
Enhanced Photovoltaic Properties of Y6 Derivatives with Asymmetric Terminal Groups: A Theoretical Insight

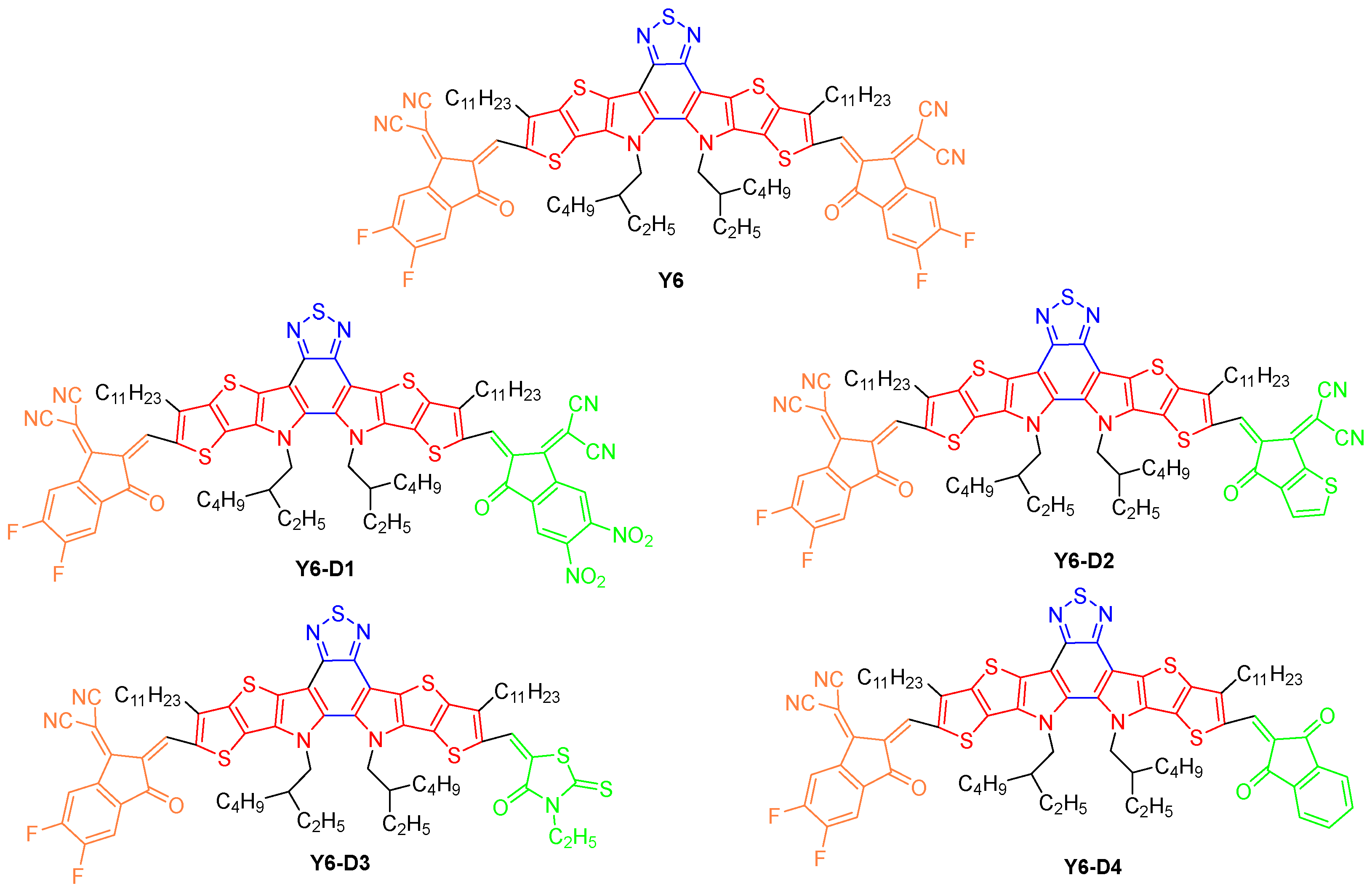{kind=link}
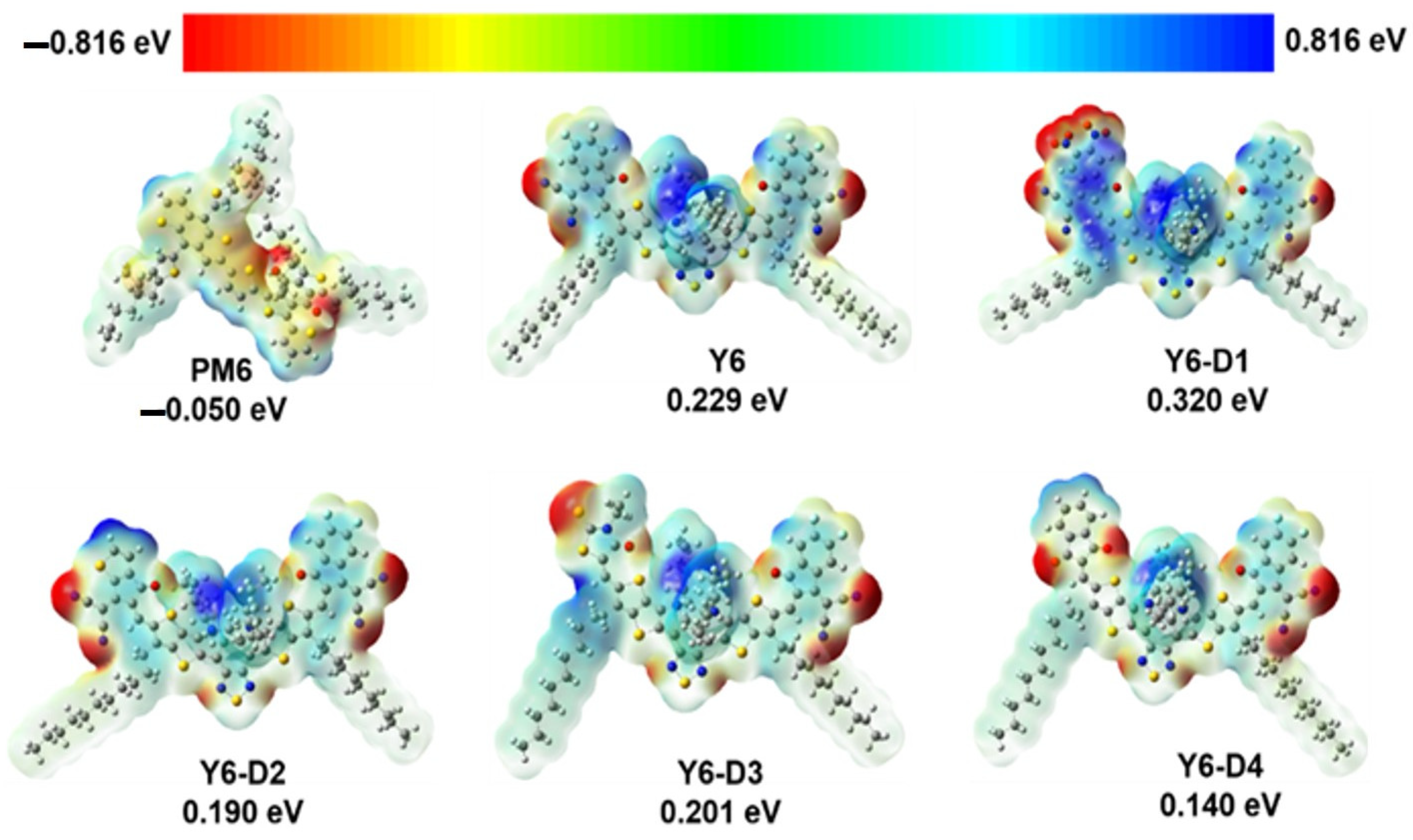{kind=link}
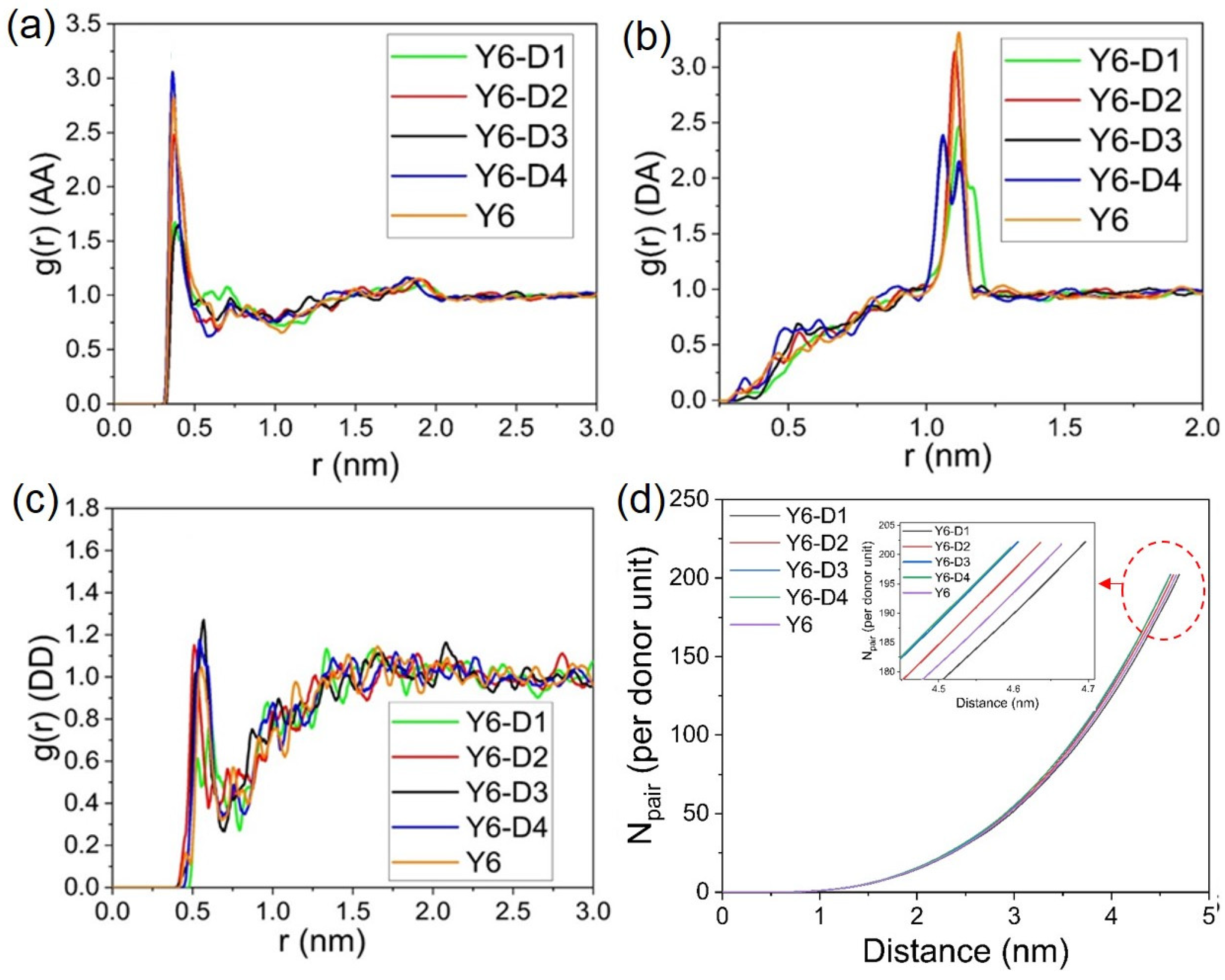{kind=link}
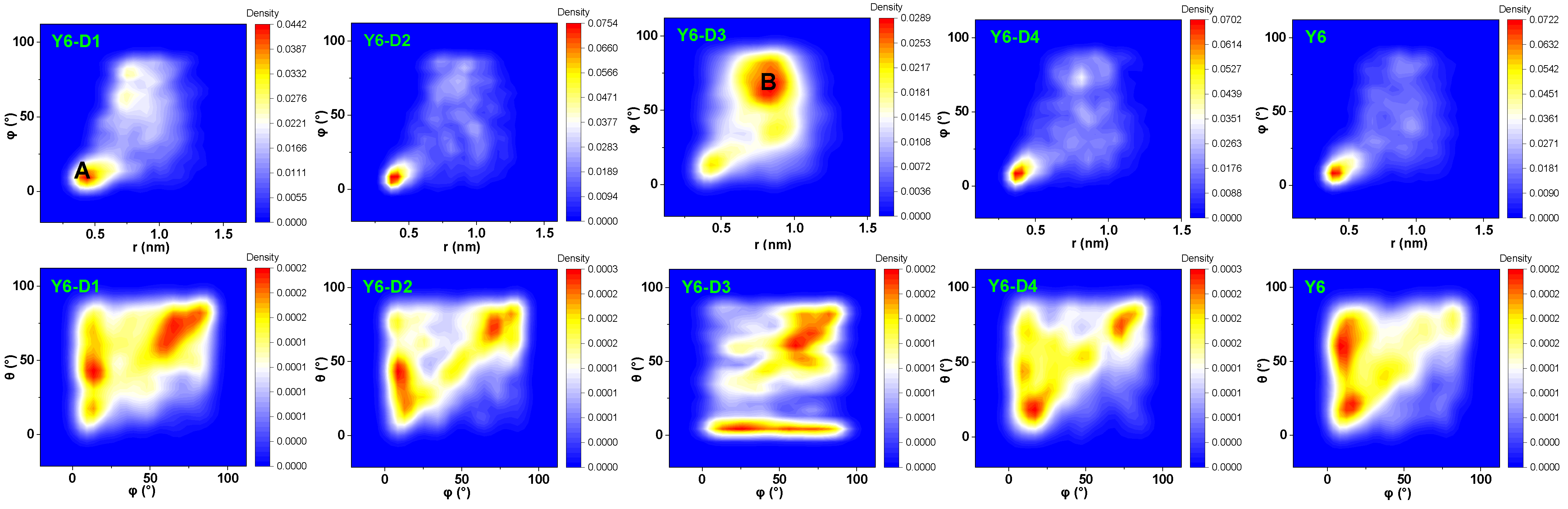{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. ESP of PM6, Y6, and Asymmetric Y6 Derivatives
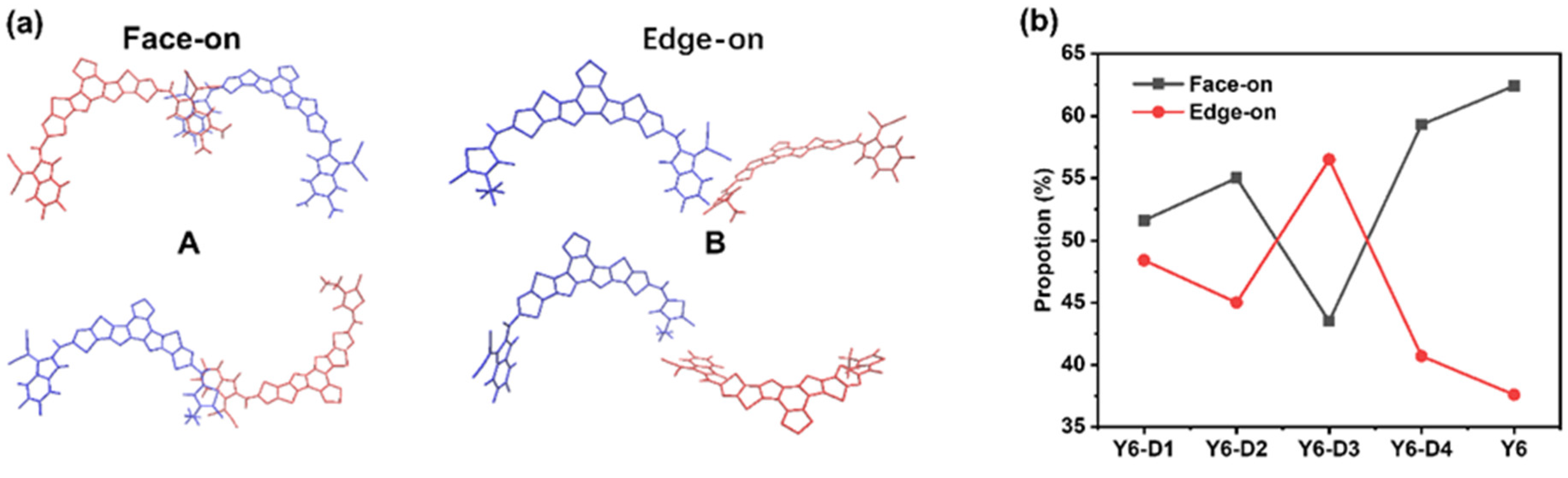
2.2. Molecular Packing
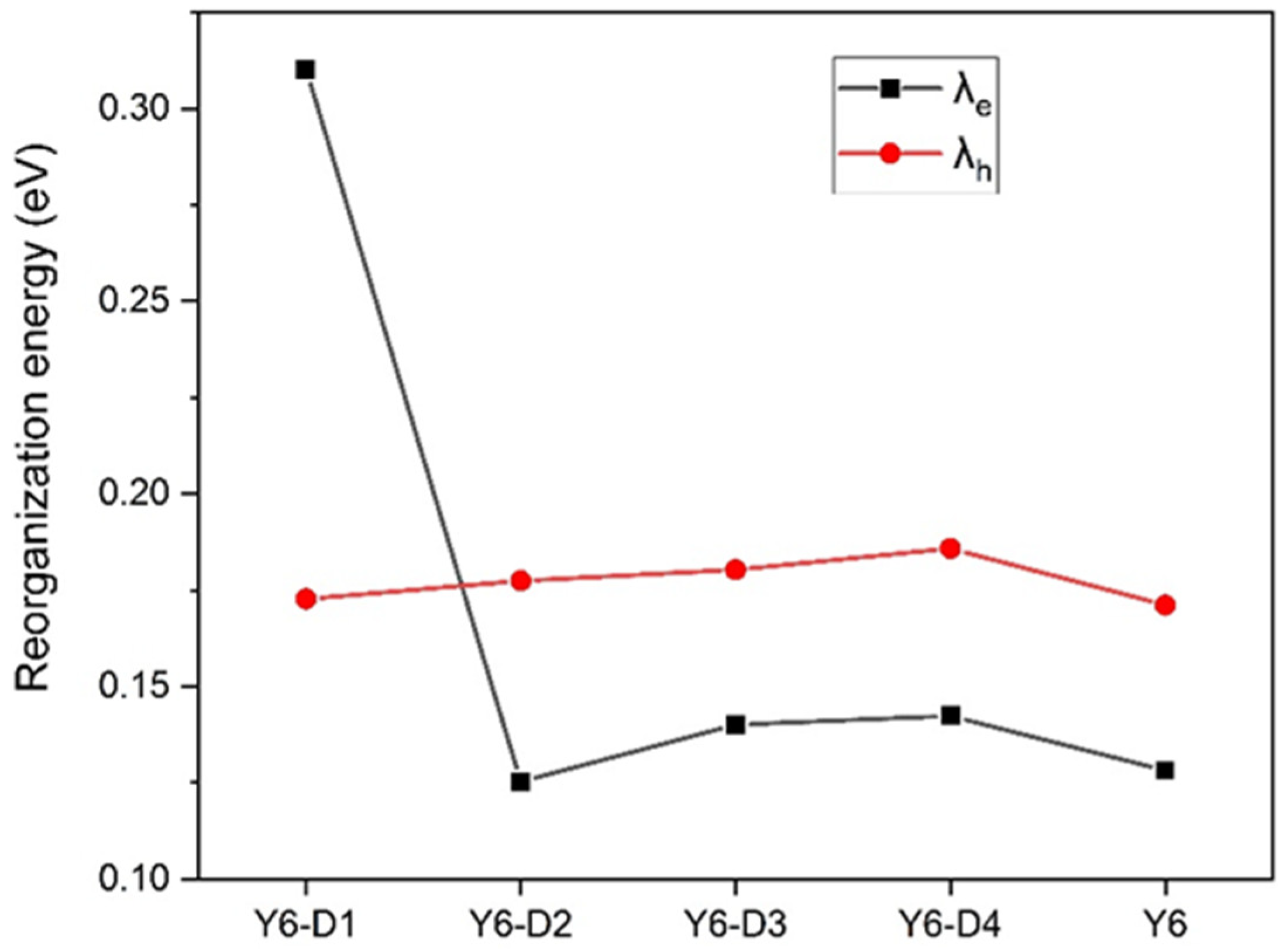
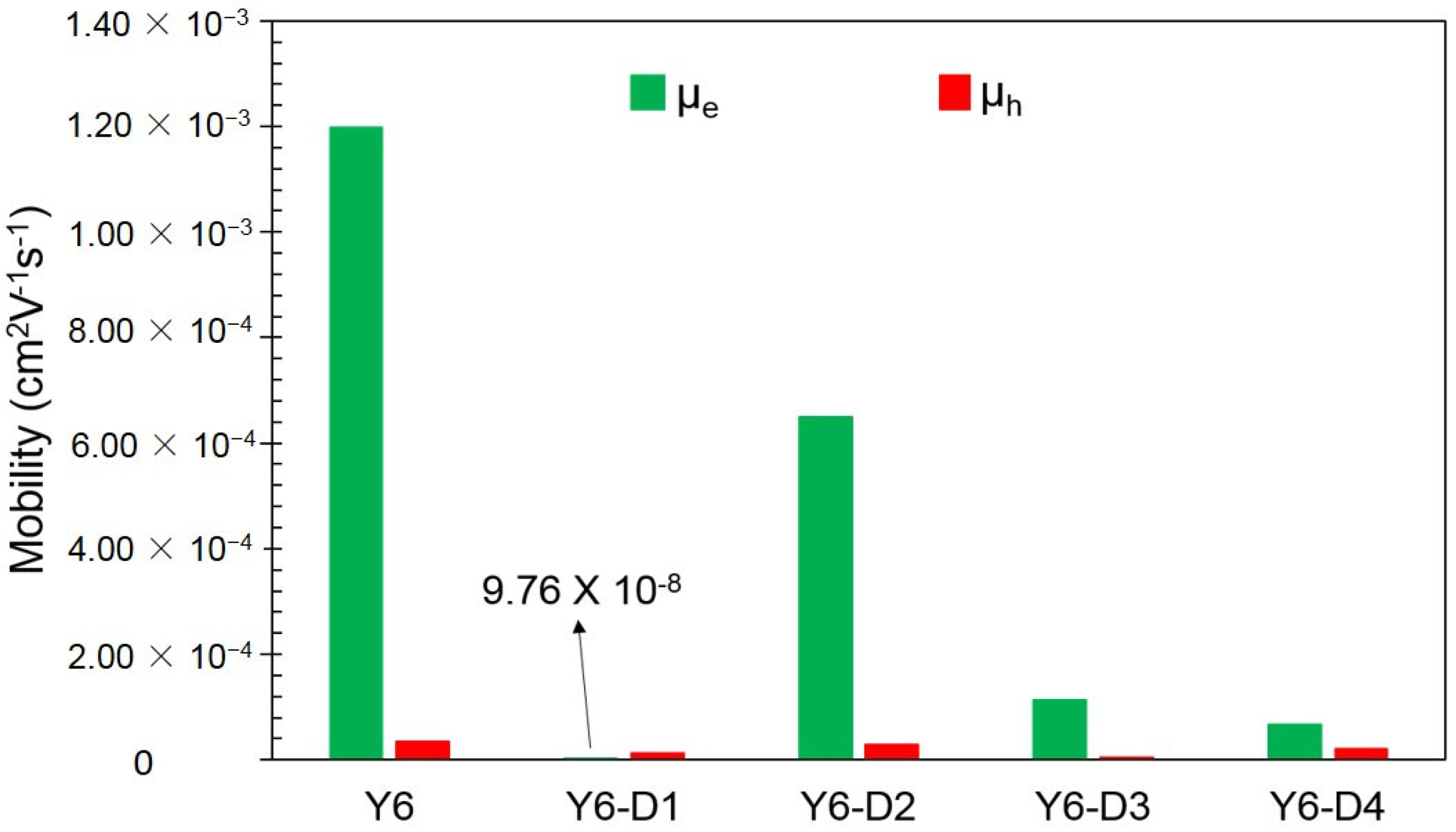
2.3. Reorganization Energy and Charge Carrier Mobility
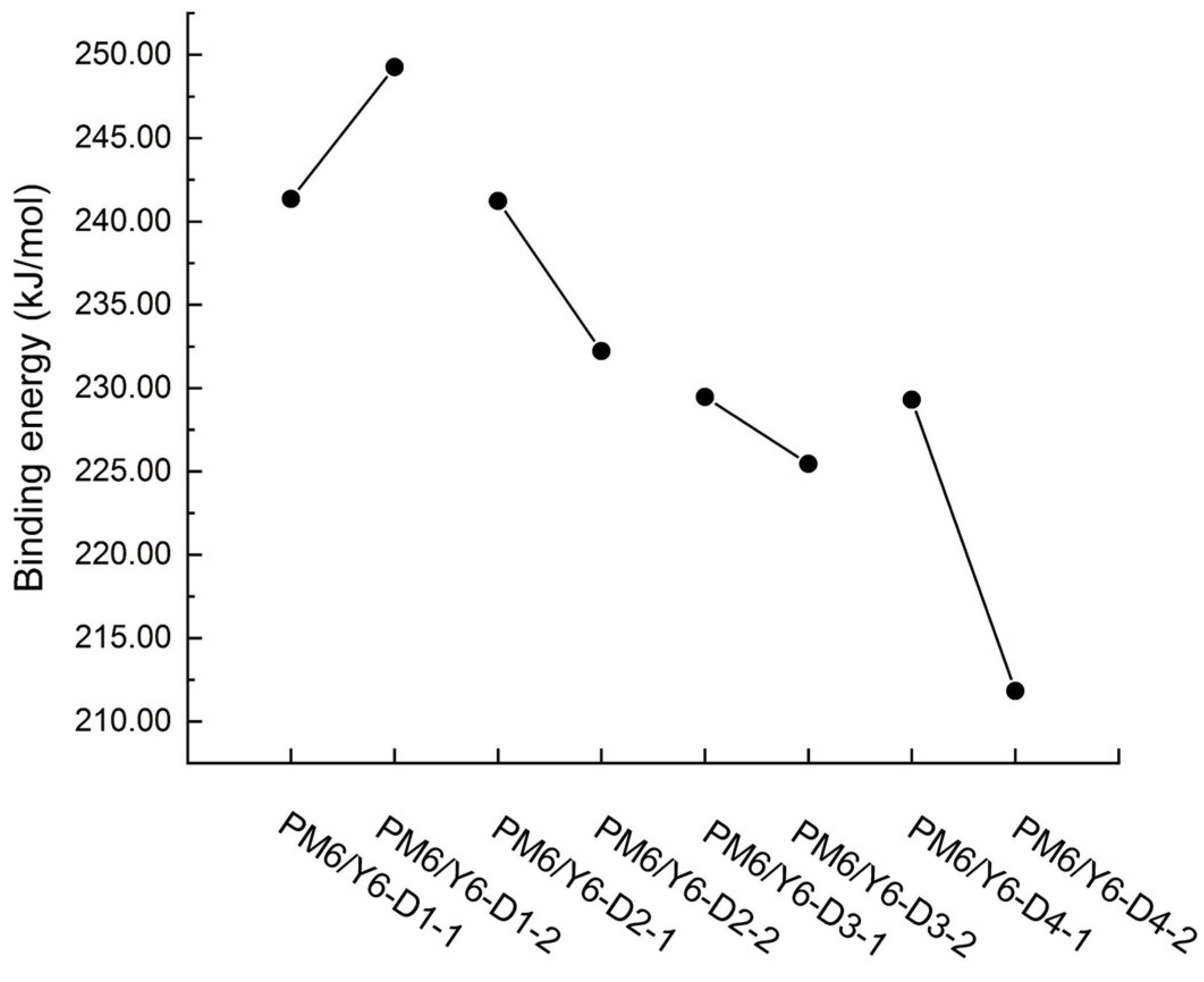
2.4. Binding Energies of PM6/Asymmetric Y6 Derivative Bimolecular Systems
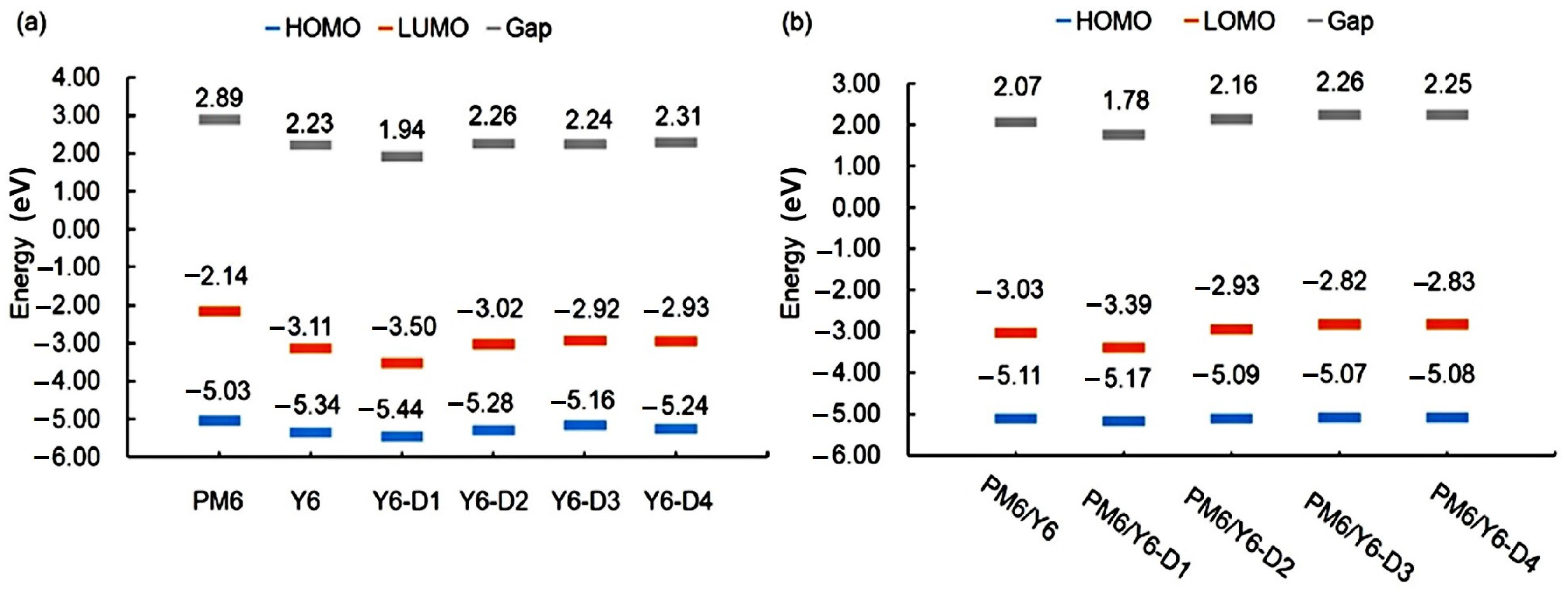
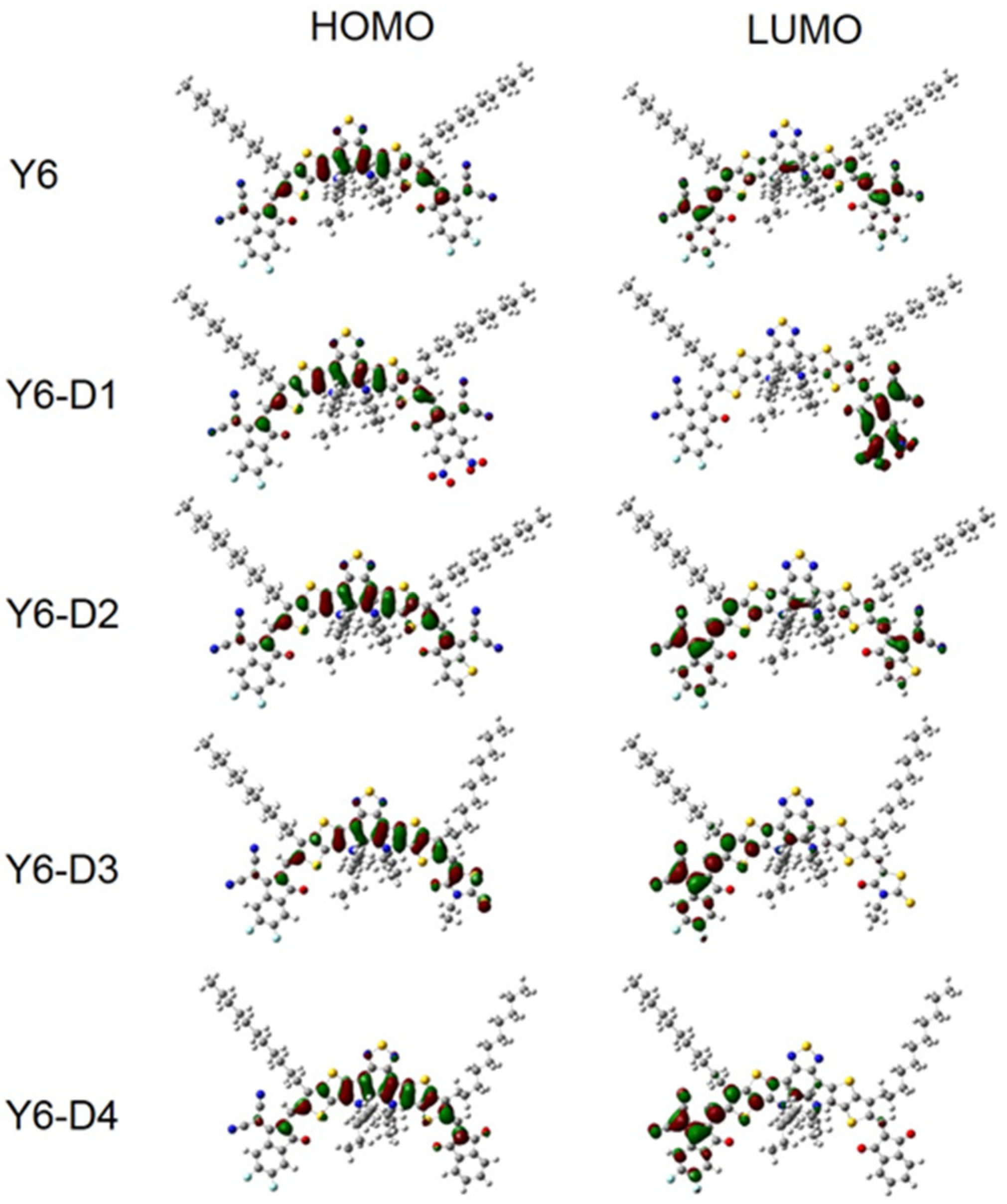
2.5. FMOs and Gap Energies of PM6/Asymmetric Y6 Derivative Bimolecular Systems
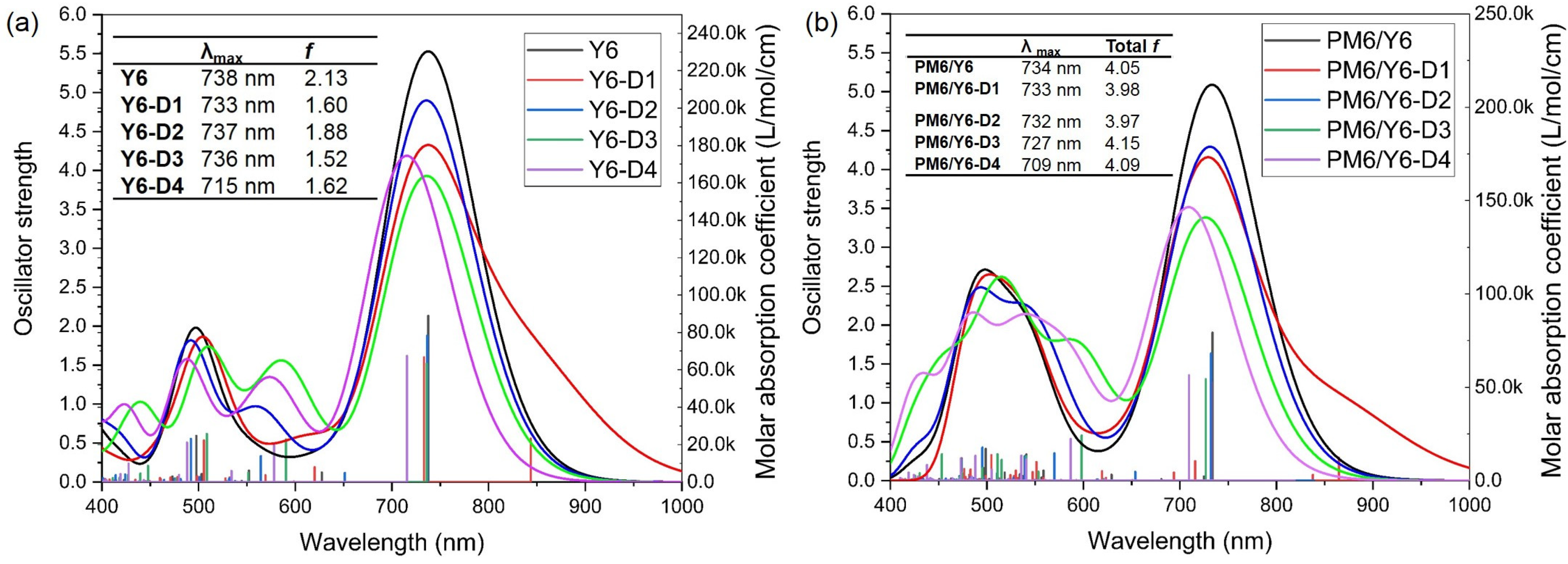
2.6. UV–Vis Spectra of Bimolecular Systems
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cui, Y.; Xu, Y.; Yao, H.; Bi, P.; Hong, L.; Zhang, J.; Zu, Y.; Zhang, T.; Qin, J.; Ren, J.; et al. Single-Junction Organic Photovoltaic Cell with 19% Efficiency. Adv. Mater. 2021, 33, e2102420. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Xu, C.; Zheng, S. Increasing Charge Carrier Mobility through Modifications of Terminal Groups of Y6: A Theoretical Study. Int. J. Mol. Sci. 2023, 24, 8610. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.U.; Iqbal, J.; Mehmood, R.F.; Akram, S.J.; El-Badry, Y.A.; Noor, S.; Khera, R.A. End-capped modification of Y-Shaped dithienothiophen[3,2-b]-pyrrolobenzothiadiazole (TPBT) based non-fullerene acceptors for high performance organic solar cells by using DFT approach. Surf. Interfaces 2022, 30, 101875. [Google Scholar] [CrossRef]
- Zhang, Y.; Ji, Y.; Zhang, Y.; Zhang, W.; Bai, H.; Du, M.; Wu, H.; Guo, Q.; Zhou, E. Recent Progress of Y6-Derived Asymmetric Fused Ring Electron Acceptors. Adv. Funct. Mater. 2022, 32, 2205115. [Google Scholar] [CrossRef]
- Li, S.; Zhan, L.; Jin, Y.; Zhou, G.; Lau, T.K.; Qin, R.; Shi, M.; Li, C.Z.; Zhu, H.; Lu, X.; et al. Asymmetric Electron Acceptors for High-Efficiency and Low-Energy-Loss Organic Photovoltaics. Adv. Mater. 2020, 32, 2001160. [Google Scholar] [CrossRef]
- Li, S.; Zhan, L.; Yao, N.; Xia, X.; Chen, Z.; Yang, W.; He, C.; Zuo, L.; Shi, M.; Zhu, H.; et al. Unveiling structure-performance relationships from multi-scales in non-fullerene organic photovoltaics. Nat. Commun. 2021, 12, 4627. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhong, Z.; Li, L.; Liu, X.; Huang, J.; Wu, H.; Li, M.; Lu, Z.; Yu, J.; Hai, J. Fused-heterocycle engineering on asymmetric non-fullerene acceptors enables organic solar cells approaching 29 mA/cm2 short-circuit current density. Chem. Eng. J. 2022, 430, 132830. [Google Scholar] [CrossRef]
- Li, C.; Zhou, J.; Song, J.; Xu, J.; Zhang, H.; Zhang, X.; Guo, J.; Zhu, L.; Wei, D.; Han, G.; et al. Non-fullerene acceptors with branched side chains and improved molecular packing to exceed 18% efficiency in organic solar cells. Nat. Energy 2021, 6, 605. [Google Scholar] [CrossRef]
- Li, X.; Duan, X.; Liang, Z.; Yan, L.; Yang, Y.; Qiao, J.; Hao, X.; Zhang, C.; Zhang, J.; Li, Y.; et al. Benzo[1,2-b:4,5-b′]difuran Based Polymer Donor for High-Efficiency (>16%) and Stable Organic Solar Cells. Adv. Energy Mater. 2022, 12, 2103684. [Google Scholar] [CrossRef]
- Kupgan, G.; Chen, X.K.; Brédas, J.L. Molecular packing of non-fullerene acceptors for organic solar cells: Distinctive local morphology in Y6 vs. ITIC derivatives. Mater. Today Adv. 2021, 11, 100154. [Google Scholar] [CrossRef]
- Müller-Buschbaum, P. The Active Layer Morphology of Organic Solar Cells Probed with Grazing Incidence Scattering Techniques. Adv. Mater. 2014, 26, 7692–7709. [Google Scholar] [CrossRef]
- Chen, Y.; Bai, F.; Peng, Z.; Zhu, L.; Zhang, J.; Zou, X.; Qin, Y.; Kim, H.K.; Yuan, J.; Ma, L.K.; et al. Asymmetric Alkoxy and Alkyl Substitution on Nonfullerene Acceptors Enabling High-Performance Organic Solar Cells. Adv. Energy Mater. 2020, 11, 2003141. [Google Scholar] [CrossRef]
- Qiu, W.; Zheng, S. Effects of functionalization of Y6 end-groups with electron-withdrawing groups on the photovoltaic properties at the donor-acceptor interfaces of PM6/Y6 OSCs: A theoretical insight. Org. Electron. 2021, 96, 106235. [Google Scholar] [CrossRef]
- Xiao, M.; Tian, Y.; Zheng, S. An insight into the relationship between morphology and open circuit voltage/electronic absorption spectrum at donor-acceptor interface in boron subphthalocyanine chloride/C70 solar cell: A DFT/TDDFT exploration. Org. Electron. 2018, 59, 279–287. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.P.; Rosenthal, K.D.; Ran, N.A.; Seifrid, M.; Bazan, G.C.; Thuc-Quyen, N. Determining the dielectric constants of organic photovoltaic materials using impedance spectroscopy. Adv. Funct. Mater. 2018, 28, 1801542. [Google Scholar] [CrossRef]
- Zheng, Z.; Egger, D.A.; Bredas, J.L.; Kronik, L.; Coropceanu, V. Effect of solid-state polarization on charge-transfer excitations and transport levels at organic interfaces from a screened range-separated hybrid functional. J. Phys. Chem. Lett. 2017, 8, 3277–3283. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab-initio calculation of vibrational absorption and circular-dichroism spectra using density-functional force-fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Tirado-Rives, J.; Jorgensen, W.L. Performance of B3LYP density functional methods for a large set of organic molecules. J. Chem. Theor. Comput. 2008, 4, 297–306. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Shukla, S.; Srivastava, A.; Srivastava, K.; Tandon, P.; Jamalis, J.; Singh, R.B. Non-covalent interactions and spectroscopic study of chalcone derivative 1-(4-chlorophenyl)-3-(5-methylfuran-2-yl) prop-2-en-1-one. J. Mol. Struct. 2020, 1201, 127145. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Dennington, R.K.T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 2012, 38, 314–323. [Google Scholar] [CrossRef]
- Reynolds, C.A.; Essex, J.W.; Richards, W.G. Atomic Charges for Variable Molecular-Conformations. J. Am. Chem. Soc. 1992, 114, 9075–9079. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.D.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges—The Resp Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef]
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Xu, C.L.; Yao, C.; Zheng, S.H. Effects of lateral-chain thiophene fluorination on morphology and charge transport of BDT-T based small molecule donors: A study with multiscale simulations. J. Mater. Chem. C 2021, 9, 14637–14647. [Google Scholar] [CrossRef]
- Han, G.C.; Guo, Y.; Duan, R.H.; Shen, X.; Yi, Y. Importance of side-chain anchoring atoms on electron donor/fullerene interfaces for high-performance organic solar cells. J. Mater. Chem. A 2017, 5, 9316–9321. [Google Scholar] [CrossRef]
- Martinez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; DiNola, A.R.H.J.; Haak, J.R. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics—Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Nose, S. A Unified Formulation of the Constant Temperature Molecular-Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Gorham-Bergeron, E.; Emin, D. Phonon-assisted hopping due to interaction with both acoustical and optical phonons. Phys. Rev. B Solid State 1977, 15, 3667. [Google Scholar] [CrossRef]
- Coropceanu, V.; Cornil, J.; da Silva Filho, D.A.; Olivier, Y.; Silbey, R.; Brédas, J.-L. Charge Transport in Organic Semiconductors. Chem. Rev. 2007, 107, 926–952. [Google Scholar] [CrossRef]
- Young, W.M.; Elcock, E.W. Monte Carlo studies of vacancy migration in binary ordered alloys: I. Proc. Phys. Soc. 1966, 89, 735. [Google Scholar] [CrossRef]
- Marcus, R.A. Electron-Transfer Reactions in Chemistry—Theory and Experiment. Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar] [CrossRef]
- Ridley, J.; Zerner, M. An intermediate neglect of differential overlap technique for spectroscopy: Pyrrole and the azines. Theor. Chim. Acta 1973, 32, 111–134. [Google Scholar] [CrossRef]
- Guilbert, A.A.Y.; Frost, J.M.; Agostinelli, T.; Pires, E.; Lilliu, S.; Macdonald, J.E.; Nelson, J. Influence of Bridging Atom and Side Chains on the Structure and Crystallinity of Cyclopentadithiophene–Benzothiadiazole Polymers. Chem. Mater. 2014, 26, 1226–1233. [Google Scholar] [CrossRef]
- Moreno, M.; Casalegno, M.; Raos, G.; Meille, S.V.; Po, R. Molecular Modeling of Crystalline Alkylthiophene Oligomers and Polymers. J. Phys. Chem. B 2010, 114, 1591–1602. [Google Scholar] [CrossRef]
- Poelking, C.; Andrienko, D. Effect of Polymorphism, Regioregularity and Paracrystallinity on Charge Transport in Poly(3-hexylthiophene) [P3HT] Nanofibers. Macromolecules 2013, 46, 8941–8956. [Google Scholar] [CrossRef]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, Y.; Cao, Z.; Zhang, X.; Zou, Z.; Zheng, S. Enhanced Photovoltaic Properties of Y6 Derivatives with Asymmetric Terminal Groups: A Theoretical Insight. Int. J. Mol. Sci. 2023, 24, 14753. https://doi.org/10.3390/ijms241914753
Xiang Y, Cao Z, Zhang X, Zou Z, Zheng S. Enhanced Photovoltaic Properties of Y6 Derivatives with Asymmetric Terminal Groups: A Theoretical Insight. International Journal of Molecular Sciences. 2023; 24(19):14753. https://doi.org/10.3390/ijms241914753
Chicago/Turabian StyleXiang, Yunjie, Zhijun Cao, Xiaolu Zhang, Zhuo Zou, and Shaohui Zheng. 2023. "Enhanced Photovoltaic Properties of Y6 Derivatives with Asymmetric Terminal Groups: A Theoretical Insight" International Journal of Molecular Sciences 24, no. 19: 14753. https://doi.org/10.3390/ijms241914753