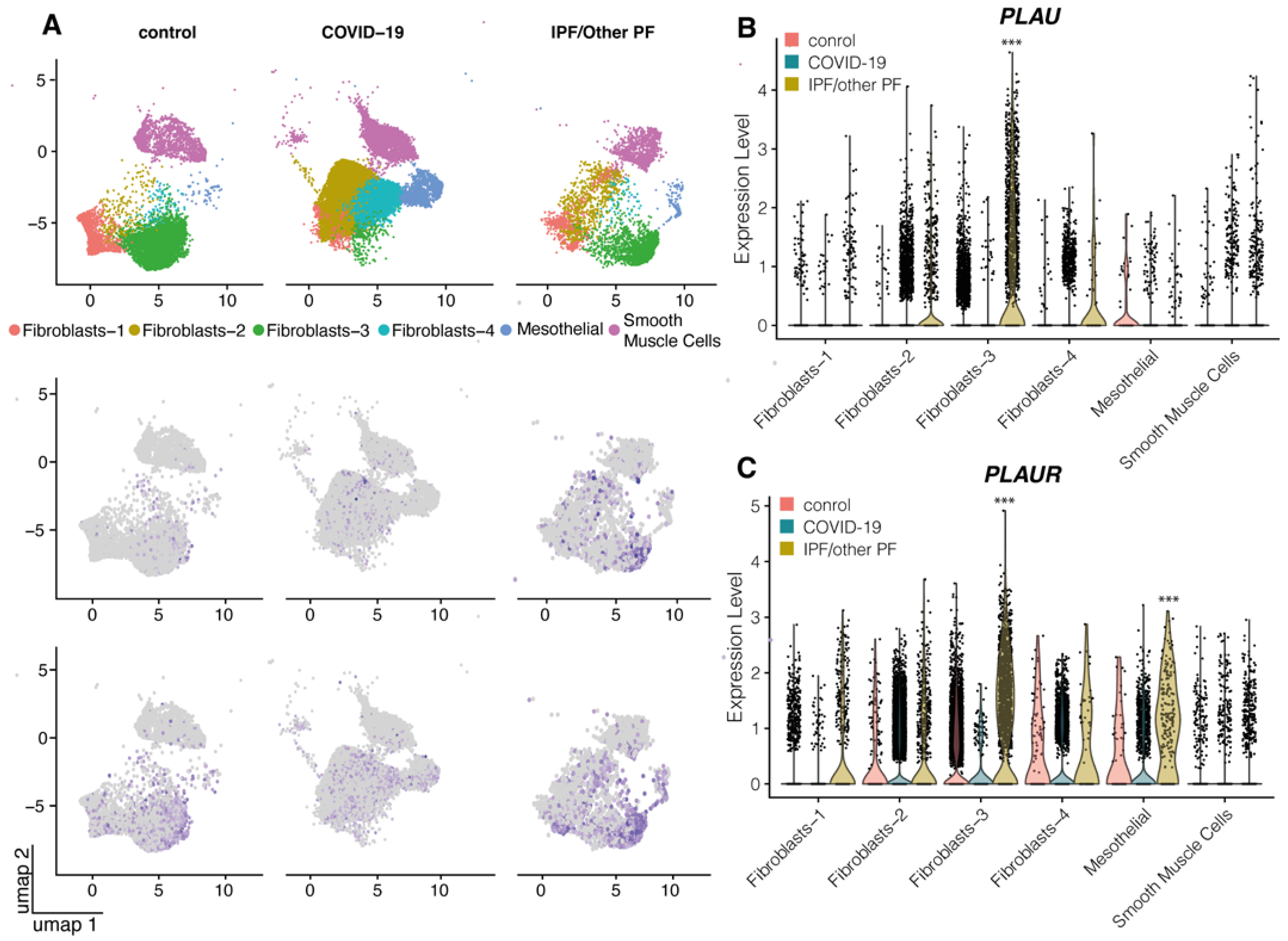
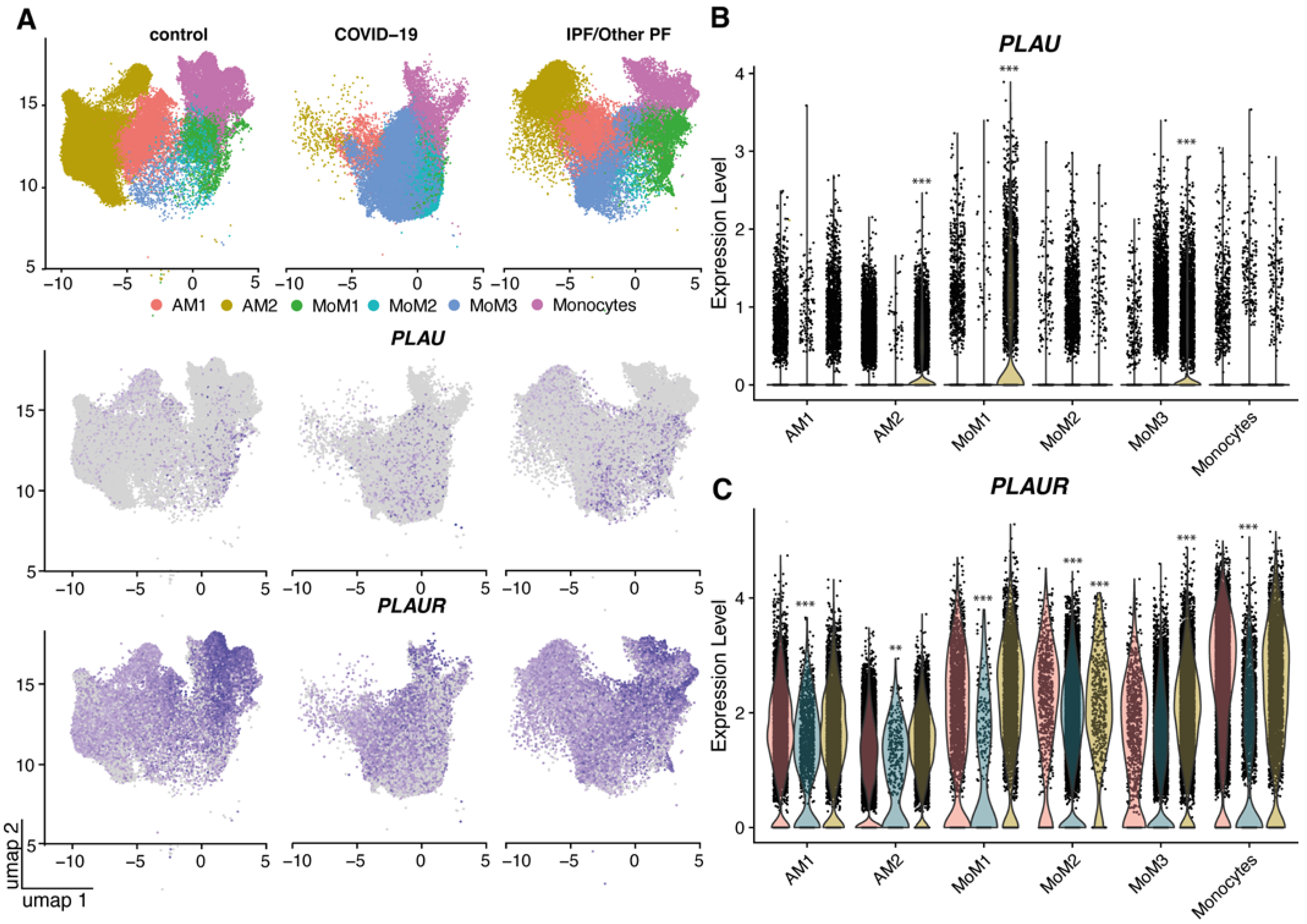
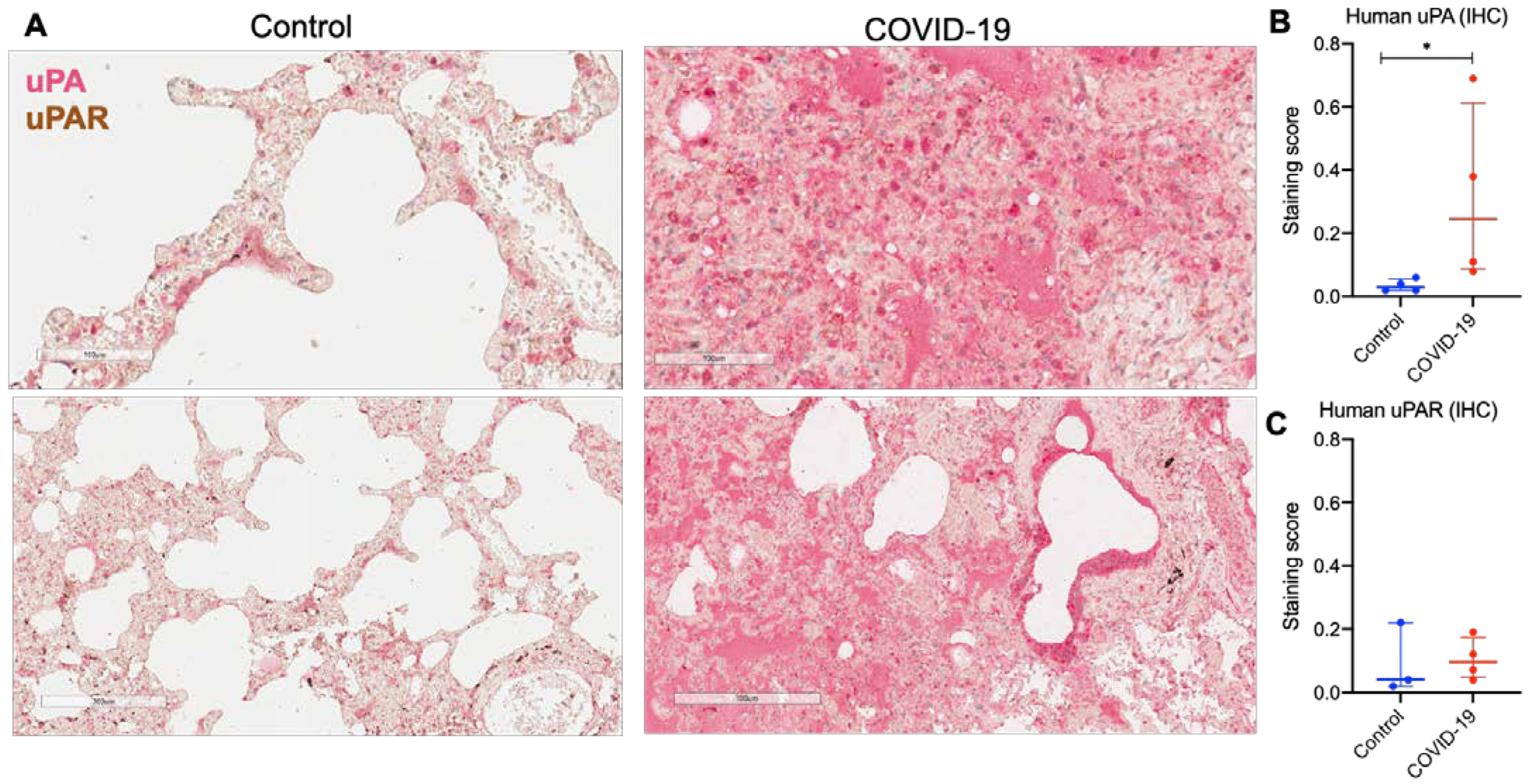
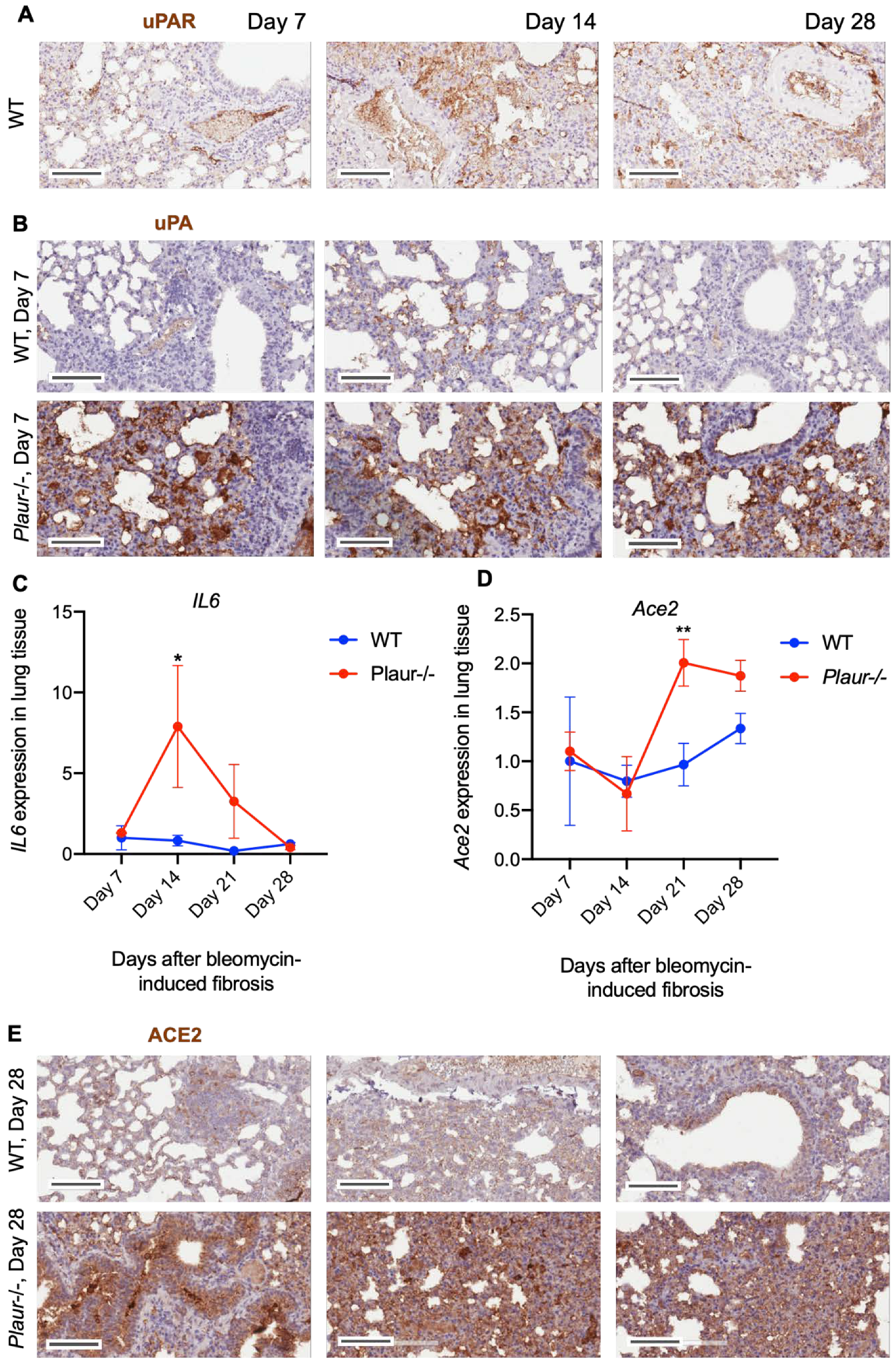
2.1. uPA and uPAR Were Differentially Expressed in Lung Tissue of COVID-19 Patients
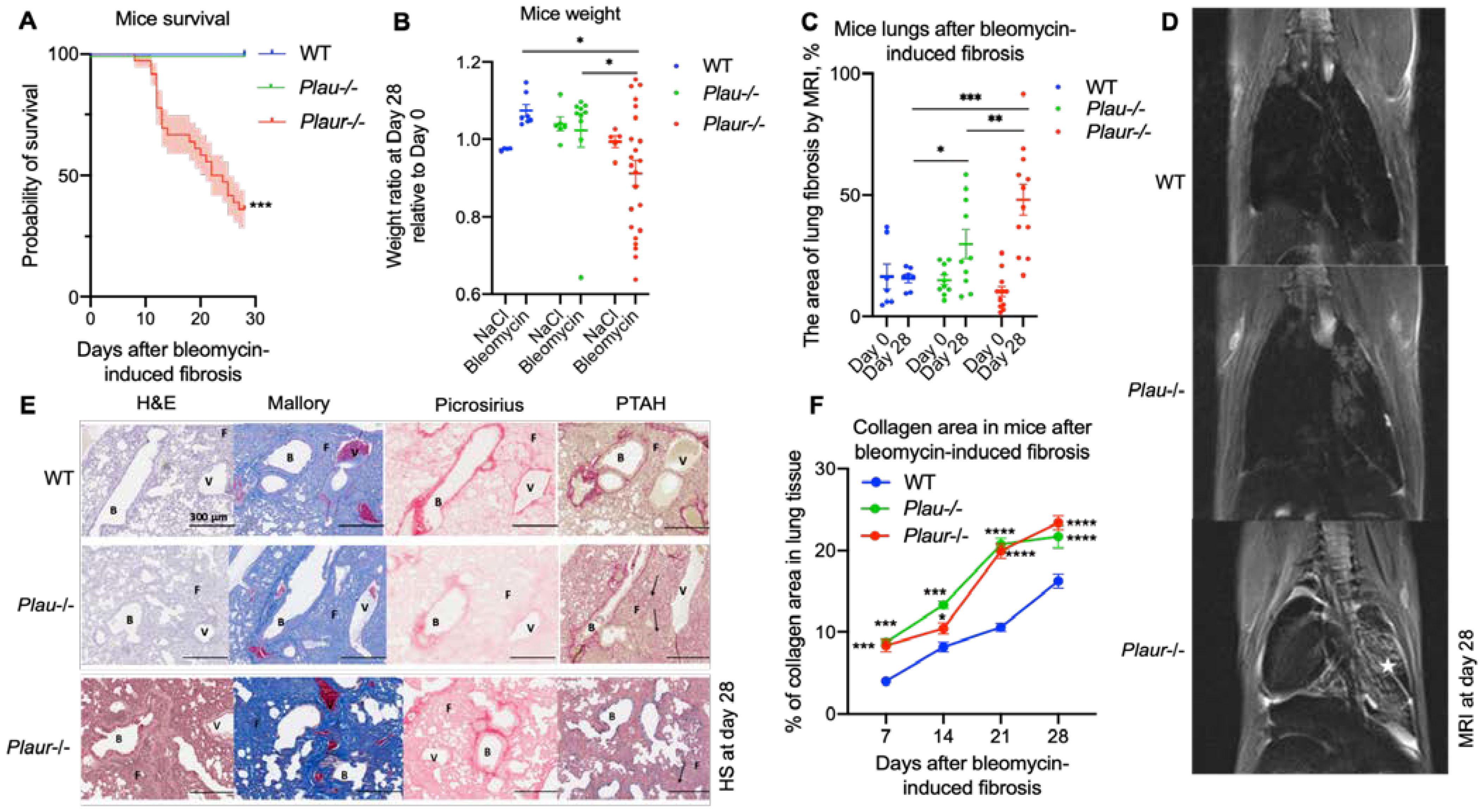
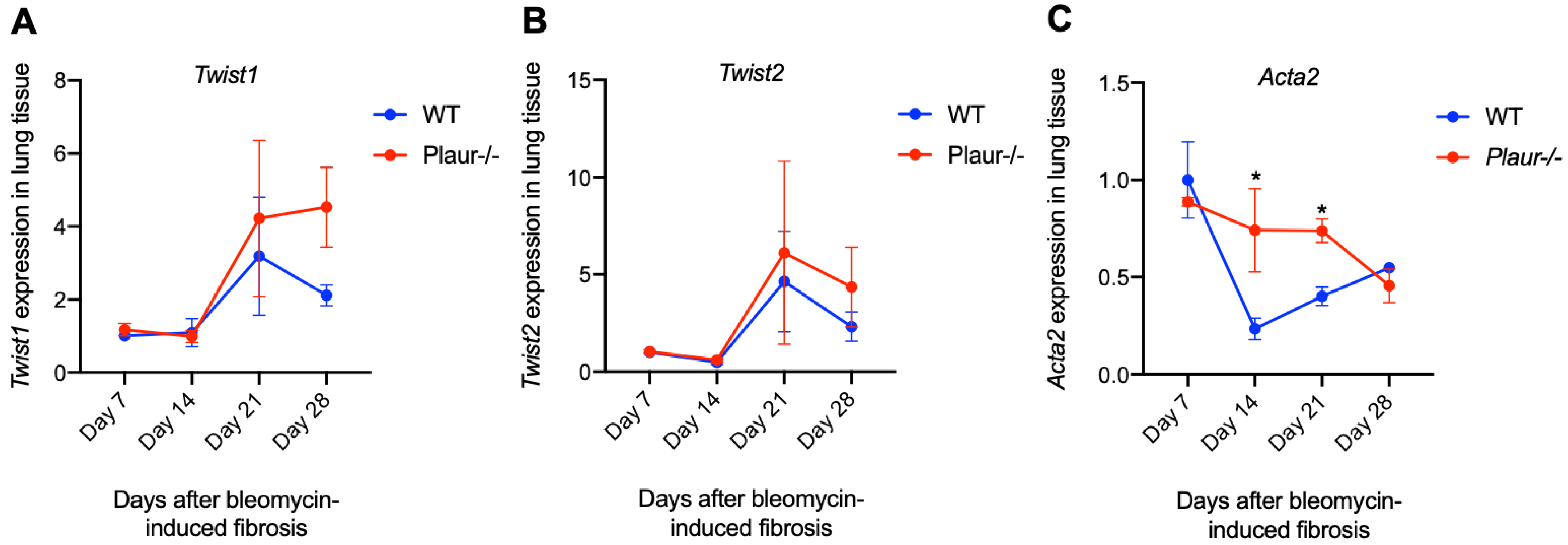
2.2. Bleomycin-Induced Pulmonary Fibrosis Was More Severe in Plaur-/- Mice
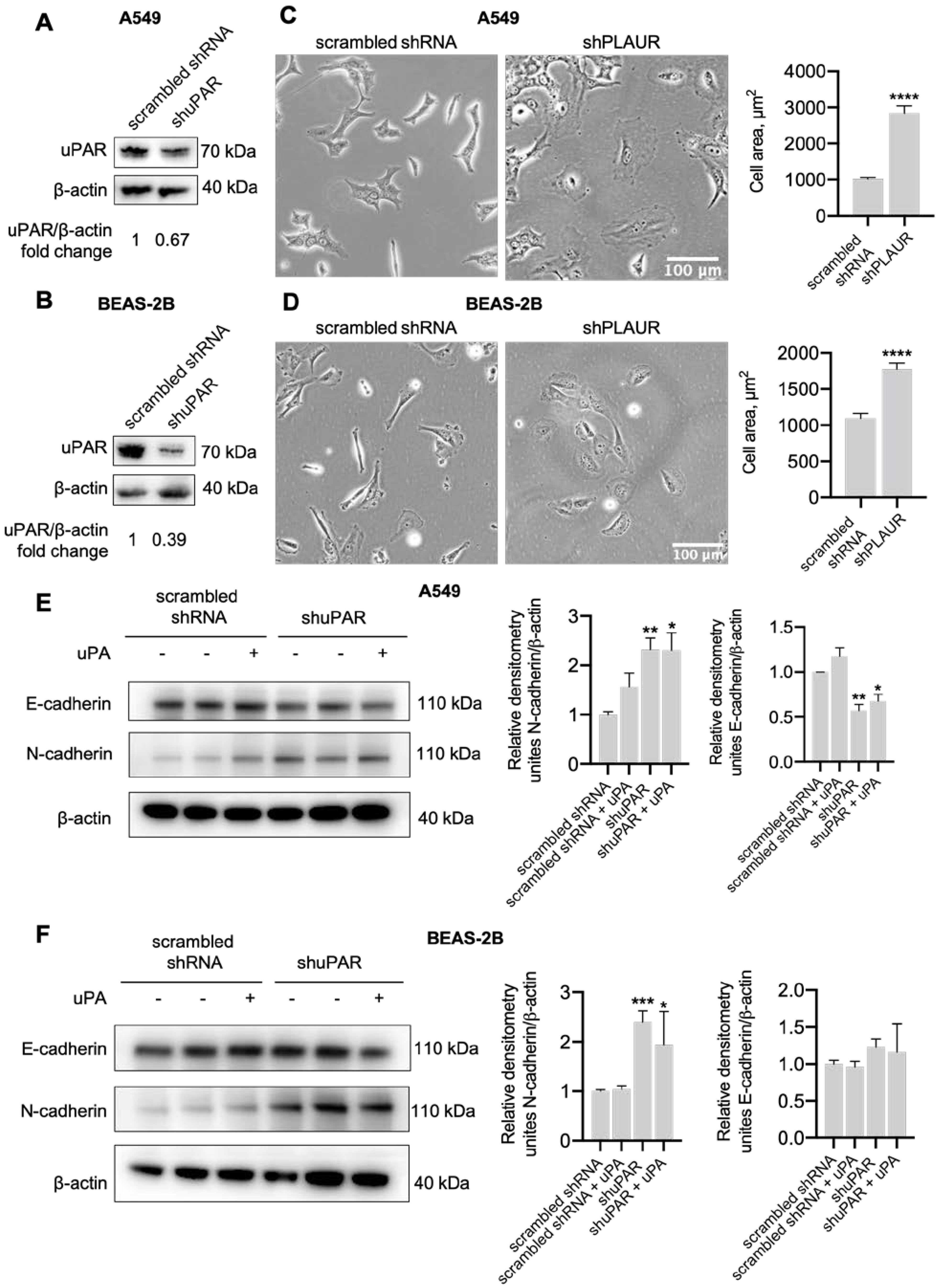
2.3. uPAR Downregulation in Lung Epithelial Cells Induced Mesenchymal Phenotype
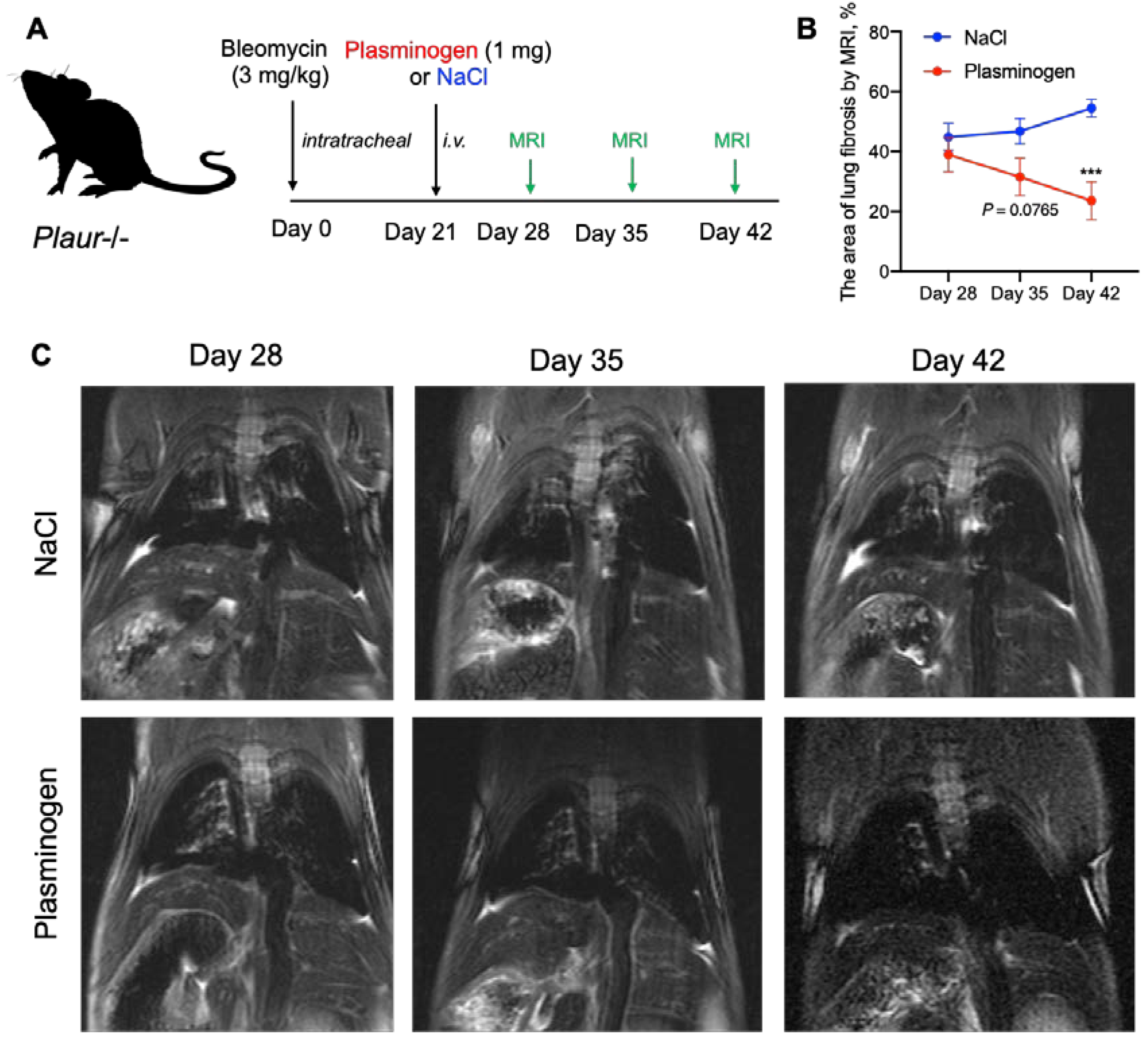
2.4. Plasminogen Administration Reversed Bleomycin-Induced Lung Fibrosis in Plaur-/- Mice
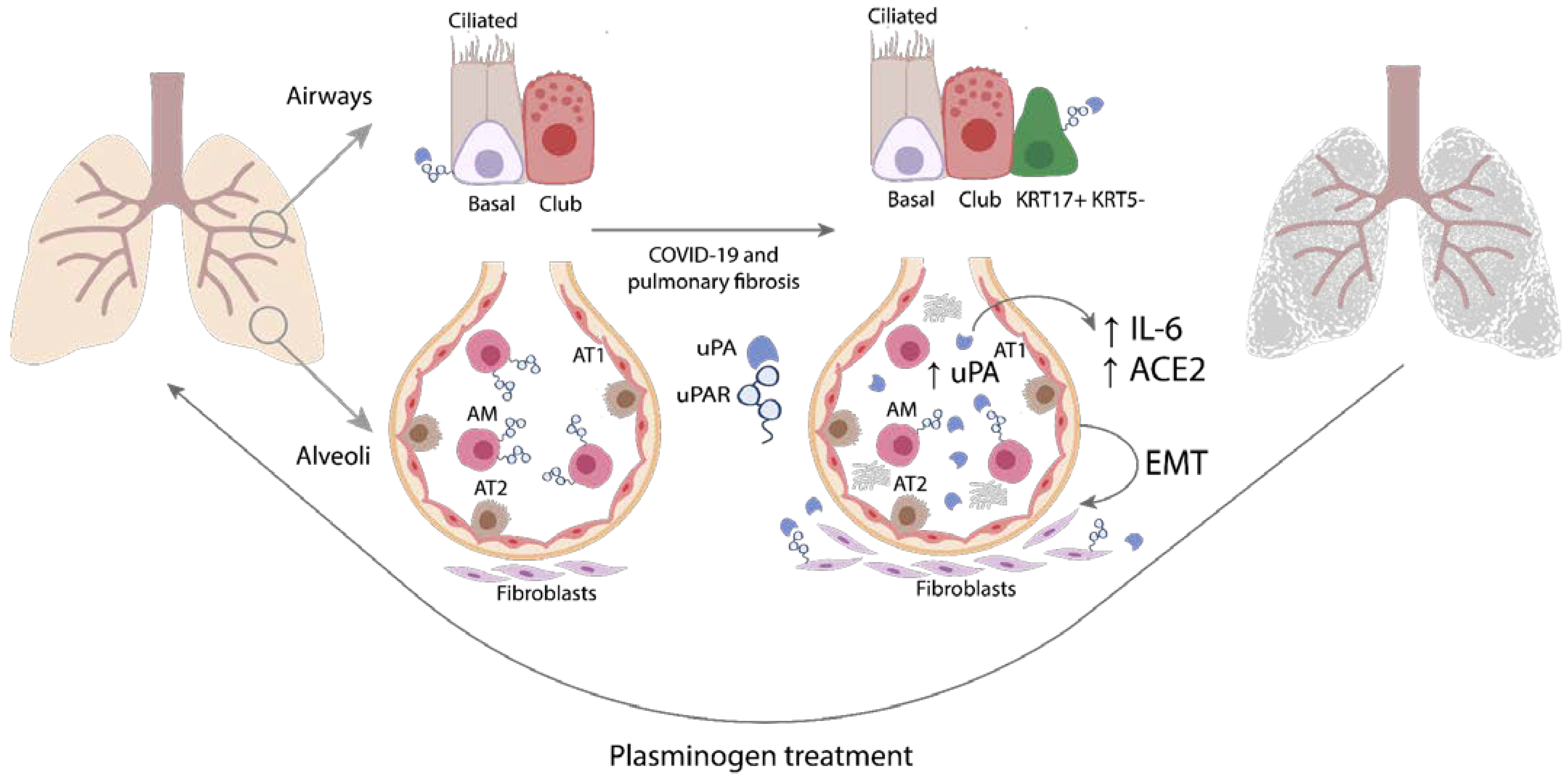
3. Discussion
4. Materials and Methods
4.1. Human Lung Tissue Samples
4.2. Animals
4.3. Bleomycin-Induced Pulmonary Fibrosis in Mice
4.4. Bolus Administration of Plasminogen to Mice
4.5. MRI for Noninvasive Evaluation of Lung Fibrosis
- The background signal of the image was adjusted (the brightness level was set so that 99.5% of pixels outside the mouse body were dark);
- Regions of interest (ROIs) covering the lung tissue but avoiding the heart and large pulmonary vessels were manually selected;
- The percentage of dark (“healthy”) pixels in the ROI based on the pre-set background brightness was calculated. The resulting value was derived by subtracting the percentage of dark pixels from 100%, which represents the proportion of fibrotic tissue in the lungs.
4.6. Mouse Lung Isolation and Sample Collection
4.7. Immunohistochemistry, Microscopy and Morphometric Analysis
4.8. Histopathologic Evaluation of Lung Fibrosis
4.9. Plasmid Cloning
4.10. Cell Culture
4.11. Cell Size Measurement
4.12. RNA Isolation, Reverse Transcription and qPCR
4.13. Protein Extraction, SDS-PAGE Electrophoresis and Western Blot
4.14. Transcriptome Analysis
4.15. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19—18 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---18-march-2020 (accessed on 24 April 2022).
- Weekly Epidemiological Update on COVID-19—20 April 2022. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---20-april-2022 (accessed on 24 April 2022).
- Fu, Z.; Tang, N.; Chen, Y.; Ma, L.; Wei, Y.; Lu, Y.; Ye, K.; Liu, H.; Tang, F.; Huang, G.; et al. CT Features of COVID-19 Patients with Two Consecutive Negative RT-PCR Tests after Treatment. Sci. Rep. 2020, 10, 11548. [Google Scholar] [CrossRef] [PubMed]
- Francone, M.; Iafrate, F.; Masci, G.M.; Coco, S.; Cilia, F.; Manganaro, L.; Panebianco, V.; Andreoli, C.; Colaiacomo, M.C.; Zingaropoli, M.A.; et al. Chest CT Score in COVID-19 Patients: Correlation with Disease Severity and Short-Term Prognosis. Eur. Radiol. 2020, 30, 6808–6817. [Google Scholar] [CrossRef] [PubMed]
- Vasarmidi, E.; Tsitoura, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K.M. Pulmonary Fibrosis in the Aftermath of the COVID-19 Era (Review). Exp. Ther. Med. 2020, 20, 2557–2560. [Google Scholar] [CrossRef] [PubMed]
- Bocchino, M.; Lieto, R.; Romano, F.; Sica, G.; Bocchini, G.; Muto, E.; Capitelli, L.; Sequino, D.; Valente, T.; Fiorentino, G.; et al. Chest CT–Based Assessment of 1-Year Outcomes after Moderate COVID-19 Pneumonia. Radiology 2022, 305, 479–485. [Google Scholar] [CrossRef]
- Bazdyrev, E.; Rusina, P.; Panova, M.; Novikov, F.; Grishagin, I.; Nebolsin, V. Lung Fibrosis after COVID-19: Treatment Prospects. Pharmaceuticals 2021, 14, 807. [Google Scholar] [CrossRef]
- Desai, A.D.; Lavelle, M.; Boursiquot, B.C.; Wan, E.Y. Long-Term Complications of COVID-19. Am. J. Physiol. Cell Physiol. 2022, 322, C1–C11. [Google Scholar] [CrossRef]
- Rai, D.K.; Sharma, P.; Kumar, R. Post Covid 19 Pulmonary Fibrosis. Is It Real Threat? Indian J. Tuberc. 2021, 68, 330–333. [Google Scholar] [CrossRef]
- Wygrecka, M.; Birnhuber, A.; Seeliger, B.; Michalick, L.; Pak, O.; Schultz, A.-S.; Schramm, F.; Zacharias, M.; Gorkiewicz, G.; David, S.; et al. Altered Fibrin Clot Structure and Dysregulated Fibrinolysis Contribute to Thrombosis Risk in Severe COVID-19. Blood Adv. 2022, 6, 1074–1087. [Google Scholar] [CrossRef]
- Baratella, E.; Ruaro, B.; Marrocchio, C.; Starvaggi, N.; Salton, F.; Giudici, F.; Quaia, E.; Confalonieri, M.; Cova, M.A. Interstitial Lung Disease at High Resolution CT after SARS-CoV-2-Related Acute Respiratory Distress Syndrome According to Pulmonary Segmental Anatomy. J. Clin. Med. 2021, 10, 3985. [Google Scholar] [CrossRef]
- Salton, F.; Confalonieri, P.; Campisciano, G.; Cifaldi, R.; Rizzardi, C.; Generali, D.; Pozzan, R.; Tavano, S.; Bozzi, C.; Lapadula, G.; et al. Cytokine Profiles as Potential Prognostic and Therapeutic Markers in SARS-CoV-2-Induced ARDS. J. Clin. Med. 2022, 11, 2951. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.L.; Zhao, R.; Matalon, S.; Matthay, M.A. Elevated Plasmin(Ogen) as a Common Risk Factor for COVID-19 Susceptibility. Physiol. Rev. 2020, 100, 1065–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokeen, K.; Pandey, S.; Shah, M.; Kumar, S. Insight towards the Effect of the Multi Basic Cleavage Site of SARS-CoV-2 Spike Protein on Cellular Proteases. Virus Res. 2022, 318, 198845. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.; Padijnayayveetil, J.; Tucker, T.; Stankowska, D.; Idell, S. The Fibrinolytic System and the Regulation of Lung Epithelial Cell Proteolysis, Signaling, and Cellular Viability. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L967–L975. [Google Scholar] [CrossRef]
- Schuliga, M.; Jaffar, J.; Harris, T.; Knight, D.A.; Westall, G.; Stewart, A.G. The Fibrogenic Actions of Lung Fibroblast-Derived Urokinase: A Potential Drug Target in IPF. Sci. Rep. 2017, 7, 41770. [Google Scholar] [CrossRef] [Green Version]
- Chambers, R.C.; Scotton, C.J. Coagulation Cascade Proteinases in Lung Injury and Fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 96–101. [Google Scholar] [CrossRef]
- De Lorenzi, V.; Sarra Ferraris, G.M.; Madsen, J.B.; Lupia, M.; Andreasen, P.A.; Sidenius, N. Urokinase Links Plasminogen Activation and Cell Adhesion by Cleavage of the RGD Motif in Vitronectin. EMBO Rep. 2016, 17, 982–998. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.W.; Marshall, C.J. Regulation of Cell Signalling by UPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23. [Google Scholar] [CrossRef]
- Blasi, F.; Carmeliet, P. UPAR: A Versatile Signalling Orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef]
- Zhou, H.-M.; Nichols, A.; Meda, P.; Vassalli, J.-D. Urokinase-Type Plasminogen Activator and Its Receptor Synergize to Promote Pathogenic Proteolysis. EMBO J. 2000, 19, 4817–4826. [Google Scholar] [CrossRef]
- Ellis, V.; Behrendt, N.; Danø, K. Plasminogen Activation by Receptor-Bound Urokinase. A Kinetic Study with Both Cell-Associated and Isolated Receptor. J. Biol. Chem. 1991, 266, 12752–12758. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Aaronson, H.; Mitola, D.J.; Leppla, S.H.; Bugge, T.H. Potent Antitumor Activity of a Urokinase-Activated Engineered Anthrax Toxin. Proc. Natl. Acad. Sci. USA 2003, 100, 657–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, L.C. Kinetics of Reciprocal Pro-Urokinase/Plasminogen Activation-Stimulation by a Template Formed by the Urokinase Receptor Bound to Poly(D-Lysine). Eur. J. Biochem. 1997, 245, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.M.; Harris, R.L.; Owen, K.A.; Bass, R.; Ghorayeb, C.; Bar-Or, A.; Ellis, V. Initiation of Plasminogen Activation on the Surface of Monocytes Expressing the Type II Transmembrane Serine Protease Matriptase. Blood 2006, 108, 2616–2623. [Google Scholar] [CrossRef] [Green Version]
- Rubina, K.; Shmakova, A.; Shabanov, A.; Andreev, Y.; Borovkova, N.; Kulabukhov, V.; Evseev, A.; Popugaev, K.; Petrikov, S.; Semina, E. Novel Prognostic Determinants of COVID-19-Related Mortality: A Pilot Study on Severely-Ill Patients in Russia. PLoS ONE 2022, 17, e0264072. [Google Scholar] [CrossRef]
- Della-Morte, D.; Pacifici, F.; Ricordi, C.; Massoud, R.; Rovella, V.; Proietti, S.; Iozzo, M.; Lauro, D.; Bernardini, S.; Bonassi, S.; et al. Low Level of Plasminogen Increases Risk for Mortality in COVID-19 Patients. Cell Death Dis. 2021, 12, 773. [Google Scholar] [CrossRef]
- Rovina, N.; Akinosoglou, K.; Eugen-Olsen, J.; Hayek, S.; Reiser, J.; Giamarellos-Bourboulis, E.J. Soluble Urokinase Plasminogen Activator Receptor (SuPAR) as an Early Predictor of Severe Respiratory Failure in Patients with COVID-19 Pneumonia. Crit. Care 2020, 24, 187. [Google Scholar] [CrossRef]
- Napolitano, F.; Di Spigna, G.; Vargas, M.; Iacovazzo, C.; Pinchera, B.; Spalletti Cernia, D.; Ricciardone, M.; Covelli, B.; Servillo, G.; Gentile, I.; et al. Soluble Urokinase Receptor as a Promising Marker for Early Prediction of Outcome in COVID-19 Hospitalized Patients. J. Clin. Med. 2021, 10, 4914. [Google Scholar] [CrossRef]
- Huang, M.; Li, L.; Shen, J.; Wang, Y.; Wang, R.; Yuan, C.; Huang, M.; Jiang, L. Plasma Levels of the Active Form of SuPAR Are Associated with COVID-19 Severity. Crit. Care 2020, 24, 704. [Google Scholar] [CrossRef]
- Oulhaj, A.; Alsuwaidi, A.R.; Suliman, A.; Gasmelseed, H.; Khan, S.; Alawi, S.; Hukan, Y.; George, J.; Alshamsi, F.; Sheikh, F.; et al. Admission Levels of Soluble Urokinase Plasminogen Activator Receptor (SuPAR) Are Associated with the Development of Severe Complications in Hospitalised COVID-19 Patients: A Prospective Cohort Study. Int. J. Infect. Dis. 2021, 107, 188–194. [Google Scholar] [CrossRef]
- Arnold, D.T.; Attwood, M.; Barratt, S.; Morley, A.; Elvers, K.T.; McKernon, J.; Donald, C.; Oates, A.; Noel, A.; MacGowan, A.; et al. Predicting Outcomes of COVID-19 from Admission Biomarkers: A Prospective UK Cohort Study. Emerg. Med. J. 2021, 38, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Nekrasova, L.A.; Shmakova, A.A.; Samokhodskaya, L.M.; Kirillova, K.I.; Stoyanova, S.S.; Mershina, E.A.; Nazarova, G.B.; Rubina, K.A.; Semina, E.V.; Kamalov, A.A. The Association of PLAUR Genotype and Soluble SuPAR Serum Level with COVID-19-Related Lung Damage Severity. Int. J. Mol. Sci. 2022, 23, 16210. [Google Scholar] [CrossRef] [PubMed]
- Bharat, A.; Querrey, M.; Markov, N.S.; Kim, S.; Kurihara, C.; Garza-Castillon, R.; Manerikar, A.; Shilatifard, A.; Tomic, R.; Politanska, Y.; et al. Lung Transplantation for Patients with Severe COVID-19. Sci. Transl. Med. 2020, 12, eabe4282. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.-I.; Taylor, C.J.; Jetter, C.; et al. Single-Cell RNA Sequencing Reveals Profibrotic Roles of Distinct Epithelial and Mesenchymal Lineages in Pulmonary Fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef] [PubMed]
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.; Ibarra, I.L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 Infection Triggers Profibrotic Macrophage Responses and Lung Fibrosis. Cell 2021, 184, 6243–6261.e27. [Google Scholar] [CrossRef] [PubMed]
- Shmakova, A.A.; Rubina, K.A.; Anokhin, K.V.; Tkachuk, V.A.; Semina, E.V. The Role of Plasminogen Activator System in the Pathogenesis of Epilepsy. Biochemistry (Moscow) 2019, 84, 979–991. [Google Scholar] [CrossRef]
- Mokuda, S.; Tokunaga, T.; Masumoto, J.; Sugiyama, E. Angiotensin-Converting Enzyme 2, a SARS-CoV-2 Receptor, Is Upregulated by Interleukin 6 through STAT3 Signaling in Synovial Tissues. J. Rheumatol. 2020, 47, 1593–1595. [Google Scholar] [CrossRef]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar Epithelial Cell Mesenchymal Transition Develops in Vivo during Pulmonary Fibrosis and Is Regulated by the Extracellular Matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [Green Version]
- Abaurrea, A.; Araujo, A.M.; Caffarel, M.M. The Role of the IL-6 Cytokine Family in Epithelial–Mesenchymal Plasticity in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 8334. [Google Scholar] [CrossRef]
- Belanger, K.K.; Tian, B.; Brasier, A.R.; Ameredes, B.T. Inhibition of IL-6 Release in Vitro by In Vivo Administration of an IKK Inhibitor in Mice with Lung Fibrosis Induced by Poly I:C. J. Allergy Clin. Immunol. 2017, 139, AB76. [Google Scholar] [CrossRef]
- Semina, E.V.; Rubina, K.A.; Shmakova, A.A.; Rysenkova, K.D.; Klimovich, P.S.; Aleksanrushkina, N.A.; Sysoeva, V.Y.; Karagyaur, M.N.; Tkachuk, V.A. Downregulation of UPAR Promotes Urokinase Translocation into the Nucleus and Epithelial to Mesenchymal Transition in Neuroblastoma. J. Cell. Physiol. 2020, 235, 6268–6286. [Google Scholar] [CrossRef] [PubMed]
- Juneja, G.K.; Castelo, M.; Yeh, C.H.; Cerroni, S.E.; Hansen, B.E.; Chessum, J.E.; Abraham, J.; Cani, E.; Dwivedi, D.J.; Fraser, D.D.; et al. Biomarkers of Coagulation, Endothelial Function, and Fibrinolysis in Critically Ill Patients with COVID-19: A Single-Center Prospective Longitudinal Study. J. Thromb. Haemost. 2021, 19, 1546–1557. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Cheng, Z.; Luo, L.; Zhu, Y.; Lin, W.; Ming, Z.; Chen, W.; Hu, Y. Incidence and Impact of Disseminated Intravascular Coagulation in COVID-19 a Systematic Review and Meta-Analysis. Thromb. Res. 2021, 201, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, R.G.; Pilling, J. Fibrinolytic Activity of Normal Urine. Nature 1947, 159, 779. [Google Scholar] [CrossRef] [PubMed]
- Ellis, V.; Scully, M.F.; Kakkar, V.V. Plasminogen Activation Initiated by Single-Chain Urokinase-Type Plasminogen Activator. Potentiation by U937 Monocytes. J. Biol. Chem. 1989, 264, 2185–2188. [Google Scholar] [CrossRef]
- Eden, G.; Archinti, M.; Furlan, F.; Murphy, R.; Degryse, B. The Urokinase Receptor Interactome. Curr. Pharm. Des. 2011, 17, 1874–1889. [Google Scholar] [CrossRef]
- Yu, S.; Sui, Y.; Wang, J.; Li, Y.; Li, H.; Cao, Y.; Chen, L.; Jiang, L.; Yuan, C.; Huang, M. Crystal Structure and Cellular Functions of UPAR Dimer. Nat. Commun. 2022, 13, 1665. [Google Scholar] [CrossRef]
- Marudamuthu, A.S.; Bhandary, Y.P.; Shetty, S.K.; Fu, J.; Sathish, V.; Prakash, Y.; Shetty, S. Role of the Urokinase-Fibrinolytic System in Epithelial–Mesenchymal Transition during Lung Injury. Am. J. Pathol. 2015, 185, 55–68. [Google Scholar] [CrossRef] [Green Version]
- FitzGerald, E.S.; Jamieson, A.M. Unique Transcriptional Changes in Coagulation Cascade Genes in SARS-CoV-2-Infected Lung Epithelial Cells: A Potential Factor in COVID-19 Coagulopathies. bioRxiv 2020, 182972. [Google Scholar] [CrossRef]
- Mast, A.E.; Wolberg, A.S.; Gailani, D.; Garvin, M.R.; Alvarez, C.; Miller, J.I.; Aronow, B.; Jacobson, D. SARS-CoV-2 Suppresses Anticoagulant and Fibrinolytic Gene Expression in the Lung. eLife 2021, 10, e64330. [Google Scholar] [CrossRef]
- Hou, Y.; Ding, Y.; Nie, H.; Ji, H.-L. Fibrinolysis Influences SARS-CoV-2 Infection in Ciliated Cells. bioRxiv 2021, 425801. [Google Scholar] [CrossRef]
- Olson, D.; Pöllänen, J.; Høyer-Hansen, G.; Rønne, E.; Sakaguchi, K.; Wun, T.C.; Appella, E.; Danø, K.; Blasi, F. Internalization of the Urokinase-Plasminogen Activator Inhibitor Type-1 Complex Is Mediated by the Urokinase Receptor. J. Biol. Chem. 1992, 267, 9129–9133. [Google Scholar] [CrossRef] [PubMed]
- Cortese, K.; Sahores, M.; Madsen, C.D.; Tacchetti, C.; Blasi, F. Clathrin and LRP-1-Independent Constitutive Endocytosis and Recycling of UPAR. PLoS ONE 2008, 3, e3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krstić, J.; Obradović, H.; Jauković, A.; Okić-Đorđević, I.; Trivanović, D.; Kukolj, T.; Mojsilović, S.; Ilić, V.; Santibañez, J.F.; Bugarski, D. Urokinase Type Plasminogen Activator Mediates Interleukin-17-Induced Peripheral Blood Mesenchymal Stem Cell Motility and Transendothelial Migration. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Res. 2015, 1853, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plekhanova, O.S.; Parfyonova, Y.V.; Bibilashvily, R.S.; Stepanova, V.V.; Erne, P.; Bobik, A.; Tkachuk, V.A. Urokinase Plasminogen Activator Enhances Neointima Growth and Reduces Lumen Size in Injured Carotid Arteries. J. Hypertens. 2000, 18, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Plekhanova, O.; Parfyonova, Y.; Bibilashvily, R.; Domogatskii, S.; Stepanova, V.; Gulba, D.C.; Agrotis, A.; Bobik, A.; Tkachuk, V. Urokinase Plasminogen Activator Augments Cell Proliferation and Neointima Formation in Injured Arteries via Proteolytic Mechanisms. Atherosclerosis 2001, 159, 297–306. [Google Scholar] [CrossRef]
- Zhang, G.; Kim, H.; Cai, X.; López-Guisa, J.M.; Alpers, C.E.; Liu, Y.; Carmeliet, P.; Eddy, A.A. Urokinase Receptor Deficiency Accelerates Renal Fibrosis in Obstructive Nephropathy. JASN 2003, 14, 1254–1271. [Google Scholar] [CrossRef] [Green Version]
- Swaisgood, C.M.; French, E.L.; Noga, C.; Simon, R.H.; Ploplis, V.A. The Development of Bleomycin-Induced Pulmonary Fibrosis in Mice Deficient for Components of the Fibrinolytic System. Am. J. Pathol. 2000, 157, 177–187. [Google Scholar] [CrossRef] [Green Version]
- Lardot, C.G.; Huaux, F.A.; Broeckaert, F.R.; Declerck, P.J.; Delos, M.; Fubini, B.; Lison, D.F. Role of Urokinase in the Fibrogenic Response of the Lung to Mineral Particles. Am. J. Respir. Crit. Care Med. 1998, 157, 617–628. [Google Scholar] [CrossRef]
- Nakatsuka, Y.; Handa, T.; Kokosi, M.; Tanizawa, K.; Puglisi, S.; Jacob, J.; Sokai, A.; Ikezoe, K.; Kanatani, K.T.; Kubo, T.; et al. The Clinical Significance of Body Weight Loss in Idiopathic Pulmonary Fibrosis Patients. Respiration 2018, 96, 338–347. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, X.; Li, Y.; Chen, H.; Chen, T.; Su, N.; Huang, F.; Zhou, J.; Zhang, B.; Yan, F.; et al. Clinical Course and Outcomes of 344 Intensive Care Patients with COVID-19. Am. J. Respir. Crit. Care Med. 2020, 201, 1430–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-B.; Hu, L.; Ming, Q.; Wei, X.-J.; Zhang, Z.-Y.; Chen, L.-D.; Wang, M.-H.; Yao, W.-Z.; Huang, Q.-F.; Ye, Z.-Q.; et al. Risk Factors for Mortality of Coronavirus Disease-2019 (COVID-19) Patients in Two Centers of Hubei Province, China: A Retrospective Analysis. PLoS ONE 2021, 16, e0246030. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M. ACE2 and COVID-19 Susceptibility and Severity. Aging Dis. 2022, 13, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, F.; Lavandero, S.; Xie, X.; Sabharwal, B.; Zheng, Y.-Y.; Correa, A.; Narula, J.; Levy, P. Manipulation of ACE2 Expression in COVID-19. Open Heart 2020, 7, e001424. [Google Scholar] [CrossRef]
- Fagyas, M.; Fejes, Z.; Sütő, R.; Nagy, Z.; Székely, B.; Pócsi, M.; Ivády, G.; Bíró, E.; Bekő, G.; Nagy, A.; et al. Circulating ACE2 Activity Predicts Mortality and Disease Severity in Hospitalized COVID-19 Patients. Int. J. Infect. Dis. 2022, 115, 8–16. [Google Scholar] [CrossRef]
- Biagioni, A.; Laurenzana, A.; Chillà, A.; Del Rosso, M.; Andreucci, E.; Poteti, M.; Bani, D.; Guasti, D.; Fibbi, G.; Margheri, F. UPAR Knockout Results in a Deep Glycolytic and OXPHOS Reprogramming in Melanoma and Colon Carcinoma Cell Lines. Cells 2020, 9, 308. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Xing, Z.-H.; Jiang, Q.-W.; Yang, Y.; Huang, J.-R.; Yuan, M.-L.; Wei, M.-N.; Li, Y.; Wang, S.-T.; Liu, K.; et al. Targeting UPAR by CRISPR/Cas9 System Attenuates Cancer Malignancy and Multidrug Resistance. Front. Oncol. 2019, 9, 80. [Google Scholar] [CrossRef] [Green Version]
- Commissioner, O. FDA Approves First Treatment for Patients with Plasminogen Deficiency, a Rare Genetic Disorder. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-patients-plasminogen-deficiency-rare-genetic-disorder (accessed on 10 July 2022).
- Carmeliet, P.; Mulligan, R.C.; Collen, D. Transgenic Animals as Tools for the Study of Fibrinolysis In Vivo. J. Intern. Med. 1994, 236, 455–459. [Google Scholar] [CrossRef]
- Vande Velde, G.; De Langhe, E.; Poelmans, J.; Dresselaers, T.; Lories, R.J.; Himmelreich, U. Magnetic Resonance Imaging for Noninvasive Assessment of Lung Fibrosis Onset and Progression: Cross-Validation and Comparison of Different Magnetic Resonance Imaging Protocols with Micro-Computed Tomography and Histology in the Bleomycin-Induced Mouse Model. Investig. Radiol. 2014, 49, 691–698. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.Y.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-Delivered Stable Gene Silencing by RNAi in Primary Cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated Analysis of Multimodal Single-Cell Data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Zhou, X.; Jiang, X.; Sun, T. Deletion of ACTA2 in Mice Promotes Angiotensin II Induced Pathogenesis of Thoracic Aortic Aneurysms and Dissections. J Thorac Dis 2018, 10, 4733–4740. [Google Scholar] [CrossRef]
- An, J.; Zheng, Y.; Dann, C.T. Mesenchymal to Epithelial Transition Mediated by CDH1 Promotes Spontaneous Reprogramming of Male Germline Stem Cells to Pluripotency. Stem Cell Rep. 2017, 8, 446–459. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shmakova, A.A.; Popov, V.S.; Romanov, I.P.; Khabibullin, N.R.; Sabitova, N.R.; Karpukhina, A.A.; Kozhevnikova, Y.A.; Kurilina, E.V.; Tsokolaeva, Z.I.; Klimovich, P.S.; et al. Urokinase System in Pathogenesis of Pulmonary Fibrosis: A Hidden Threat of COVID-19. Int. J. Mol. Sci. 2023, 24, 1382. https://doi.org/10.3390/ijms24021382
Shmakova AA, Popov VS, Romanov IP, Khabibullin NR, Sabitova NR, Karpukhina AA, Kozhevnikova YA, Kurilina EV, Tsokolaeva ZI, Klimovich PS, et al. Urokinase System in Pathogenesis of Pulmonary Fibrosis: A Hidden Threat of COVID-19. International Journal of Molecular Sciences. 2023; 24(2):1382. https://doi.org/10.3390/ijms24021382
Chicago/Turabian StyleShmakova, Anna A., Vladimir S. Popov, Iliya P. Romanov, Nikita R. Khabibullin, Nailya R. Sabitova, Anna A. Karpukhina, Yana A. Kozhevnikova, Ella V. Kurilina, Zoya I. Tsokolaeva, Polina S. Klimovich, and et al. 2023. "Urokinase System in Pathogenesis of Pulmonary Fibrosis: A Hidden Threat of COVID-19" International Journal of Molecular Sciences 24, no. 2: 1382. https://doi.org/10.3390/ijms24021382