
Exploring the Complex and Multifaceted Interplay between Melanoma Cells and the Tumor Microenvironment


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Melanocyte Function and the Emergence of Melanoma
3. Melanoma Diagnosis
4. Melanoma Susceptibility
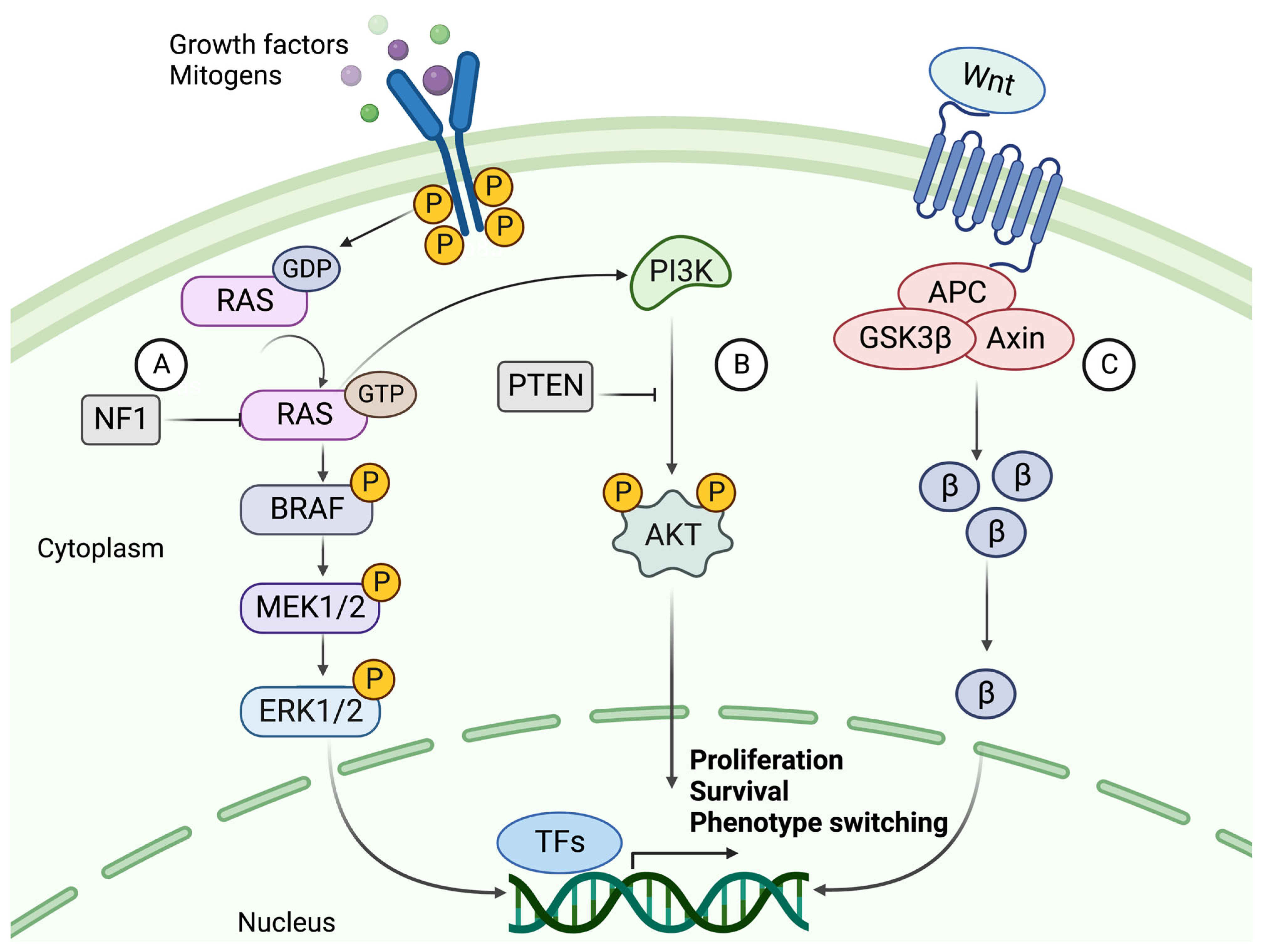
5. Dysregulated Signaling Pathways in Melanoma
5.1. The Mitogen-Activated Protein Kinase (MAPK) Pathway
5.2. The PI3K/AKT Signaling Pathway
5.3. Canonical and Noncanonical WNT Signaling
5.4. The Role of KIT, NF1, TERT, and TP53 in Melanoma
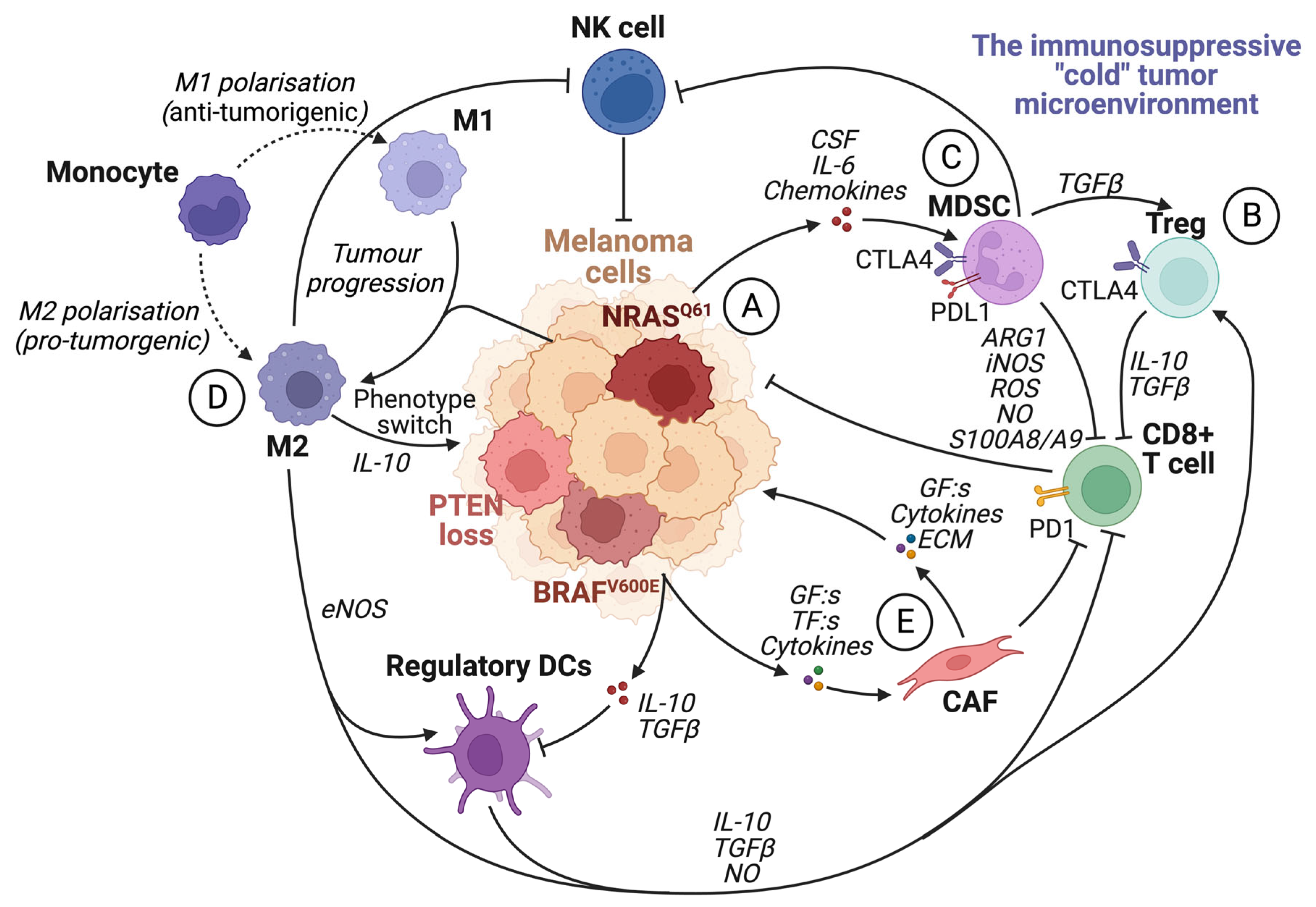
6. The Tumor Microenvironment
6.1. Metabolic Reprogramming of the Tumor Microenvironment
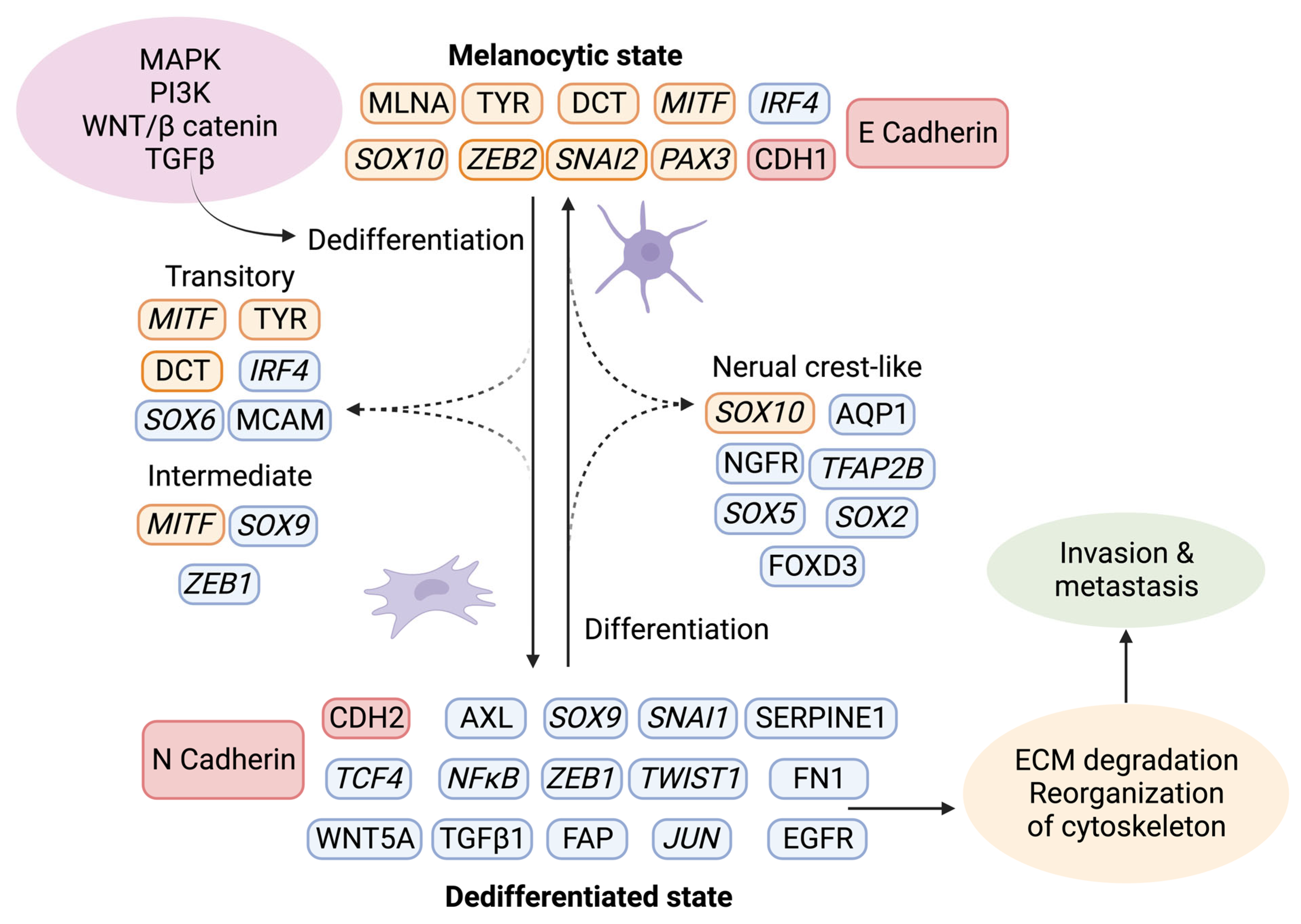
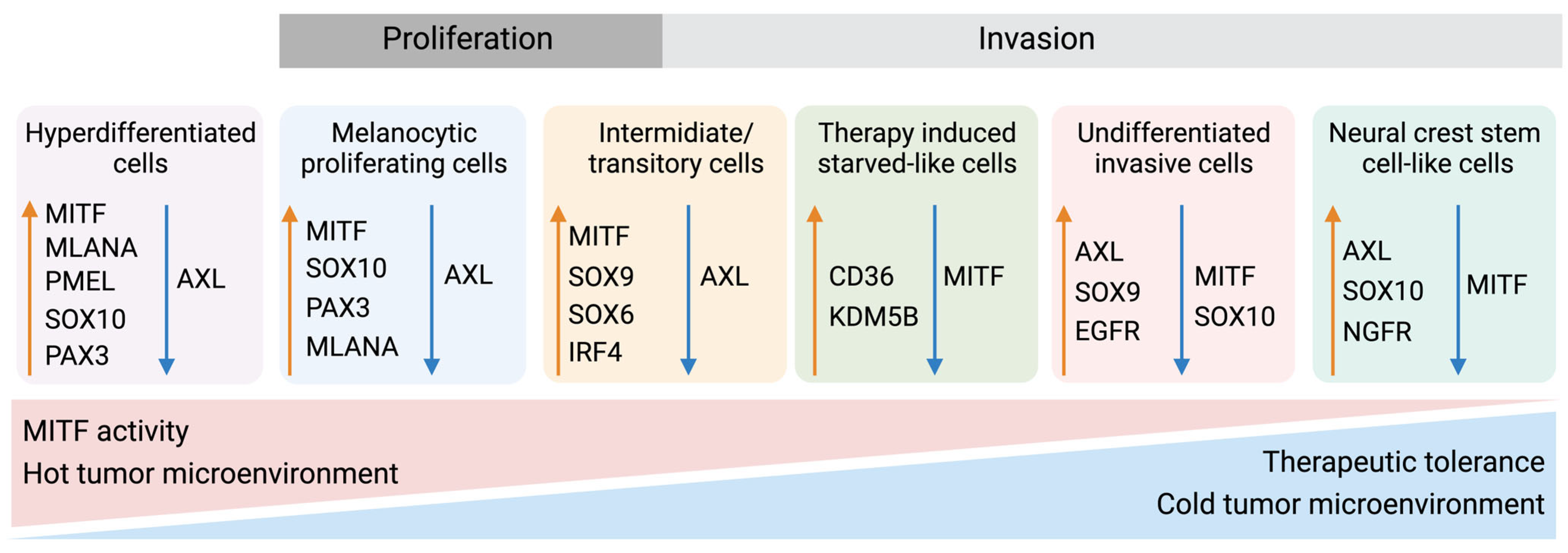
6.2. Phenotype Switching of Melanoma Cells
7. The Immune System
7.1. The Roles of the Different Immune Cells in Melanoma Development
7.2. The Spatial Architecture of the Tumor Immune Microenvironment
8. Molecular Classification of Melanoma—A Way Forward
9. The Relationship between Molecular Mechanisms and Treatment Response
9.1. Immunotherapy
9.2. Targeted Therapy
9.3. Novel Treatments
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATT | Adoptive T-cell transfer |
| CAF | Cancer-associated fibroblast |
| CTLA4 | Cytotoxic T-lymphocyte-associated antigen 4 |
| DC | Dendritic cell |
| ECM | Extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| GF | Growth factor |
| HLA | Human leukocyte antigen |
| ICI | Immune checkpoint inhibitors |
| IF | Immunofluorescence |
| IHC | Immunohistochemistry |
| IL | Interleukin |
| MAPK | Mitogen-activated protein kinase |
| MAPKi | Mitogen-activated protein kinase inhibitors |
| MCR1 | Melanocortin receptor 1 |
| MDSC | Myeloid-derived suppressor cells |
| MHC | Major histocompatibility complex |
| MITF | Microphthalmia-associated transcription factor |
| NCSC | Neural crest stem cell |
| NO | Nitric oxide |
| OIS | Oncogene-induced senescence |
| OXPHOS | Oxidative phosphorylation |
| PD1 | Programmed cell death 1 |
| PD-L1 | Programmed cell death ligand 1 |
| PI3K | Phosphoinositide 3-kinases |
| PRAME | Preferentially expressed antigen in melanoma |
| ROS | Reactive oxygen species |
| RTK | Receptor tyrosine kinase |
| TAM | Tumor-associated macrophage |
| TCGA | The Cancer Genome Atlas Network |
| TF | Transcription factor |
| TIL | Tumor-infiltrating lymphocyte |
| TLS | Tertiary lymphoid structure |
| TME | Tumor microenvironment |
References
- Lallier, T.E. Cell lineage and cell migration in the neural crest. Ann. N. Y. Acad. Sci. 1991, 615, 158–171. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Paskal, W.; Wlodarski, P.K. Intratumor and Intertumor Heterogeneity in Melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef] [PubMed]
- Coricovac, D.; Dehelean, C.; Moaca, E.A.; Pinzaru, I.; Bratu, T.; Navolan, D.; Boruga, O. Cutaneous Melanoma-A Long Road from Experimental Models to Clinical Outcome: A Review. Int. J. Mol. Sci. 2018, 19, 1566. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Singh, D.; Laversanne, M.; Vignat, J.; Vaccarella, S.; Meheus, F.; Cust, A.E.; de Vries, E.; Whiteman, D.C.; Bray, F. Global Burden of Cutaneous Melanoma in 2020 and Projections to 2040. JAMA Dermatol. 2022, 158, 495–503. [Google Scholar] [CrossRef]
- Miller, A.J.; Mihm, M.C. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef]
- Naik, P.P. Cutaneous Malignant Melanoma: A Review of Early Diagnosis and Management. World J. Oncol. 2021, 12, 7–19. [Google Scholar] [CrossRef]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Kollias, N.; Sayre, R.M.; Zeise, L.; Chedekel, M.R. Photoprotection by melanin. J. Photochem. Photobiol. B Biol. 1991, 9, 135–160. [Google Scholar] [CrossRef]
- Raposo, G.; Marks, M.S. Melanosomes--dark organelles enlighten endosomal membrane transport. Nat. Rev. Mol. Cell Biol. 2007, 8, 786–797. [Google Scholar] [CrossRef]
- Young, A.R.; Potten, C.S.; Nikaido, O.; Parsons, P.G.; Boenders, J.; Ramsden, J.M.; Chadwick, C.A. Human melanocytes and keratinocytes exposed to UVB or UVA in vivo show comparable levels of thymine dimers. J. Investig. Dermatol. 1998, 111, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Fadadu, R.P.; Wei, M.L. Ultraviolet A radiation exposure and melanoma: A review. Melanoma Res. 2022, 32, 405–410. [Google Scholar] [CrossRef]
- Țăpoi, D.A.; Gheorghișan-Gălățeanu, A.-A.; Dumitru, A.V.; Ciongariu, A.M.; Furtunescu, A.R.; Marin, A.; Costache, M. Primary Undifferentiated/Dedifferentiated Cutaneous Melanomas—A Review on Histological, Immunohistochemical, and Molecular Features with Emphasis on Prognosis and Treatment. Int. J. Mol. Sci. 2023, 24, 9985. [Google Scholar]
- Gheoca Mutu, D.E.; Avino, A.; Balcangiu-Stroescu, A.E.; Mehedințu, M.; Bălan, D.G.; Brîndușe, L.A.; Popescu, A.M.; Ionescu, D.; Cristea, B.M.; Tomescu, L.F.; et al. Histopathological evaluation of cutaneous malignant melanoma: A retrospective study. Exp. Ther. Med. 2022, 23, 402. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dréno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 1: Diagnostics: Update 2022. Eur. J. Cancer 2022, 170, 236–255. [Google Scholar] [CrossRef]
- Lopes, J.; Rodrigues, C.M.P.; Gaspar, M.M.; Reis, C.P. Melanoma Management: From Epidemiology to Treatment and Latest Advances. Cancers 2022, 14, 4652. [Google Scholar] [CrossRef]
- Weinstein, D.; Leininger, J.; Hamby, C.; Safai, B. Diagnostic and prognostic biomarkers in melanoma. J. Clin. Aesthetic Dermatol. 2014, 7, 13–24. [Google Scholar]
- Banerjee, S.S.; Harris, M. Morphological and immunophenotypic variations in malignant melanoma. Histopathology 2000, 36, 387–402. [Google Scholar] [CrossRef]
- Nwafor, J.N.; Torere, B.E.; Agu, E.; Kadiku, L.; Ogunyemi, T.; Akinsanya, P.A.; Araromi, O.O.; Akahara, D.E.; Okobi, O.E. The Role of Biomarkers in the Diagnosis and Prognosis of Different Stages of Melanoma. Cureus 2023, 15, e38693. [Google Scholar] [CrossRef] [PubMed]
- Cazzato, G.; Mangialardi, K.; Falcicchio, G.; Colagrande, A.; Ingravallo, G.; Arezzo, F.; Giliberti, G.; Trilli, I.; Loizzi, V.; Lettini, T.; et al. Preferentially Expressed Antigen in Melanoma (PRAME) and Human Malignant Melanoma: A Retrospective Study. Genes 2022, 13, 545. [Google Scholar] [CrossRef] [PubMed]
- Sondermann, W.; Zimmer, L.; Schadendorf, D.; Roesch, A.; Klode, J.; Dissemond, J. Initial misdiagnosis of melanoma located on the foot is associated with poorer prognosis. Medicine 2016, 95, e4332. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Flores, A.; Singh, R.; Cassarino, D.S. Top 10 Differential Diagnoses for Desmoplastic Melanoma. Head Neck Pathol. 2023, 17, 143–153. [Google Scholar] [CrossRef]
- Ikeda, H.; Lethé, B.; Lehmann, F.; van Baren, N.; Baurain, J.F.; de Smet, C.; Chambost, H.; Vitale, M.; Moretta, A.; Boon, T.; et al. Characterization of an antigen that is recognized on a melanoma showing partial HLA loss by CTL expressing an NK inhibitory receptor. Immunity 1997, 6, 199–208. [Google Scholar] [CrossRef]
- Lezcano, C.; Jungbluth, A.A.; Nehal, K.S.; Hollmann, T.J.; Busam, K.J. PRAME Expression in Melanocytic Tumors. Am. J. Surg. Pathol. 2018, 42, 1456–1465. [Google Scholar] [CrossRef]
- Ricci, C.; Franceschini, T.; Giunchi, F.; Grillini, M.; Ambrosi, F.; Massari, F.; Mollica, V.; Colecchia, M.; Fiorentino, M. Immunohistochemical Expression of Preferentially Expressed Antigen in Melanoma (PRAME) in the Uninvolved Background Testis, Germ Cell Neoplasia In Situ, and Germ Cell Tumors of the Testis. Am. J. Clin. Pathol. 2022, 157, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Arndt, I.; Sharma, R.; Riesenberg, S.; Jostes, S.; Schneider, S.; Hölzel, M.; Kristiansen, G.; Schorle, H. The cancer/testis-antigen PRAME supports the pluripotency network and represses somatic and germ cell differentiation programs in seminomas. Br. J. Cancer 2016, 115, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Lezcano, C.; Jungbluth, A.A.; Busam, K.J. Comparison of Immunohistochemistry for PRAME With Cytogenetic Test Results in the Evaluation of Challenging Melanocytic Tumors. Am. J. Surg. Pathol. 2020, 44, 893–900. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef]
- Soura, E.; Eliades, P.J.; Shannon, K.; Stratigos, A.J.; Tsao, H. Hereditary melanoma: Update on syndromes and management: Genetics of familial atypical multiple mole melanoma syndrome. J. Am. Acad. Dermatol. 2016, 74, 395–407; quiz 408–410. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ek, W.E.; Whiteman, D.; Vaughan, T.L.; Spurdle, A.B.; Easton, D.F.; Pharoah, P.D.; Thompson, D.J.; Dunning, A.M.; Hayward, N.K.; et al. Most common ‘sporadic’ cancers have a significant germline genetic component. Hum. Mol. Genet. 2014, 23, 6112–6118. [Google Scholar] [CrossRef]
- Read, J.; Wadt, K.A.; Hayward, N.K. Melanoma genetics. J. Med. Genet. 2016, 53, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; Delancey, J.O.; Raskin, L.; Everett, J.; Jeter, J.; Begg, C.B.; Orlow, I.; Berwick, M.; Armstrong, B.K.; Kricker, A.; et al. Risk of non-melanoma cancers in first-degree relatives of CDKN2A mutation carriers. J. Natl. Cancer Inst. 2012, 104, 953–956. [Google Scholar] [CrossRef]
- Bandarchi, B.; Ma, L.; Navab, R.; Seth, A.; Rasty, G. From melanocyte to metastatic malignant melanoma. Dermatol. Res. Pract. 2010, 2010, 583748. [Google Scholar] [CrossRef]
- Tagliabue, E.; Gandini, S.; Bellocco, R.; Maisonneuve, P.; Newton-Bishop, J.; Polsky, D.; Lazovich, D.; Kanetsky, P.A.; Ghiorzo, P.; Gruis, N.A.; et al. MC1R variants as melanoma risk factors independent of at-risk phenotypic characteristics: A pooled analysis from the M-SKIP project. Cancer Manag. Res. 2018, 10, 1143–1154. [Google Scholar] [CrossRef]
- Fargnoli, M.C.; Gandini, S.; Peris, K.; Maisonneuve, P.; Raimondi, S. MC1R variants increase melanoma risk in families with CDKN2A mutations: A meta-analysis. Eur. J. Cancer 2010, 46, 1413–1420. [Google Scholar] [CrossRef]
- Cao, J.; Wan, L.; Hacker, E.; Dai, X.; Lenna, S.; Jimenez-Cervantes, C.; Wang, Y.; Leslie, N.R.; Xu, G.X.; Widlund, H.R.; et al. MC1R is a potent regulator of PTEN after UV exposure in melanocytes. Mol. Cell 2013, 51, 409–422. [Google Scholar] [CrossRef]
- Shtivelman, E.; Davies, M.Q.; Hwu, P.; Yang, J.; Lotem, M.; Oren, M.; Flaherty, K.T.; Fisher, D.E. Pathways and therapeutic targets in melanoma. Oncotarget 2014, 5, 1701–1752. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.; Lok, H.C.; Sahni, S.; Lane, D.J.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta 2016, 1863, 770–784. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Cyclin D1 multitasks. Nature 2011, 474, 171–172. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- van Tuyn, J.; Jaber-Hijazi, F.; MacKenzie, D.; Cole, J.J.; Mann, E.; Pawlikowski, J.S.; Rai, T.S.; Nelson, D.M.; McBryan, T.; Ivanov, A.; et al. Oncogene-Expressing Senescent Melanocytes Up-Regulate MHC Class II, a Candidate Melanoma Suppressor Function. J. Investig. Dermatol. 2017, 137, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Tchernitsa, O.I.; Hinzmann, B.; Schmitz, A.C.; Grips, M.; Hellriegel, M.; Sers, C.; Rosenthal, A.; Schäfer, R. A genome-wide survey of RAS transformation targets. Nat. Genet. 2000, 24, 144–152. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wang, H.; Li, C. Signal pathways of melanoma and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 424. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Z.; Dhillon, A.S.; Anderson, R.L.; McArthur, G.; Ferrao, P.T. Phenotype switching in melanoma: Implications for progression and therapy. Front. Oncol. 2015, 5, 31. [Google Scholar] [CrossRef]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904.e5. [Google Scholar] [CrossRef]
- Campbell, P.M.; Der, C.J. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 2004, 14, 105–114. [Google Scholar] [CrossRef]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S17–S27. [Google Scholar] [CrossRef]
- Randic, T.; Kozar, I.; Margue, C.; Utikal, J.; Kreis, S. NRAS mutant melanoma: Towards better therapies. Cancer Treat. Rev. 2021, 99, 102238. [Google Scholar] [CrossRef]
- Cheung, M.; Sharma, A.; Madhunapantula, S.V.; Robertson, G.P. Akt3 and mutant V600E B-Raf cooperate to promote early melanoma development. Cancer Res. 2008, 68, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Vredeveld, L.C.; Possik, P.A.; Smit, M.A.; Meissl, K.; Michaloglou, C.; Horlings, H.M.; Ajouaou, A.; Kortman, P.C.; Dankort, D.; McMahon, M.; et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev. 2012, 26, 1055–1069. [Google Scholar] [CrossRef] [PubMed]
- Delmas, V.; Beermann, F.; Martinozzi, S.; Carreira, S.; Ackermann, J.; Kumasaka, M.; Denat, L.; Goodall, J.; Luciani, F.; Viros, A.; et al. Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev. 2007, 21, 2923–2935. [Google Scholar] [CrossRef]
- Cai, J.; Guan, H.; Fang, L.; Yang, Y.; Zhu, X.; Yuan, J.; Wu, J.; Li, M. MicroRNA-374a activates Wnt/β-catenin signaling to promote breast cancer metastasis. J. Clin. Investig. 2013, 123, 566–579. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.D.M.; Guhan, S.; Tsao, H. KIT and Melanoma: Biological Insights and Clinical Implications. Yonsei Med. J. 2020, 61, 562–571. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef]
- Yuan, Y.; Jiang, Y.C.; Sun, C.K.; Chen, Q.M. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep. 2016, 35, 2499–2515. [Google Scholar] [CrossRef]
- Kimm, M.A.; Klenk, C.; Alunni-Fabbroni, M.; Kästle, S.; Stechele, M.; Ricke, J.; Eisenblätter, M.; Wildgruber, M. Tumor-Associated Macrophages-Implications for Molecular Oncology and Imaging. Biomedicines 2021, 9, 374. [Google Scholar] [CrossRef]
- van Pelt, G.W.; Sandberg, T.P.; Morreau, H.; Gelderblom, H.; van Krieken, J.; Tollenaar, R.; Mesker, W.E. The tumour-stroma ratio in colon cancer: The biological role and its prognostic impact. Histopathology 2018, 73, 197–206. [Google Scholar] [CrossRef]
- Neagu, M. Metabolic Traits in Cutaneous Melanoma. Front. Oncol. 2020, 10, 851. [Google Scholar] [CrossRef]
- Trotta, A.P.; Gelles, J.D.; Serasinghe, M.N.; Loi, P.; Arbiser, J.L.; Chipuk, J.E. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J. Biol. Chem. 2017, 292, 11727–11739. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Danhier, P.; Bański, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.R.; Thomas, K.J. The role of mitochondria in the development and progression of lung cancer. Comput. Struct. Biotechnol. J. 2013, 6, e201303019. [Google Scholar] [CrossRef]
- Zhang, X.; Tai, Z.; Miao, F.; Huang, H.; Zhu, Q.; Bao, L.; Chen, Z. Metabolism heterogeneity in melanoma fuels deactivation of immunotherapy: Predict before protect. Front. Oncol. 2022, 12, 1046102. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Ortenberg, R.; Varanasi, S.K.; Mangalhara, K.C.; Mardamshina, M.; Markovits, E.; Baruch, E.N.; Tripple, V.; Arama-Chayoth, M.; Greenberg, E.; et al. Proteomics of Melanoma Response to Immunotherapy Reveals Mitochondrial Dependence. Cell 2019, 179, 236–250.e18. [Google Scholar] [CrossRef]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-associated fibroblasts: Overview, progress, challenges, and directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Venning, F.A.; Wullkopf, L.; Erler, J.T. Targeting ECM Disrupts Cancer Progression. Front. Oncol. 2015, 5, 224. [Google Scholar] [CrossRef]
- Martinez-Vidal, L.; Murdica, V.; Venegoni, C.; Pederzoli, F.; Bandini, M.; Necchi, A.; Salonia, A.; Alfano, M. Causal contributors to tissue stiffness and clinical relevance in urology. Commun. Biol. 2021, 4, 1011. [Google Scholar] [CrossRef]
- Mesker, W.E.; van Pelt, G.W.; Tollenaar, R. Tumor stroma as contributing factor in the lymph node metastases process? Oncotarget 2019, 10, 922–923. [Google Scholar] [CrossRef]
- van Pelt, G.W.; Kjær-Frifeldt, S.; van Krieken, J.; Al Dieri, R.; Morreau, H.; Tollenaar, R.; Sørensen, F.B.; Mesker, W.E. Scoring the tumor-stroma ratio in colon cancer: Procedure and recommendations. Virchows Arch. Int. J. Pathol. 2018, 473, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Pedri, D.; Karras, P.; Landeloos, E.; Marine, J.C.; Rambow, F. Epithelial-to-mesenchymal-like transition events in melanoma. FEBS J. 2022, 289, 1352–1368. [Google Scholar] [CrossRef]
- Cham, J.; Shavit, A.; Ebrahimi, A.; Viray, M.; Gibbs, P.; Bhangoo, M.S. Malignant Melanoma With Neuroendocrine Differentiation: A Case Report and Literature Review. Front. Oncol. 2021, 11, 763992. [Google Scholar] [CrossRef] [PubMed]
- Nettersheim, D.; Schorle, H. The plasticity of germ cell cancers and its dependence on the cellular microenvironment. J. Cell. Mol. Med. 2017, 21, 1463–1467. [Google Scholar] [CrossRef]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef]
- Mort, R.L.; Jackson, I.J.; Patton, E.E. The melanocyte lineage in development and disease. Development 2015, 142, 1387. [Google Scholar] [CrossRef]
- Hoek, K.; Rimm, D.L.; Williams, K.R.; Zhao, H.; Ariyan, S.; Lin, A.; Kluger, H.M.; Berger, A.J.; Cheng, E.; Trombetta, E.S.; et al. Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res. 2004, 64, 5270–5282. [Google Scholar] [CrossRef]
- Pearlman, R.L.; Montes de Oca, M.K.; Pal, H.C.; Afaq, F. Potential therapeutic targets of epithelial-mesenchymal transition in melanoma. Cancer Lett. 2017, 391, 125–140. [Google Scholar] [CrossRef]
- Hodorogea, A.; Calinescu, A.; Antohe, M.; Balaban, M.; Nedelcu, R.I.; Turcu, G.; Ion, D.A.; Badarau, I.A.; Popescu, C.M.; Popescu, R.; et al. Epithelial-Mesenchymal Transition in Skin Cancers: A Review. Anal. Cell. Pathol. 2019, 2019, 3851576. [Google Scholar] [CrossRef]
- Sanna, A.; Phung, B.; Mitra, S.; Lauss, M.; Choi, J.; Zhang, T.; Njauw, C.N.; Cordero, E.; Harbst, K.; Rosengren, F.; et al. DNA promoter hypermethylation of melanocyte lineage genes determines melanoma phenotype. JCI Insight 2022, 7, e156577. [Google Scholar] [CrossRef]
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006, 19, 290–302. [Google Scholar] [CrossRef]
- Rodriguez, M.; Aladowicz, E.; Lanfrancone, L.; Goding, C.R. Tbx3 represses E-cadherin expression and enhances melanoma invasiveness. Cancer Res. 2008, 68, 7872–7881. [Google Scholar] [CrossRef]
- Huang, F.; Santinon, F.; Flores González, R.E.; Del Rincón, S.V. Melanoma Plasticity: Promoter of Metastasis and Resistance to Therapy. Front. Oncol. 2021, 11, 756001. [Google Scholar] [CrossRef]
- Pagliuca, C.; Di Leo, L.; De Zio, D. New Insights into the Phenotype Switching of Melanoma. Cancers 2022, 14, 6118. [Google Scholar] [CrossRef]
- Hossain, S.M.; Eccles, M.R. Phenotype Switching and the Melanoma Microenvironment; Impact on Immunotherapy and Drug Resistance. Int. J. Mol. Sci. 2023, 24, 1601. [Google Scholar] [CrossRef]
- Carlino, M.S.; Larkin, J.; Long, G.V. Immune checkpoint inhibitors in melanoma. Lancet 2021, 398, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Kalaora, S.; Nagler, A.; Wargo, J.A.; Samuels, Y. Mechanisms of immune activation and regulation: Lessons from melanoma. Nat. Rev. Cancer 2022, 22, 195–207. [Google Scholar] [CrossRef]
- Marzagalli, M.; Ebelt, N.D.; Manuel, E.R. Unraveling the crosstalk between melanoma and immune cells in the tumor microenvironment. Semin. Cancer Biol. 2019, 59, 236–250. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Lee, N.; Zakka, L.R.; Mihm, M.C., Jr.; Schatton, T. Tumour-infiltrating lymphocytes in melanoma prognosis and cancer immunotherapy. Pathology 2016, 48, 177–187. [Google Scholar] [CrossRef]
- Ohue, Y.; Nishikawa, H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019, 110, 2080–2089. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pagès, F.; Sautès-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.K.-K.; Chung, J.Y.-F.; Tang, P.C.-T.; Chan, A.S.-W.; Ho, J.Y.-Y.; Lin, T.P.-T.; Chen, J.; Leung, K.-T.; To, K.-F.; Lan, H.-Y.; et al. TGF-β signaling networks in the tumor microenvironment. Cancer Lett. 2022, 550, 215925. [Google Scholar] [CrossRef]
- Komi, D.E.A.; Redegeld, F.A. Role of Mast Cells in Shaping the Tumor Microenvironment. Clin. Rev. Allergy Immunol. 2020, 58, 313–325. [Google Scholar] [CrossRef]
- Gunaydin, G. CAFs Interacting With TAMs in Tumor Microenvironment to Enhance Tumorigenesis and Immune Evasion. Front. Oncol. 2021, 11, 668349. [Google Scholar] [CrossRef]
- Grisaru-Tal, S.; Rothenberg, M.E.; Munitz, A. Eosinophil–lymphocyte interactions in the tumor microenvironment and cancer immunotherapy. Nat. Immunol. 2022, 23, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Pulluri, B.; Kumar, A.; Shaheen, M.; Jeter, J.; Sundararajan, S. Tumor microenvironment changes leading to resistance of immune checkpoint inhibitors in metastatic melanoma and strategies to overcome resistance. Pharmacol. Res. 2017, 123, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Dai, L.-J.; Wu, S.-Y.; Xiao, Y.; Ma, D.; Jiang, Y.-Z.; Shao, Z.-M. Spatial architecture of the immune microenvironment orchestrates tumor immunity and therapeutic response. J. Hematol. Oncol. 2021, 14, 98. [Google Scholar] [CrossRef]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.S.; Rodman, C.; Su, M.J.; Melms, J.C.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997.e24. [Google Scholar] [CrossRef]
- Bravo, A.I.; Aris, M.; Panouillot, M.; Porto, M.; Dieu-Nosjean, M.C.; Teillaud, J.L.; Barrio, M.M.; Mordoh, J. HEV-associated dendritic cells are observed in metastatic tumor-draining lymph nodes of cutaneous melanoma patients with longer distant metastasis-free survival after adjuvant immunotherapy. Front. Immunol. 2023, 14, 1231734. [Google Scholar] [CrossRef]
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schüffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, W.-C.; Budiarto, B.R.; Wang, Y.-F.; Lin, C.-Y.; Gwo, M.-C.; So, D.K.; Tzeng, Y.-S.; Chen, S.-Y. Spatial multi-omics analyses of the tumor immune microenvironment. J. Biomed. Sci. 2022, 29, 96. [Google Scholar] [CrossRef]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L., Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef]
- Koh, J.; Kwak, Y.; Kim, J.; Kim, W.H. High-Throughput Multiplex Immunohistochemical Imaging of the Tumor and Its Microenvironment. Cancer Res. Treat. 2019, 52, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Capelozzi, V.L.; Parra, E.R. Editorial: Multiplexed image analysis for translational research project applications. Front. Oncol. 2022, 12, 1089265. [Google Scholar] [CrossRef]
- Sobottka, B.; Nowak, M.; Frei, A.L.; Haberecker, M.; Merki, S.; Levesque, M.P.; Dummer, R.; Moch, H.; Koelzer, V.H. Establishing standardized immune phenotyping of metastatic melanoma by digital pathology. Lab. Investig. A J. Tech. Methods Pathol. 2021, 101, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Attrill, G.H.; Ferguson, P.M.; Palendira, U.; Long, G.V.; Wilmott, J.S.; Scolyer, R.A. The tumour immune landscape and its implications in cutaneous melanoma. Pigment Cell Melanoma Res. 2021, 34, 529–549. [Google Scholar] [CrossRef]
- Kuczkiewicz-Siemion, O.; Sokół, K.; Puton, B.; Borkowska, A.; Szumera-Ciećkiewicz, A. The Role of Pathology-Based Methods in Qualitative and Quantitative Approaches to Cancer Immunotherapy. Cancers 2022, 14, 119. [Google Scholar] [CrossRef]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Nyberg, R.; Wu, Y.; Bernard, B.; Redmond, W.L. Developing an enhanced 7-color multiplex IHC protocol to dissect immune infiltration in human cancers. PLoS ONE 2021, 16, e0247238. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, F.; Pasqualini, E.; Simi, S.; Baroni, G.; Massi, D. Bright-Field Multiplex Immunohistochemistry Assay for Tumor Microenvironment Evaluation in Melanoma Tissues. Cancers 2022, 14, 3682. [Google Scholar] [CrossRef] [PubMed]
- Yaseen, Z.; Gide, T.N.; Conway, J.W.; Potter, A.J.; Quek, C.; Hong, A.M.; Long, G.V.; Scolyer, R.A.; Wilmott, J.S. Validation of an Accurate Automated Multiplex Immunofluorescence Method for Immuno-Profiling Melanoma. Front. Mol. Biosci. 2022, 9, 810858. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef]
- Lee, H.; Ferguson, A.L.; Quek, C.; Vergara, I.A.; Pires daSilva, I.; Allen, R.; Gide, T.N.; Conway, J.W.; Koufariotis, L.T.; Hayward, N.K.; et al. Intratumoral CD16+ Macrophages Are Associated with Clinical Outcomes of Patients with Metastatic Melanoma Treated with Combination Anti-PD-1 and Anti-CTLA-4 Therapy. Clin. Cancer Res. 2023, 29, 2513–2524. [Google Scholar] [CrossRef]
- Massi, D.; Rulli, E.; Cossa, M.; Valeri, B.; Rodolfo, M.; Merelli, B.; De Logu, F.; Nassini, R.; Del Vecchio, M.; Di Guardo, L.; et al. The density and spatial tissue distribution of CD8+ and CD163+ immune cells predict response and outcome in melanoma patients receiving MAPK inhibitors. J. Immunother. Cancer 2019, 7, 308. [Google Scholar] [CrossRef]
- Situm, M.; Buljan, M.; Kolić, M.; Vučić, M. Melanoma—Clinical, dermatoscopical, and histopathological morphological characteristics. Acta Dermatovenerol. Croat. 2014, 22, 1–12. [Google Scholar]
- Duncan, L.M. The classification of cutaneous melanoma. Hematol. Oncol. Clin. N. Am. 2009, 23, 501–513. [Google Scholar] [CrossRef]
- Keung, E.Z.; Gershenwald, J.E. The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: Implications for melanoma treatment and care. Expert Rev. Anticancer Ther. 2018, 18, 775–784. [Google Scholar] [CrossRef]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef]
- Jonsson, G.; Busch, C.; Knappskog, S.; Geisler, J.; Miletic, H.; Ringner, M.; Lillehaug, J.R.; Borg, A.; Lonning, P.E. Gene expression profiling-based identification of molecular subtypes in stage IV melanomas with different clinical outcome. Clin. Cancer Res. 2010, 16, 3356–3367. [Google Scholar] [CrossRef] [PubMed]
- Harbst, K.; Staaf, J.; Lauss, M.; Karlsson, A.; Måsbäck, A.; Johansson, I.; Bendahl, P.O.; Vallon-Christersson, J.; Törngren, T.; Ekedahl, H.; et al. Molecular profiling reveals low- and high-grade forms of primary melanoma. Clin. Cancer Res. 2012, 18, 4026–4036. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855.e819. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef]
- Lim, S.Y.; Pedersen, B.; Rizos, H. Protein-based classification of melanoma differentiation subtypes. Pigment Cell Melanoma Res. 2022, 35, 471–473. [Google Scholar] [CrossRef]
- Arozarena, I.; Wellbrock, C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat. Rev. Cancer 2019, 19, 377–391. [Google Scholar] [CrossRef] [PubMed]
- Wouters, J.; Kalender-Atak, Z.; Minnoye, L.; Spanier, K.I.; De Waegeneer, M.; Bravo González-Blas, C.; Mauduit, D.; Davie, K.; Hulselmans, G.; Najem, A.; et al. Robust gene expression programs underlie recurrent cell states and phenotype switching in melanoma. Nat. Cell Biol. 2020, 22, 986–998. [Google Scholar] [CrossRef]
- Kozar, I.; Margue, C.; Rothengatter, S.; Haan, C.; Kreis, S. Many ways to resistance: How melanoma cells evade targeted therapies. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 313–322. [Google Scholar] [CrossRef]
- Garbe, C.; Amaral, T.; Peris, K.; Hauschild, A.; Arenberger, P.; Basset-Seguin, N.; Bastholt, L.; Bataille, V.; Del Marmol, V.; Dréno, B.; et al. European consensus-based interdisciplinary guideline for melanoma. Part 2: Treatment-Update 2022. Eur. J. Cancer 2022, 170, 256–284. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chiarion-Sileni, V.; Lewis, K.D.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Long-term survival in advanced melanoma for patients treated with nivolumab plus ipilimumab in CheckMate 067. J. Clin. Oncol. 2022, 40, 9522. [Google Scholar] [CrossRef]
- Queirolo, P.; Boutros, A.; Tanda, E.; Spagnolo, F.; Quaglino, P. Immune-checkpoint inhibitors for the treatment of metastatic melanoma: A model of cancer immunotherapy. Semin. Cancer Biol. 2019, 59, 290–297. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.-j.; Zhang, Z.-z.; Ge, M.-j.; Chen, J.-y.; Zhuo, W. Immune-based combination therapy to convert immunologically cold tumors into hot tumors: An update and new insights. Acta Pharmacol. Sin. 2022, 44, 288–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Nathan, P. Cutaneous Melanoma-A Review of Systemic Therapies. Acta Derm. Venereol. 2020, 100, adv00141. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Shklovskaya, E.; Lim, S.Y.; Carlino, M.S.; Menzies, A.M.; Stewart, A.; Pedersen, B.; Irvine, M.; Alavi, S.; Yang, J.Y.H.; et al. Transcriptional downregulation of MHC class I and melanoma de- differentiation in resistance to PD-1 inhibition. Nat. Commun. 2020, 11, 1897. [Google Scholar] [CrossRef] [PubMed]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Sucker, A.; Zhao, F.; Real, B.; Heeke, C.; Bielefeld, N.; Maβen, S.; Horn, S.; Moll, I.; Maltaner, R.; Horn, P.A.; et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin. Cancer Res. 2014, 20, 6593–6604. [Google Scholar] [CrossRef]
- Johnson, D.B.; Nixon, M.J.; Wang, Y.; Wang, D.Y.; Castellanos, E.; Estrada, M.V.; Ericsson-Gonzalez, P.I.; Cote, C.H.; Salgado, R.; Sanchez, V.; et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight 2018, 3, e120360. [Google Scholar] [CrossRef]
- Donia, M.; Andersen, R.; Kjeldsen, J.W.; Fagone, P.; Munir, S.; Nicoletti, F.; Andersen, M.H.; Thor Straten, P.; Svane, I.M. Aberrant Expression of MHC Class II in Melanoma Attracts Inflammatory Tumor-Specific CD4+ T- Cells, Which Dampen CD8+ T-cell Antitumor Reactivity. Cancer Res. 2015, 75, 3747–3759. [Google Scholar] [CrossRef]
- Goodall, J.; Carreira, S.; Denat, L.; Kobi, D.; Davidson, I.; Nuciforo, P.; Sturm, R.A.; Larue, L.; Goding, C.R. Brn-2 represses microphthalmia-associated transcription factor expression and marks a distinct subpopulation of microphthalmia-associated transcription factor-negative melanoma cells. Cancer Res. 2008, 68, 7788–7794. [Google Scholar] [CrossRef]
- Boyle, G.M.; Woods, S.L.; Bonazzi, V.F.; Stark, M.S.; Hacker, E.; Aoude, L.G.; Dutton-Regester, K.; Cook, A.L.; Sturm, R.A.; Hayward, N.K. Melanoma cell invasiveness is regulated by miR-211 suppression of the BRN2 transcription factor. Pigment Cell Melanoma Res. 2011, 24, 525–537. [Google Scholar] [CrossRef]
- Thurber, A.E.; Douglas, G.; Sturm, E.C.; Zabierowski, S.E.; Smit, D.J.; Ramakrishnan, S.N.; Hacker, E.; Leonard, J.H.; Herlyn, M.; Sturm, R.A. Inverse expression states of the BRN2 and MITF transcription factors in melanoma spheres and tumour xenografts regulate the NOTCH pathway. Oncogene 2011, 30, 3036–3048. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2017, 168, 542. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.P.; Marchbank, K.; Webster, M.R.; Valiga, A.A.; Kaur, A.; Vultur, A.; Li, L.; Herlyn, M.; Villanueva, J.; Liu, Q.; et al. Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov. 2013, 3, 1378–1393. [Google Scholar] [CrossRef]
- Hou, L.; Pavan, W.J. Transcriptional and signaling regulation in neural crest stem cell-derived melanocyte development: Do all roads lead to Mitf? Cell Res. 2008, 18, 1163–1176. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Beiu, C.; Giurcaneanu, C.; Grumezescu, A.M.; Holban, A.M.; Popa, L.G.; Mihai, M.M. Nanosystems for Improved Targeted Therapies in Melanoma. J. Clin. Med. 2020, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Verma, J.; Warsame, C.; Seenivasagam, R.K.; Katiyar, N.K.; Aleem, E.; Goel, S. Nanoparticle-mediated cancer cell therapy: Basic science to clinical applications. Cancer Metastasis Rev. 2023, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Pereira, I.; Monteiro, C.; Pereira-Silva, M.; Peixoto, D.; Nunes, C.; Reis, S.; Veiga, F.; Hamblin, M.R.; Paiva-Santos, A.C. Nanodelivery systems for cutaneous melanoma treatment. Eur. J. Pharm. Biopharm. 2023, 184, 214–247. [Google Scholar] [CrossRef] [PubMed]
- Rohaan, M.W.; Borch, T.H.; van den Berg, J.H.; Met, Ö.; Kessels, R.; Geukes Foppen, M.H.; Stoltenborg Granhøj, J.; Nuijen, B.; Nijenhuis, C.; Jedema, I.; et al. Tumor-Infiltrating Lymphocyte Therapy or Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2022, 387, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Van Tine, B.A.; Biswas, S.; McAlpine, C.; Johnson, M.L.; Olszanski, A.J.; Clarke, J.M.; Araujo, D.; Blumenschein, G.R., Jr.; Kebriaei, P.; et al. Autologous T cell therapy for MAGE-A4(+) solid cancers in HLA-A*02(+) patients: A phase 1 trial. Nat. Med. 2023, 29, 104–114. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef]
- Rezaei, R.; Esmaeili Gouvarchin Ghaleh, H.; Farzanehpour, M.; Dorostkar, R.; Ranjbar, R.; Bolandian, M.; Mirzaei Nodooshan, M.; Ghorbani Alvanegh, A. Combination therapy with CAR T cells and oncolytic viruses: A new era in cancer immunotherapy. Cancer Gene Ther. 2022, 29, 647–660. [Google Scholar] [CrossRef]
- Kasakovski, D.; Skrygan, M.; Gambichler, T.; Susok, L. Advances in Targeting Cutaneous Melanoma. Cancers 2021, 13, 90. [Google Scholar] [CrossRef]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef]
- Pan, Y.; Tian, T.; Park, C.O.; Lofftus, S.Y.; Mei, S.; Liu, X.; Luo, C.; O’Malley, J.T.; Gehad, A.; Teague, J.E.; et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 2017, 543, 252–256. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuras, M. Exploring the Complex and Multifaceted Interplay between Melanoma Cells and the Tumor Microenvironment. Int. J. Mol. Sci. 2023, 24, 14403. https://doi.org/10.3390/ijms241814403
Kuras M. Exploring the Complex and Multifaceted Interplay between Melanoma Cells and the Tumor Microenvironment. International Journal of Molecular Sciences. 2023; 24(18):14403. https://doi.org/10.3390/ijms241814403
Chicago/Turabian StyleKuras, Magdalena. 2023. "Exploring the Complex and Multifaceted Interplay between Melanoma Cells and the Tumor Microenvironment" International Journal of Molecular Sciences 24, no. 18: 14403. https://doi.org/10.3390/ijms241814403