
Unscrambling the Role of Redox-Active Biometals in Dopaminergic Neuronal Death and Promising Metal Chelation-Based Therapy for Parkinson’s Disease
 ,
, 
 , and
, and
Abstract
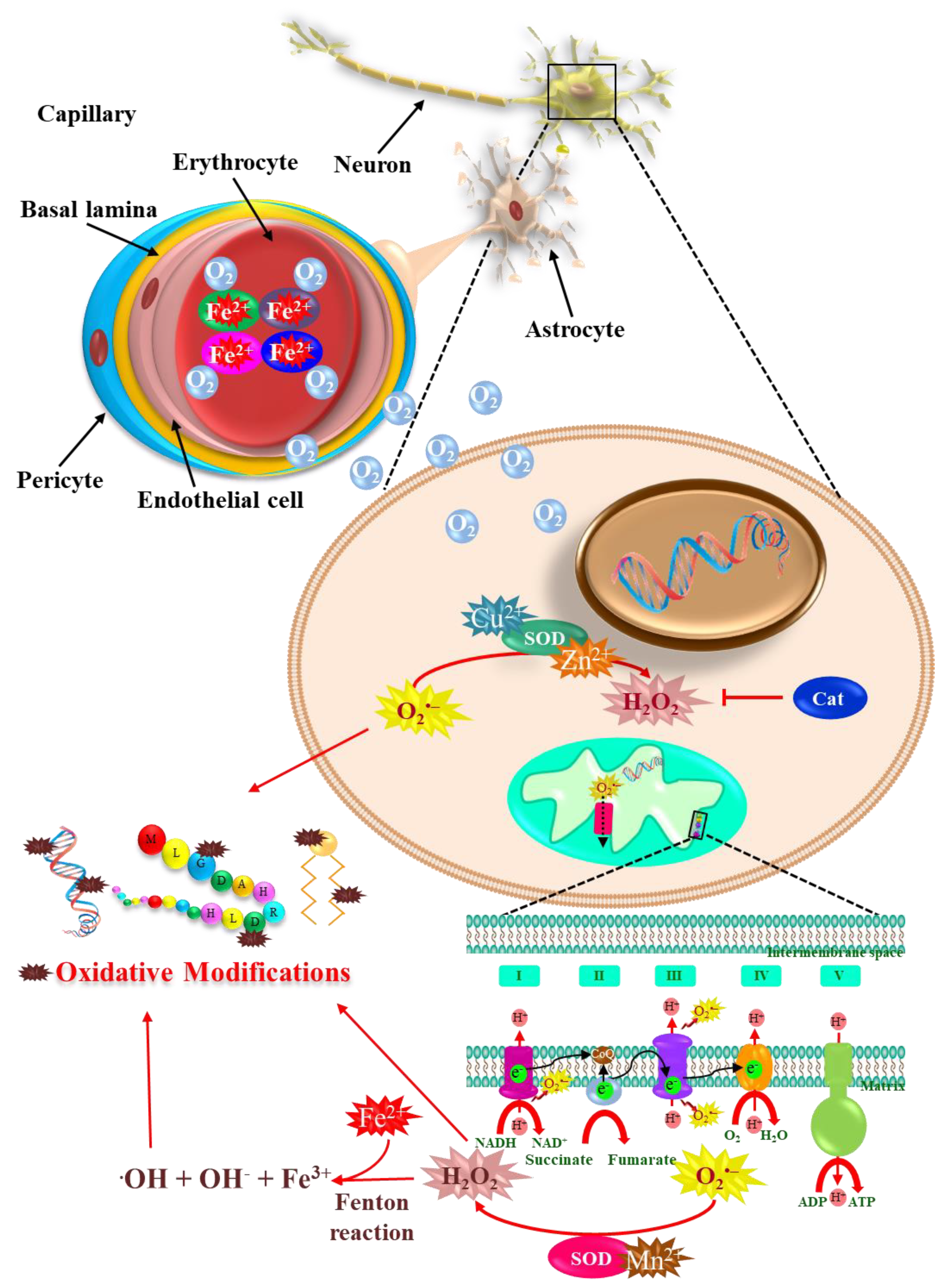
:1. Introduction
2. Metals, Parkinson’s Disease, and Oxidative Stress
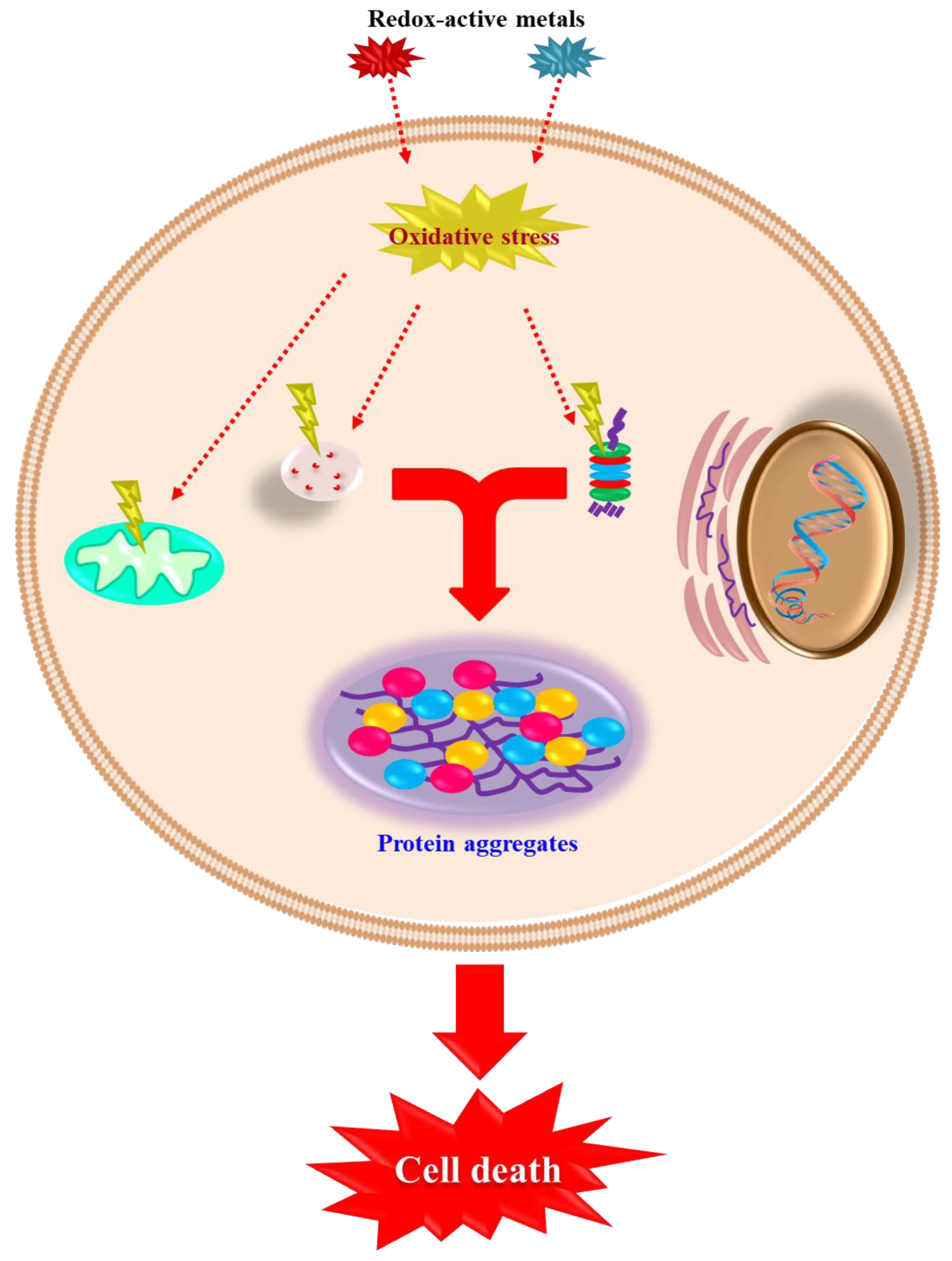
3. How Cells Die: Classical Mechanisms of Cell Death
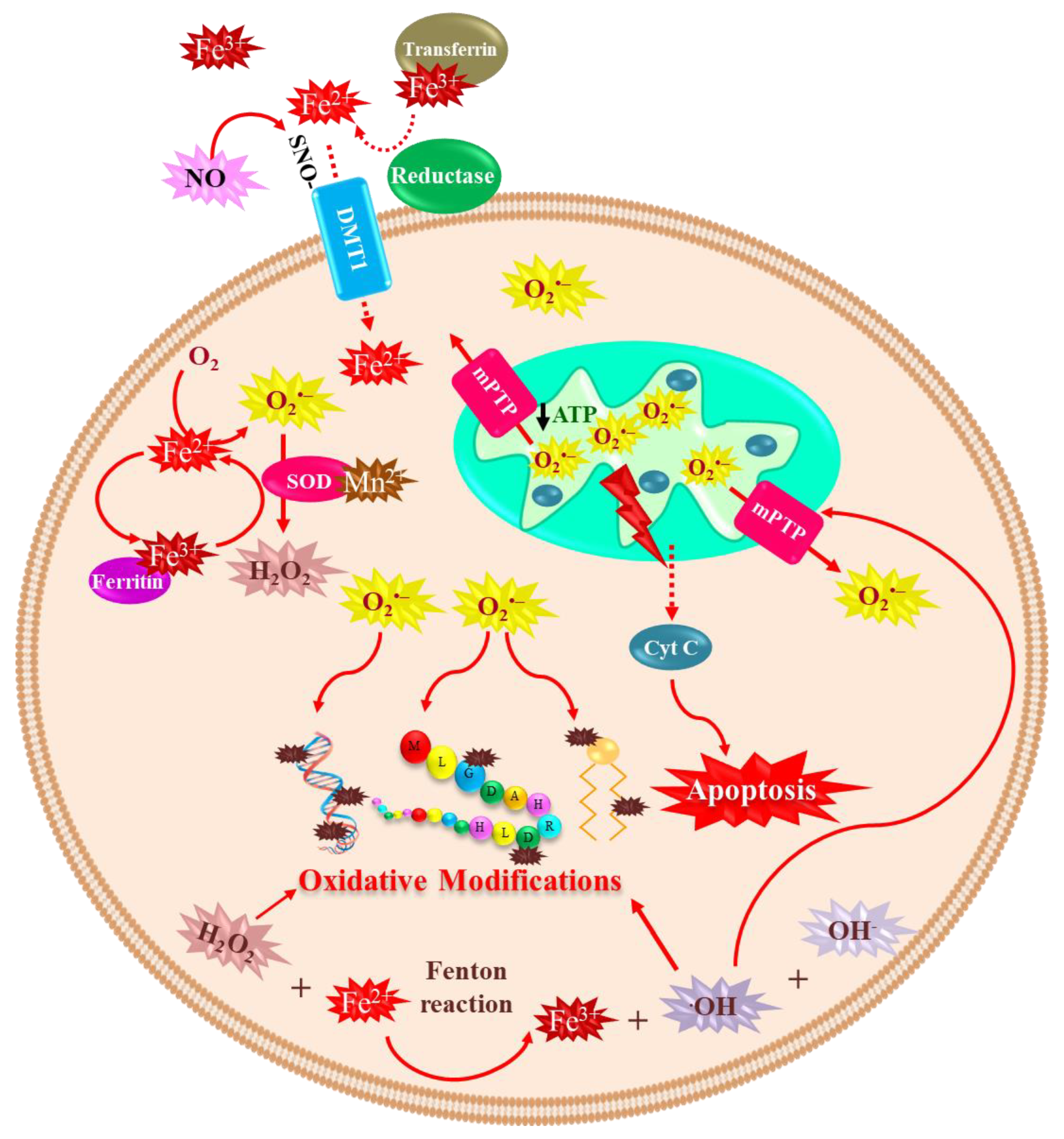
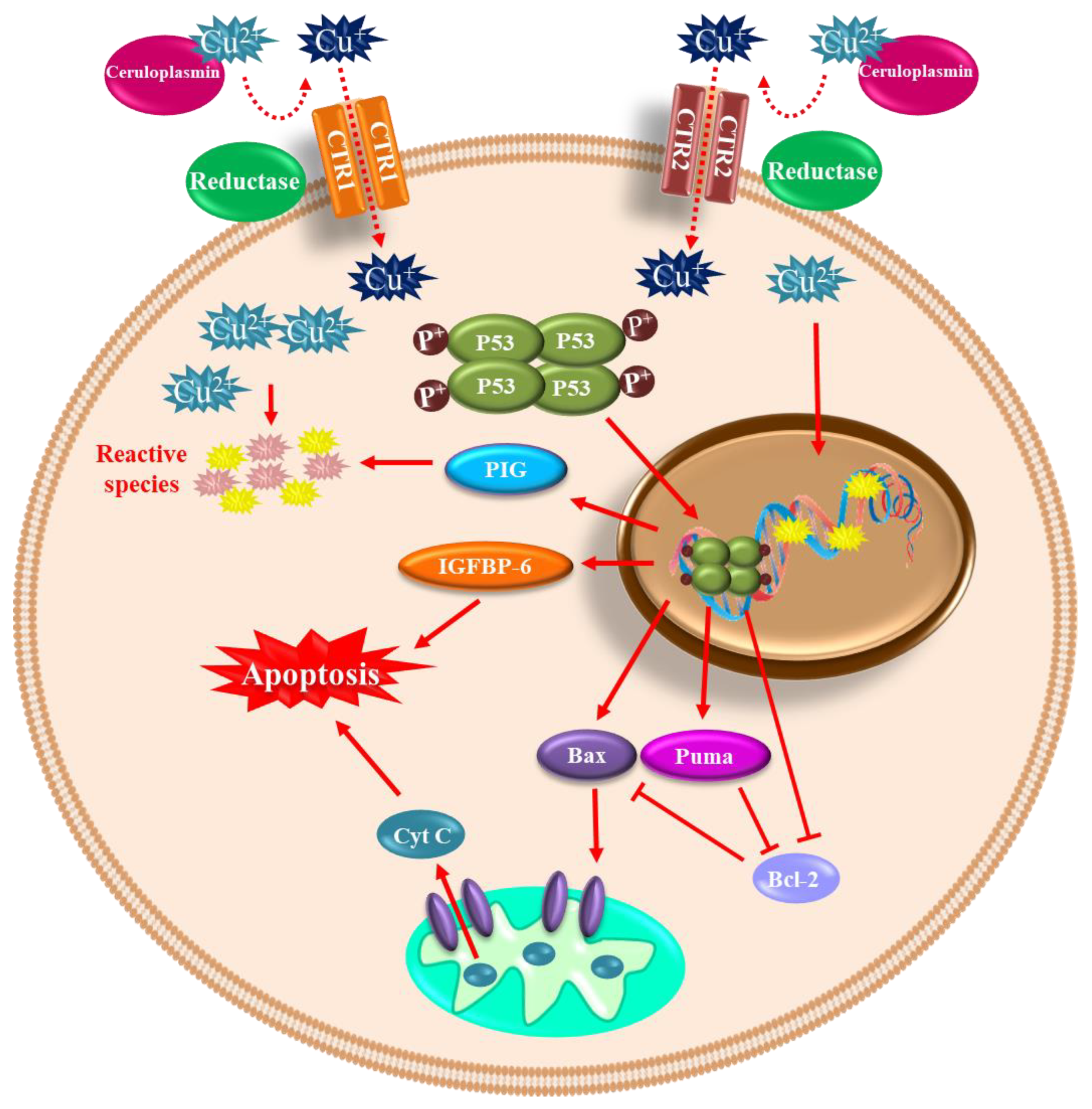
4. Redox-Active Metals’ Role in Dopaminergic Neuronal Death
4.1. Iron
Current Status of Iron Chelation Therapeutic Effect on PD Patients

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Design | Clinical Trial | Subjects Male:Female (m:f) | Outcomes | Reference |
|---|---|---|---|---|
| Randomized, double-blinded, placebo-controlled clinical trial | Phase 2 | 22 subjects: •8 placebo (m:f) 3:5 •7 DFP 20 mg/kg/day (m:f) 4:3 •7 DFP 30 mg/kg/day (m:f) 5:2 | Brain iron chelation by DFP therapy was well-tolerated; there was an associated reduced dentate and caudate nucleus iron content with a trend for improvement in motor-UPDRS scores and quality of life, not statistical significance. | [102] |
| Randomized, placebo-controlled clinical trial | Phase 1 | 40 subjects: •21 early start DFP 30 mg/kg/day (m:f) 12:9 •19 delayed start DFP 30 mg/kg/day (m:f) 13:6 | Most DFP-treated patients displayed clinical and radiological improvements. Those with lower CP activity appeared to respond better to iron chelation. | [103] |
| Randomized, double-blind, placebo-controlled, parallel-group, single-center trial | Phase 2 | 40 subjects: •21 early start DFP 30 mg/kg/day (m:f) 12:9 •19 delayed start DFP 30 mg/kg/day (m:f) 13:6 | SN iron levels and UPDRS motor scores were reduced in patients with higher CP-ferroxidase activity in serum and CSF. | [101,110] |
| A multicentric, parallel-group, placebo-controlled, randomized clinical trial | Phase 2 | 372 subjects: •186 placebo •186 DFP 30 mg/kg/day | DFP without dopaminergic treatment worsened the handicap at the PD diagnosis time compared with placebo over 36 weeks. This finding provides evidence that the iron accumulation in the nigrostriatal pathway is a powerful short-term compensatory mechanism for increasing dopamine synthesis but possibly at the expense of long-term worsening iron-related cell death. | [104] |
4.2. Copper
| Metal-Protein Attenuating Compound | Metal Ions Binding | Neurodegenerative Disease | Outcomes | Reference |
|---|---|---|---|---|
| X1INH 1-methyl-1H-imidazole-2-carboxaldehyde isonicotinoyl hydrazone | Cu+ Cu2+ | Parkinson’s disease | X1INH increased the number of smaller, less compact inclusions in a well-established model of α-Syn aggregation. | [142] |
| INHHQ 8-hydroxyquinoline-2-carboxaldehyde isonicotinoyl hydrazone INHHQ | Cu2+ Zn2+ | Alzheimer’s disease, Parkinson’s disease | INHHQ can disrupt, in vitro, anomalous copper-α-Syn interactions through a mechanism probably involving metal ions sequestering. | [141] |
| Clioquinol (CQ) 5-chloro-7-iodo-8-quinolinol | Fe3+, Cu2+ Zn2+ | Parkinson’s disease | CQ remarkably improved the motor and non-motor deficits based on reduced iron content and ROS level in the SN. | [138] |
| HPCIH, HPCFur pyridine-2-carboxaldehyde isonicotinoyl hydrazone pyridine-2-car-boxaldehyde 2-furoyl hydrazone | Cu2+ | Misfolded prion protein | HPCFur has a protective effect on methionine and histidine oxidation, which is related to physiological and pathological aging. | [147] |
5. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ortega, R.; Carmona, A. Neurotoxicity of Environmental Metal Toxicants: Special Issue. Toxics 2022, 10, 382. [Google Scholar] [CrossRef]
- Andreini, C.; Bertini, I.; Cavallaro, G.; Holliday, G.L.; Thornton, J.M. Metal ions in biological catalysis: From enzyme databases to general principles. J. Biol. Inorg. Chem. 2008, 13, 1205–1218. [Google Scholar] [CrossRef]
- Shanker, A.K. Mode of Action and Toxicity of Trace Elements. In Trace Elements as Contaminants and Nutrients: Consequences in Ecosystems and Human Health; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Pospíšil, P.; Prasad, A.; Rác, M. Mechanism of the Formation of Electronically Excited Species by Oxidative Metabolic Processes: Role of Reactive Oxygen Species. Biomolecules 2019, 9, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell. 2021, 81, 3691–3707. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Baros, S.; Valko, M. Redox active metal-induced oxidative stress in biological systems. Transit. Met. Chem. 2012, 37, 127–134. [Google Scholar] [CrossRef]
- DeBenedictis, C.A.; Raab, A.; Ducie, E.; Howley, S.; Feldmann, J.; Grabrucker, A.M. Concentrations of Essential Trace Metals in the Brain of Animal Species-A Comparative Study. Brain Sci. 2020, 10, 460. [Google Scholar] [CrossRef]
- Chang, C.J. Searching for harmony in transition-metal signaling. Nat. Chem. Biol. 2015, 11, 744–747. [Google Scholar] [CrossRef]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front. Mol. Neurosci. 2018, 11, 216. [Google Scholar] [CrossRef] [Green Version]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in neurobiology: Probing their chemistry and biology with molecular imaging. Chem. Rev. 2008, 108, 1517–1549. [Google Scholar] [CrossRef]
- Borisova, T. Nervous System Injury in Response to Contact With Environmental, Engineered and Planetary Micro- and Nano-Sized Particles. Front. Physiol. 2018, 9, 728. [Google Scholar] [CrossRef]
- Ullah, I.; Zhao, L.; Hai, Y.; Fahim, M.; Alwayli, D.; Wang, X.; Li, H. Metal elements and pesticides as risk factors for Parkinson’s disease—A review. Toxicol. Rep. 2021, 8, 607–616. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M. The evolution of free radicals and oxidative stress. Am. J. Med. 2000, 108, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Martemucci, G.; Costagliola, C.; Mariano, M.; D’Andrea, L.; Napolitano, P.; D’Alessandro, A.G. Free Radical Properties, Source and Targets, Antioxidant Consumption and Health. Oxygen 2022, 2, 48–78. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [Green Version]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; del Razo, L.M.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.; Franco, R. Oxidative stress, redox signaling, and autophagy: Cell death versus survival. Antioxid. Redox Signal. 2014, 21, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef] [PubMed]
- Stykel, M.G.; Ryan, S.D. Nitrosative stress in Parkinson’s disease. NPJ Parkinsons Dis. 2022, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A. Postmortem studies in Parkinson’s disease. Dialogues Clin. Neurosci. 2004, 6, 281–293. [Google Scholar] [CrossRef]
- Farzam, A.; Chohan, K.; Strmiskova, M.; Hewitt, S.J.; Park, D.S.; Pezacki, J.P.; Özcelik, D. A functionalized hydroxydopamine quinone links thiol modification to neuronal cell death. Redox Biol. 2020, 28, 101377. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Kish, S.J.; Morito, C.; Hornykiewicz, O. Glutathione peroxidase activity in Parkinson’s disease brain. Neurosci. Lett. 1985, 58, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ball, N.; Teo, W.-P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef] [Green Version]
- Dexter, D.T.; Wells, F.R.; Lees, A.J.; Agid, F.; Agid, Y.; Jenner, P.; Marsden, C.D. Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J. Neurochem. 1989, 52, 1830–1836. [Google Scholar] [CrossRef]
- Nakamura, K.; Bindokas, V.P.; Marks, J.D.; Wright, D.A.; Frim, D.M.; Miller, R.J.; Kang, U.J. The selective toxicity of 1-methyl-4-phenylpyridinium to dopaminergic neurons: The role of mitochondrial complex I and reactive oxygen species revisited. Mol. Pharm. 2000, 58, 271–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obata, T. Dopamine efflux by MPTP and hydroxyl radical generation. J. Neural Transm. 2002, 109, 1159–1180. [Google Scholar] [CrossRef]
- Hoffman, C.; Aballay, A. Role of neurons in the control of immune defense. Curr. Opin. Immunol. 2019, 60, 30–36. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.L.; Franz, K.J. Phosphorylation-dependent metal binding by alpha-synuclein peptide fragments. J. Biol. Inorg. Chem. 2007, 12, 234–247. [Google Scholar] [CrossRef]
- Liu, L.L.; Franz, K.J. Phosphorylation of an alpha-synuclein peptide fragment enhances metal binding. J. Am. Chem. Soc. 2005, 127, 9662–9663. [Google Scholar] [CrossRef]
- Garcia-Garcia, A.; Rodriguez-Rocha, H.; Madayiputhiya, N.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Biomarkers of protein oxidation in human disease. Curr. Mol. Med. 2012, 12, 681–697. [Google Scholar] [CrossRef]
- Duarte-Jurado, A.P.; Gopar-Cuevas, Y.; Saucedo-Cardenas, O.; de Jesus Loera-Arias, M.; Montes-de-Oca-Luna, R.; Garcia-Garcia, A.; Rodriguez-Rocha, H. Antioxidant Therapeutics in Parkinson’s Disease: Current Challenges and Opportunities. Antioxidants 2021, 10, 453. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Izzo, V.; Bravo San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W., II.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Čepelak, I.; Dodig, S.; Dodig, D. Ferroptosis: Regulated cell death. Arh. Za Hig. Rada I Toksikol. 2020, 71, 99–109. [Google Scholar] [CrossRef]
- Dlouhy, A.C.; Outten, C.E. The iron metallome in eukaryotic organisms. Met. Ions Life Sci. 2013, 12, 241–278. [Google Scholar] [PubMed] [Green Version]
- Casey, J.L.; Hentze, M.W.; Koeller, D.M.; Caughman, S.W.; Rouault, T.A.; Klausner, R.D.; Harford, J.B. Iron-responsive elements: Regulatory RNA sequences that control mRNA levels and translation. Science 1988, 240, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arosio, P.; Elia, L.; Poli, M. Ferritin, cellular iron storage and regulation. IUBMB Life 2017, 69, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Bresgen, N.; Eckl, P.M. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siah, C.W.; Ombiga, J.; Adams, L.A.; Trinder, D.; Olynyk, J.K. Normal iron metabolism and the pathophysiology of iron overload disorders. Clin. Biochem. Rev. 2006, 27, 5–16. [Google Scholar]
- Núñez, M.T.; Urrutia, P.; Mena, N.; Aguirre, P.; Tapia, V.; Salazar, J. Iron toxicity in neurodegeneration. Biometals 2012, 25, 761–776. [Google Scholar] [CrossRef]
- Cotruvo, J.J.A.; Stubbe, J. Metallation and mismetallation of iron and manganese proteins in vitro and in vivo: The class I ribonucleotide reductases as a case study. Metallomics 2012, 4, 1020–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Li, X.; Xie, W.; Luo, F.; Kaur, D.; Andersen, J.K.; Jankovic, J.; Le, W. Genetic iron chelation protects against proteasome inhibition-induced dopamine neuron degeneration. Neurobiol. Dis. 2010, 37, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, C.-W.; Lo, S.Q.; Ang, S.T.; Chew, K.C.M.; Yu, D.; Chai, B.H.; Tan, B.; Tsang, F.; Tai, Y.K.; et al. S-Nitrosylation of Divalent Metal Transporter 1 Enhances Iron Uptake to Mediate Loss of Dopaminergic Neurons and Motoric Deficit. J. Neurosci. 2018, 38, 8364–8377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarjanli, Z.; Ghaedi, K.; Esmaeili, A.; Rahgozar, S.; Zarrabi, A. Iron oxide nanoparticles may damage to the neural tissue through iron accumulation, oxidative stress, and protein aggregation. BMC Neurosci. 2017, 18, 51. [Google Scholar] [CrossRef] [Green Version]
- Ramos, P.; Santos, A.; Pinto, N.R.; Mendes, R.; Magalhães, T.; Almeida, A. Iron levels in the human brain: A post-mortem study of anatomical region differences and age-related changes. J. Trace Elem. Med. Biol. 2014, 28, 13–17. [Google Scholar] [CrossRef]
- Linert, W.; Jameson, G.N. Redox reactions of neurotransmitters possibly involved in the progression of Parkinson’s Disease. J. Inorg. Biochem. 2000, 79, 319–326. [Google Scholar] [CrossRef]
- Youdim, M.B.; Riederer, P.F. A review of the mechanisms and role of monoamine oxidase inhibitors in Parkinson’s disease. Neurology 2004, 63 (Suppl. 2), S32-5. [Google Scholar] [CrossRef]
- Lu, H.; Chen, J.; Huang, H.; Zhou, M.; Zhu, Q.; Yao, S.Q.; Chai, Z.; Hu, Y. Iron modulates the activity of monoamine oxidase B in SH-SY5Y cells. Biometals 2017, 30, 599–607. [Google Scholar] [CrossRef]
- Napolitano, A.; Manini, P.; d’Ischia, M. Oxidation chemistry of catecholamines and neuronal degeneration: An update. Curr. Med. Chem. 2011, 18, 1832–1845. [Google Scholar] [CrossRef] [Green Version]
- Barbusinski, K. Toxicity of Industrial Wastewater Treated by Fenton’s Reagent. Pol. J. Environ. Stud. 2005, 14, 11–16. [Google Scholar]
- Kehrer, J.P. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 2000, 149, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Gilman, C.P.; Chan, S.L.; Guo, Z.; Zhu, X.; Greig, N.; Mattson, M.P. p53 is present in synapses where it mediates mitochondrial dysfunction and synaptic degeneration in response to DNA damage, and oxidative and excitotoxic insults. Neuromolecular Med. 2003, 3, 159–172. [Google Scholar] [CrossRef]
- Koskenkorva-Frank, T.S.; Weiss, G.; Koppenol, W.H.; Burckhardt, S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: Insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic. Biol. Med. 2013, 65, 1174–1194. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Et Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannielli, A.; Bido, S.; Folladori, L.; Segnali, A.; Cancellieri, C.; Maresca, A.; Massimino, L.; Rubio, A.; Morabito, G.; Caporali, L.; et al. Pharmacological Inhibition of Necroptosis Protects from Dopaminergic Neuronal Cell Death in Parkinson’s Disease Models. Cell Rep. 2018, 22, 2066–2079. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, G.; Grúz, J.; D’Acunto, C.W.; Kaňovský, P.; Strnad, M. Cytokinin Plant Hormones Have Neuroprotective Activity in In Vitro Models of Parkinson’s Disease. Molecules 2021, 26, 361. [Google Scholar] [CrossRef]
- Ashraf, A.; Clark, M.; So, P.-W. The Aging of Iron Man. Front. Aging Neurosci. 2018, 10, 65. [Google Scholar] [CrossRef] [Green Version]
- McAllum, E.J.; Hare, D.J.; Volitakis, I.; McLean, C.A.; Bush, A.I.; Finkelstein, D.; Roberts, B.R. Regional iron distribution and soluble ferroprotein profiles in the healthy human brain. Prog. Neurobiol. 2020, 186, 101744. [Google Scholar] [CrossRef]
- Zhang, Z.; Wei, T.; Hou, J.; Li, G.; Yu, S.; Xin, W. Iron-induced oxidative damage and apoptosis in cerebellar granule cells: Attenuation by tetramethylpyrazine and ferulic acid. Eur. J. Pharm. 2003, 467, 41–47. [Google Scholar] [CrossRef]
- Hare, D.; Aror, M.; Jenkins, N.; Finkelstein, D.; Doble, P.; Bush, A. Is early-life iron exposure critical in neurodegeneration? Nat. Rev. Neurol. 2015, 11, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Lei, P.; Hare, D.; Duce, J.; George, J.; Adlard, P.; McLean, C.; Rogers, J.; Cherny, R.; Finkelstein, D.; et al. Parkinson’s disease iron deposition caused by nitric oxide-induced loss of β-amyloid precursor protein. J. Neurosci. 2015, 35, 3591–3597. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Lei, P. Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. Biomed Res. Int. 2014, 2014, 581256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenner, P.; Dexter, D.T.; Sian, J.; Schapira, A.H.; Marsden, C.D. Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann. Neurol. 1992, 32, S82-7. [Google Scholar] [CrossRef]
- Costello, D.J.; Walsh, S.L.; Harrington, H.J.; Walsh, C.H. Concurrent hereditary haemochromatosis and idiopathic Parkinson’s disease: A case report series. J. Neurol. Neurosurg. Psychiatry 2004, 75, 631–633. [Google Scholar] [CrossRef]
- Miyajima, H.; Takahashi, Y.; Kono, S. Aceruloplasminemia, an inherited disorder of iron metabolism. Biometals 2003, 16, 205–213. [Google Scholar] [CrossRef]
- Nielsen, J.E.; Jensen, L.N.; Krabbe, K. Hereditary haemochromatosis: A case of iron accumulation in the basal ganglia associated with a parkinsonian syndrome. J. Neurol. Neurosurg. Psychiatry 1995, 59, 318–321. [Google Scholar] [CrossRef] [Green Version]
- Borie, C.; Gasparini, F.; Verpillat, P.; Bonnet, A.-M.; Agid, Y.; Hetet, G.; Brice, A.; Dürr, A.; Grandchamp, B. French Parkinson’s disease genetic study group. Association study between iron-related genes polymorphisms and Parkinson’s disease. J. Neurol. 2002, 249, 801–804. [Google Scholar] [CrossRef]
- Rhodes, S.; Buchanan, D.; Ahmed, I.; Taylor, K.; Loriot, M.-A.; Sinsheimer, J.; Bronstein, J.; Elbaz, A.; Mellick, G.; Rotter, J.; et al. Pooled analysis of iron-related genes in Parkinson’s disease: Association with transferrin. Neurobiol. Dis. 2014, 62, 172–178. [Google Scholar] [CrossRef] [Green Version]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, P.; Ayton, S.; I Finkelstein, D.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.W.; A Adlard, P.; A Cherny, R.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Schulte, E.C.; Fukumori, A.; Mollenhauer, B.; Hor, H.; Arzberger, T.; Perneczky, R.; Kurz, A.; Diehl-Schmid, J.; Hüll, M.; Lichtner, P.; et al. Rare variants in β-Amyloid precursor protein (APP) and Parkinson’s disease. Eur. J. Hum. Genet. 2015, 23, 1328–1333. [Google Scholar] [CrossRef] [Green Version]
- Edwards-Lee, T.; Ringman, J.M.; Chung, J.; Werner, J.; Morgan, A.; Hyslop, P.S.G.; Thompson, P.; Dutton, R.; Mlikotic, A.; Rogaeva, E.; et al. An African American family with early-onset Alzheimer disease and an APP (T714I) mutation. Neurology 2005, 64, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.; Brooks, W.; Arthur, H.; Creasey, H.; Broe, G. Further evidence for an association between a mutation in the APP gene and Lewy body formation. Neurosci. Lett. 1997, 227, 49–52. [Google Scholar] [CrossRef]
- Rosenberg, C.K.; Pericak-Vance, M.A.; Saunders, A.M.; Gilbert, J.R.; Gaskell, P.C.; Hulette, C.M. Lewy body and Alzheimer pathology in a family with the amyloid-beta precursor protein APP717 gene mutation. Acta Neuropathol. 2000, 100, 145–152. [Google Scholar] [CrossRef]
- Ayton, S.; Lei, P.; Duce, J.A.; Wong, B.X.; Sedjahtera, A.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I. Ceruloplasmin dysfunction and therapeutic potential for Parkinson’s disease. Ann. Neurol. 2013, 73, 554–559. [Google Scholar] [CrossRef]
- Osaki, S.; Johnson, D.A.; Frieden, E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J. Biol. Chem. 1966, 241, 2746–2751. [Google Scholar] [CrossRef]
- Hochstrasser, H.; Bauer, P.; Walter, U.; Behnke, S.; Spiegel, J.; Csoti, I.; Zeiler, B.; Bornemann, A.; Pahnke, J.; Becker, G.; et al. Ceruloplasmin gene variations and substantia nigra hyperechogenicity in Parkinson’s disease. Neurology 2004, 63, 1912–1917. [Google Scholar] [CrossRef] [PubMed]
- Kohno, S.; Miyajima, H.; Takahashi, Y.; Inoue, Y. Aceruloplasminemia with a novel mutation associated with parkinsonism. Neurogenetics 2000, 2, 237–238. [Google Scholar] [CrossRef]
- Nagayama, H.; Hamamoto, M.; Ueda, M.; Nito, C.; Yamaguchi, H.; Katayama, Y. The effect of ascorbic acid on the pharmacokinetics of levodopa in elderly patients with Parkinson disease. Clin. Neuropharmacol. 2004, 27, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.-H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef]
- Sun, Y.; Pham, A.N.; Waite, T.D. The effect of vitamin C and iron on dopamine-mediated free radical generation: Implications to Parkinson’s disease. Dalton Trans 2018, 47, 4059–4069. [Google Scholar] [CrossRef] [PubMed]
- Aronow, W.S. Management of cardiac hemochromatosis. Arch. Med. Sci. 2018, 14, 560–568. [Google Scholar] [CrossRef]
- Taher, A.; Hershko, C.; Cappellini, M.D. Iron overload in thalassaemia intermedia: Reassessment of iron chelation strategies. Br. J. Haematol. 2009, 147, 634–640. [Google Scholar] [CrossRef]
- Febbraro, F.; Andersen, K.J.; Sanchez-Guajardo, V.; Tentillier, N.; Romero-Ramos, M. Chronic intranasal deferoxamine ameliorates motor defects and pathology in the α-synuclein rAAV Parkinson’s model. Exp. Neurol. 2013, 247, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Dexter, D.T.; Statton, S.A.; Whitmore, C.; Freinbichler, W.; Weinberger, P.; Tipton, K.F.; Della Corte, L.; Ward, R.J.; Crichton, R.R. Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson’s disease after peripheral administration. J. Neural. Transm. Vienna 2011, 118, 223–231. [Google Scholar] [CrossRef]
- Kaur, D.; Yantiri, F.; Rajagopalan, S.; Kumar, J.; Mo, J.Q.; Boonplueang, R.; Viswanath, V.; Jacobs, R.; Yang, L.; Beal, M.F.; et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: A novel therapy for Parkinson’s disease. Neuron 2003, 37, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal 2014, 21, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [Green Version]
- Grolez, G.; Moreau, C.; Sablonnière, B.; Garçon, G.; Devedjian, J.C.; Meguig, S.; Gelé, P.; Delmaire, C.; Bordet, R.; Defebvre, L.; et al. Ceruloplasmin activity and iron chelation treatment of patients with Parkinson’s disease. BMC Neurol. 2015, 15, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- University Hospital, Lille; European Commission; ApoPharma. Conservative Iron Chelation as a Disease-Modifying Strategy in Parkinson’s Disease. 2016. Available online: https://ClinicalTrials.gov/show/NCT02655315 (accessed on 17 October 2022).
- Cheli, V.T.; Correale, J.; Paez, P.M.; Pasquini, J.M. Iron Metabolism in Oligodendrocytes and Astrocytes, Implications for Myelination and Remyelination. ASN Neuro 2020, 12, 1759091420962681. [Google Scholar] [CrossRef]
- Ballas, S.K.; Zeidan, A.M.; Duong, V.H.; DeVeaux, M.; Heeney, M.M. A Systematic Review of the Effect of Iron Chelation Therapy on Survival in Patients with Sickle Cell Disease and β-Thalassemia. Blood 2017, 130, 3743. [Google Scholar]
- Li, Y.Q.; Guo, C. A Review on Lactoferrin and Central Nervous System Diseases. Cells 2021, 10, 1810. [Google Scholar] [CrossRef]
- Xu, S.-F.; Zhang, Y.-H.; Wang, S.; Pang, Z.-Q.; Fan, Y.-G.; Li, J.-Y.; Wang, Z.-Y.; Guo, C. Lactoferrin ameliorates dopaminergic neurodegeneration and motor deficits in MPTP-treated mice. Redox Biol. 2019, 21, 101090. [Google Scholar] [CrossRef]
- Kopaeva, M.Y.; Cherepov, A.B.; Nesterenko, M.V.; Zarayskaya, I.Y. Pretreatment with Human Lactoferrin Had a Positive Effect on the Dynamics of Mouse Nigrostriatal System Recovery after Acute MPTP Exposure. Biology 2021, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- University Hospital, Lille. Efficacy and Safety of the Iron Chelator Deferiprone in Parkinson’s Disease. 2009. Available online: https://ClinicalTrials.gov/show/NCT00943748 (accessed on 17 October 2022).
- Focarelli, F.; Giachino, A.; Waldron, K.J. Copper microenvironments in the human body define patterns of copper adaptation in pathogenic bacteria. PLoS Pathog. 2022, 18, e1010617. [Google Scholar] [CrossRef] [PubMed]
- Tsang, T.; Davis, C.I.; Brady, D.C. Copper biology. Curr. Biol. 2021, 31, R421–R427. [Google Scholar] [CrossRef]
- Tapiero, H.; Townsend, D.M.; Tew, K.D. Trace elements in human physiology and pathology. Copper. Biomed Pharm. 2003, 57, 386–398. [Google Scholar] [CrossRef]
- Lutsenko, S.; Bhattacharjee, A.; Hubbard, A.L. Copper handling machinery of the brain. Metallomics 2010, 2, 596–608. [Google Scholar] [CrossRef] [PubMed]
- Tarnacka, B.; Jopowicz, A.; Maślińska, M. Copper, Iron, and Manganese Toxicity in Neuropsychiatric Conditions. Int. J. Mol. Sci. 2021, 22, 7820. [Google Scholar] [CrossRef] [PubMed]
- Amorós, R.; Murcia, M.; González, L.; Soler-Blasco, R.; Rebagliato, M.; Iñiguez, C.; Carrasco, P.; Vioque, J.; Broberg, K.; Levi, M.; et al. Maternal copper status and neuropsychological development in infants and preschool children. Int. J. Hyg. Environ. Health 2019, 222, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef] [Green Version]
- Da, S.; Rashed, L. Oxidative stress and genotoxicity among workers exposed to copper in a factory for non-ferrous industry in Egypt. Egypt. J. Occup. Med. 2019, 43, 1–15. [Google Scholar] [CrossRef]
- Santos, S.; Silva, A.M.; Matos, M.; Monteiro, S.M.; Álvaro, A.R. Copper induced apoptosis in Caco-2 and Hep-G2 cells: Expression of caspases 3, 8 and 9, AIF and p53. Comp. Biochem. Physiol. Part C Toxicol. Pharm. 2016, 185–186, 138–146. [Google Scholar] [CrossRef]
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell. Biol. 2003, 23, 8576–8585. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, V.S.; Fitch, C.A.; Levenson, C.W. Tumor suppressor protein p53 mRNA and subcellular localization are altered by changes in cellular copper in human Hep G2 cells. J. Nutr. 2001, 131, 1427–1432. [Google Scholar] [CrossRef] [Green Version]
- Gromadzka, G.; Tarnacka, B.; Flaga, A.; Adamczyk, A. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 9259. [Google Scholar] [CrossRef]
- Hozumi, I.; Hasegawa, T.; Honda, A.; Ozawa, K.; Hayashi, Y.; Hashimoto, K.; Yamada, M.; Koumura, A.; Sakurai, T.; Kimura, A.; et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J. Neurol. Sci. 2011, 303, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Pall, H.S.; Williams, A.C.; Blake, D.R.; Lunec, J.; Gutteridge, J.M.; Hall, M.; Taylor, A. Raised cerebrospinal-fluid copper concentration in Parkinson’s disease. Lancet 1987, 2, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Genoud, S.; Senior, A.M.; Hare, D.J.; Double, K.L. Meta-Analysis of Copper and Iron in Parkinson’s Disease Brain and Biofluids. Mov. Disord. 2020, 35, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Santner, A.; Uversky, V.N. Metalloproteomics and metal toxicology of α-synuclein. Metallomics 2010, 2, 378–392. [Google Scholar] [CrossRef]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology 1999, 20, 239–247. [Google Scholar]
- Rybicki, B.A.; Johnson, C.C.; Uman, J.; Gorell, J.M. Parkinson’s disease mortality and the industrial use of heavy metals in Michigan. Mov. Disord. 1993, 8, 87–92. [Google Scholar] [CrossRef]
- VanLandingham, J.W.; Tassabehji, N.M.; Somers, R.C.; Levenson, C.W. Expression profiling of p53-target genes in copper-mediated neuronal apoptosis. Neuromolecular Med. 2005, 7, 311–324. [Google Scholar] [CrossRef]
- Lu, Y.; Prudent, M.; Fauvet, B.; Lashuel, H.A.; Girault, H.H. Phosphorylation of α-Synuclein at Y125 and S129 alters its metal binding properties: Implications for understanding the role of α-Synuclein in the pathogenesis of Parkinson’s Disease and related disorders. ACS Chem. Neurosci. 2011, 2, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Miotto, M.C.; Rodriguez, E.E.; Valiente-Gabioud, A.A.; Torres-Monserrat, V.; Binolfi, A.; Quintanar, L.; Zweckstetter, M.; Griesinger, C.; Fernández, C.O. Site-specific copper-catalyzed oxidation of α-synuclein: Tightening the link between metal binding and protein oxidative damage in Parkinson’s disease. Inorg. Chem. 2014, 53, 4350–4358. [Google Scholar] [CrossRef]
- Anandhan, A.; Rodriguez-Rocha, H.; Bohovych, I.; Griggs, A.M.; Zavala-Flores, L.; Reyes-Reyes, E.M.; Seravalli, J.; Stanciu, L.A.; Lee, J.; Rochet, J.-C.; et al. Overexpression of alpha-synuclein at non-toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol. Dis. 2015, 81, 76–92. [Google Scholar] [CrossRef] [Green Version]
- Letelier, M.E.; FaúnNdez, M.; Jara-Sandoval, J.; Molina-Berrã os, A.; Cortãés-Troncoso, J.; Aracena-Parks, P.; Marín-Catalán, R. Mechanisms underlying the inhibition of the cytochrome P450 system by copper ions. J. Appl. Toxicol. 2009, 29, 695–702. [Google Scholar] [CrossRef]
- Letelier, M.E.; Lepe, A.M.; Faúndez, M.; Salazar, J.; Marín, R.; Aracena, P.; Speisky, H. Possible mechanisms underlying copper-induced damage in biological membranes leading to cellular toxicity. Chem. Biol. Interact 2005, 151, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Oh, S.B.; Kim, J.; Kim, K.; Ryu, H.S.; Kim, M.S.; Ayton, S.; Bush, A.I.; Lee, J.Y.; Chung, S.J. Association of metals with the risk and clinical characteristics of Parkinson’s disease. Park. Relat Disord 2018, 55, 117–121. [Google Scholar] [CrossRef]
- Doguer, C.; Ha, J.H.; Collins, J.F. Intersection of Iron and Copper Metabolism in the Mammalian Intestine and Liver. Compr. Physiol. 2018, 8, 1433–1461. [Google Scholar]
- Shi, L.; Huang, C.; Luo, Q.; Xia, Y.; Liu, W.; Zeng, W.; Cheng, A.; Shi, R.; Zhengli, C. Clioquinol improves motor and non-motor deficits in MPTP-induced monkey model of Parkinson’s disease through AKT/mTOR pathway. Aging Albany NY 2020, 12, 9515–9533. [Google Scholar] [CrossRef]
- Zhao, J.; Shi, Q.; Tian, H.; Li, Y.; Liu, Y.; Xu, Z.; Robert, A.; Liu, Q.; Meunier, B. TDMQ20, a Specific Copper Chelator, Reduces Memory Impairments in Alzheimer’s Disease Mouse Models. ACS Chem. Neurosci. 2021, 12, 140–149. [Google Scholar] [CrossRef]
- Barnham, K.J.; Cherny, R.A.; Cappai, R.; Melov, S.; Masters, C.L.; Bush, A.I. Metal-Protein Attenuating Compounds (MPACs) for the Treatment of Alzheimer’s Disease. Drug Des. Rev.-Online Discontin. 2004, 1, 75–82. [Google Scholar]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid Redox Signal 2016, 24, 376–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cukierman, D.S.; Lázaro, D.F.; Sacco, P.; Ferreira, P.R.; Diniz, R.; Fernández, C.O.; Outeirobef, T.F.; Nicolás, A. ReyX1INH, an improved next-generation affinity-optimized hydrazonic ligand, attenuates abnormal copper(I)/copper(II)-α-Syn interactions and affects protein aggregation in a cellular model of synucleinopathy. Dalton Trans. 2020, 49, 16252–16267. [Google Scholar] [CrossRef]
- Cukierman, D.S.; Pinheiro, A.B.; Castiñeiras-Filho, S.L.; da Silva, A.S.; Miotto, M.C.; De Falco, A.; de PRibeiro, T.; Maisonette, S.; da Cunha, A.L.; Hauser-Davis, R.A.; et al. A moderate metal-binding hydrazone meets the criteria for a bioinorganic approach towards Parkinson’s disease: Therapeutic potential, blood-brain barrier crossing evaluation and preliminary toxicological studies. J. Inorg. Biochem. 2017, 170, 160–168. [Google Scholar] [CrossRef]
- Youdim, M.B.; Grünblatt, E.; Mandel, S. The copper chelator, D-penicillamine, does not attenuate MPTP induced dopamine depletion in mice. J. Neural. Transm. Vienna 2007, 114, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Bisaglia, M.; Bubacco, L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules 2020, 10, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios, C.; Alvarez-Vega, R.; Rojas, P. Depletion of copper and manganese in brain after MPTP treatment of mice. Pharm. Toxicol. 1995, 76, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Cukierman, D.S.; Bodnár, N.; Evangelista, B.N.; Nagy, L.; Kállay, C.; Rey, N.A. Impact of pyridine-2-carboxaldehyde-derived aroylhydrazones on the copper-catalyzed oxidation of the M112A PrP103–112 mutant fragment. J. Biol. Inorg. Chem. 2019, 24, 1231–1244. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Alcocer, A.; Duarte-Jurado, A.P.; Soto-Dominguez, A.; Loera-Arias, M.d.J.; Villarreal-Silva, E.E.; Saucedo-Cardenas, O.; de Oca-Luna, R.M.; Garcia-Garcia, A.; Rodriguez-Rocha, H. Unscrambling the Role of Redox-Active Biometals in Dopaminergic Neuronal Death and Promising Metal Chelation-Based Therapy for Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 1256. https://doi.org/10.3390/ijms24021256
Gonzalez-Alcocer A, Duarte-Jurado AP, Soto-Dominguez A, Loera-Arias MdJ, Villarreal-Silva EE, Saucedo-Cardenas O, de Oca-Luna RM, Garcia-Garcia A, Rodriguez-Rocha H. Unscrambling the Role of Redox-Active Biometals in Dopaminergic Neuronal Death and Promising Metal Chelation-Based Therapy for Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(2):1256. https://doi.org/10.3390/ijms24021256
Chicago/Turabian StyleGonzalez-Alcocer, Alfredo, Ana Patricia Duarte-Jurado, Adolfo Soto-Dominguez, Maria de Jesus Loera-Arias, Eliud Enrique Villarreal-Silva, Odila Saucedo-Cardenas, Roberto Montes de Oca-Luna, Aracely Garcia-Garcia, and Humberto Rodriguez-Rocha. 2023. "Unscrambling the Role of Redox-Active Biometals in Dopaminergic Neuronal Death and Promising Metal Chelation-Based Therapy for Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 2: 1256. https://doi.org/10.3390/ijms24021256