
Genetic Parameter and Hyper-Parameter Estimation Underlie Nitrogen Use Efficiency in Bread Wheat

Abstract
:1. Introduction
1.1. GS Model Definition
1.2. Feature Selection in GS Model
1.3. Regularization of GS Model
2. Results
2.1. Genetic Parameters and Hyper-Parameter Estimation
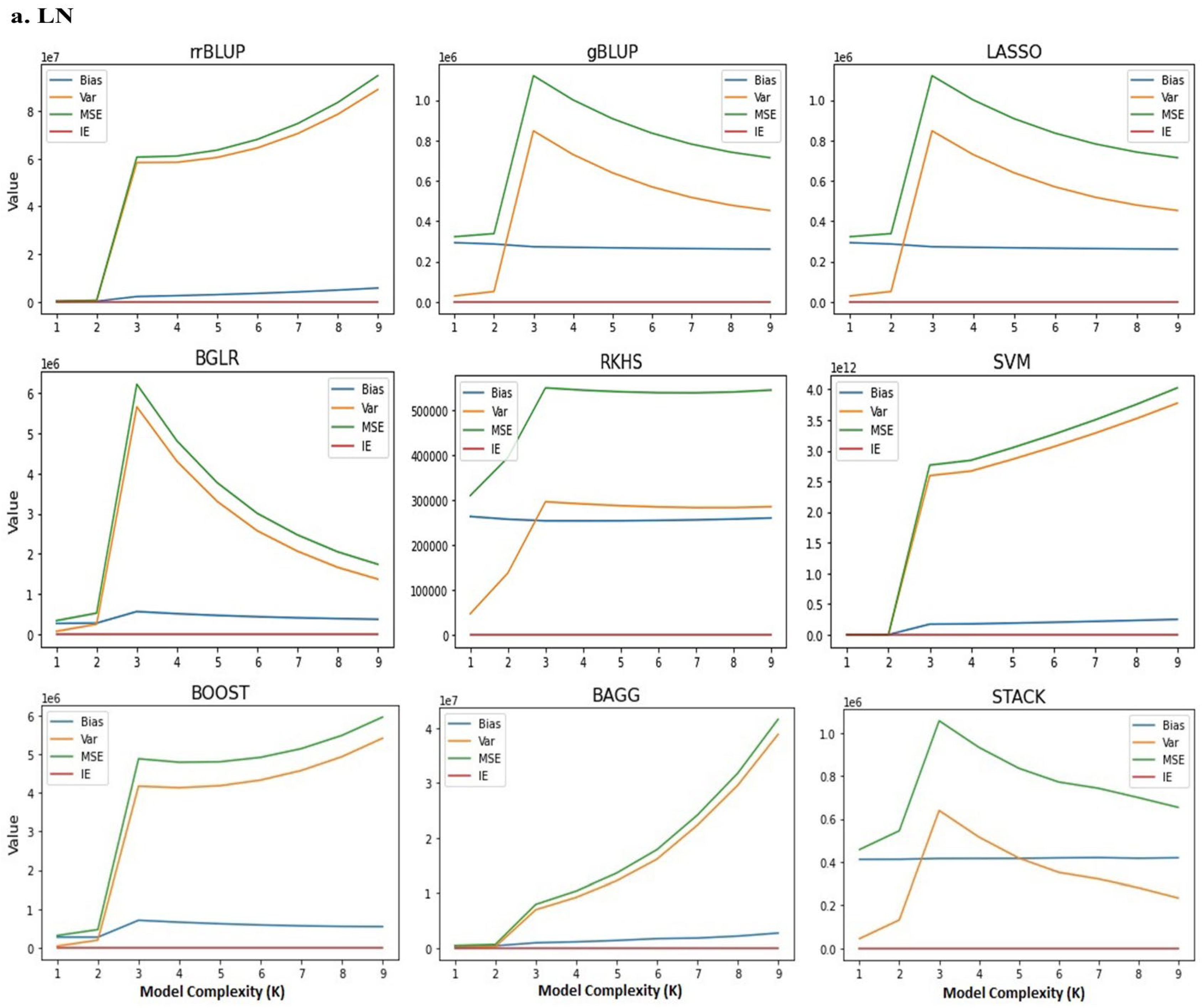
2.2. Bias–Variance Tradeoff in GS Models
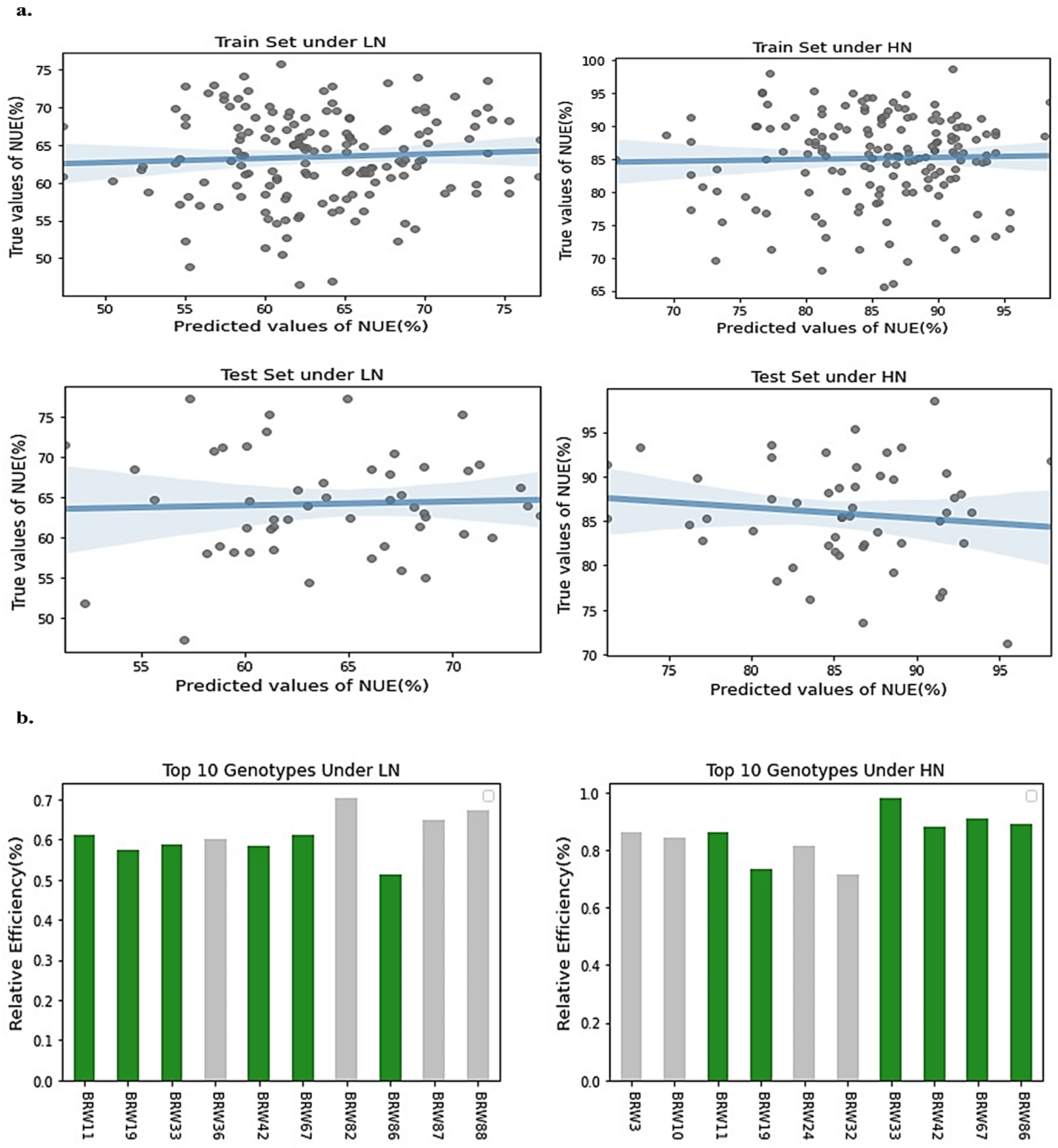
2.3. Error Measurement of GS Models
2.4. Genetic Selection Gain Estimation Based on Selected Model
3. Discussion
4. Materials and Method
4.1. Phenotypic Data
4.2. Genotypic Data
4.3. Construction of GRM
4.4. Genomic Selection Models
4.5. Genetic Parameters and Hyper-Parameters Estimation
4.6. Bias–Variance Tradeoff in GS Models
4.7. Error Measurement between GS Models
4.8. Genetic Selection Gain Estimation Based on the Selected Model
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdollahi-Arpanahi, R.; Gianola, D.; Peñagaricano, F. Deep learning versus parametric and ensemble methods for genomic prediction of complex phenotypes. Genet. Sel. Evol. 2020, 52, 1–15. [Google Scholar] [CrossRef]
- Wang, X.; Xu, Y.; Hu, Z.; Xu, C. Genomic selection methods for crop improvement: Current status and prospects. Crop J. 2018, 6, 330–340. [Google Scholar] [CrossRef]
- Crossa, J.; Pérez-Rodríguez, P.; Cuevas, J.; Montesinos-López, O.; Jarquín, D.; de los Campos, G.; Burgueño, J.; González-Camacho, J.M.; Pérez-Elizalde, S.; Beyene, Y.; et al. Genomic selection in plant breeding: Methods, models, and perspectives. Trends Plant Sci. 2017, 22, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Bhat, J.A.; Ali, S.; Salgotra, R.K.; Mir, Z.A.; Dutta, S.; Jadon, V.; Tyagi, A.; Mushtaq, M.; Jain, N.; Singh, P.K.; et al. Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Front. Genet. 2016, 7, 221. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, R. Bandwagons I, too, have known. Theor. Appl. Genet. 2016, 129, 2323–2332. [Google Scholar] [CrossRef] [PubMed]
- Sneller, C.H.; Mather, D.E.; Crepieux, S. Analytical approaches and population types for finding and utilizing QTL in complex plant populations. Crop Sci. 2009, 49, 363–380. [Google Scholar] [CrossRef]
- Schön, C.C.; Utz, H.F.; Groh, S.; Truberg, B.; Openshaw, S.; Melchinger, A.E. Quantitative trait locus mapping based on resampling in a vast maize Testcross experiment and its relevance to quantitative genetics for complex traits. Genetics 2004, 167, 485–498. [Google Scholar] [CrossRef]
- Montesinos-López, O.A.; Martín-Vallejo, J.; Crossa, J.; Gianola, D.; Hernández-Suárez, C.M.; Montesinos-López, A.; Juliana, P.; Singh, R. A benchmarking between deep learning, support Vector Machine and Bayesian threshold best linear unbiased prediction for predicting ordinal traits in plant breeding. G3 Genes Genomes Genet. 2019, 9, 601–618. [Google Scholar] [CrossRef]
- Werner, C.R.; Gaynor, R.C.; Gorjanc, G.; Hickey, J.M.; Kox, T.; Abbadi, A.; Leckband, G.; Snowdon, R.J.; Stahl, A. How population structure impacts genomic selection accuracy in cross-validation: Implications for practical breeding. Front. Plant Sci. 2020, 11, 592977. [Google Scholar] [CrossRef]
- Delfini, J.; Moda-Cirino, V.; dos Santos Neto, J.; Ruas, P.M.; Sant’ana, G.C.; Gepts, P.; Gonçalves, L.S. Population structure, genetic diversity and genomic selection signatures among a Brazilian common bean germplasm. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef]
- Lyra, D.H.; Galli, G.; Alves, F.C.; Granato, Í.S.; Vidotti, M.S.; Bandeira e Sousa, M.; Morosini, J.S.; Crossa, J.; Fritsche-Neto, R. Modeling copy number variation in the genomic prediction of maize hybrids. Theor. Appl. Genet. 2018, 132, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Park, J.-E.; Son, J.-H.; Lee, S.-H.; Park, B.H.; Park, M.; Park, W.-C.; Chai, H.-H.; Kim, H.; Lee, J.; et al. Genomic prediction accuracy using haplotypes defined by size and hierarchical clustering based on linkage disequilibrium. Front. Genet. 2020, 11, 134. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Gondro, C.; Reid, K.; Steibel, J.P. Heuristic hyperparameter optimization of deep learning models for genomic prediction. G3 Genes Genomes Genet. 2021, 11, jkab032. [Google Scholar] [CrossRef]
- Okut, H. Deep learning algorithms for complex traits genomic prediction. Hayvan Bilim. ve Ürünleri Derg. 2021, 4, 225–239. [Google Scholar] [CrossRef]
- Jannink, J.-L.; Lorenz, A.J.; Iwata, H. Genomic selection in plant breeding: From theory to practice. Brief. Funct. Genom. 2010, 9, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 2014, 11, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Pérez-Rodríguez, P.; Veturi, Y.; Simianer, H.; los Campos, G. Effectiveness of shrinkage and variable selection methods for the prediction of complex human traits using data from distantly related individuals. Ann. Hum. Genet. 2015, 79, 122–135. [Google Scholar] [CrossRef]
- Guo, P.; Zhu, B.; Niu, H.; Wang, Z.; Liang, Y.; Chen, Y.; Zhang, L.; Ni, H.; Guo, Y.; Hay, E.H.; et al. Fast genomic prediction of breeding values using parallel Markov chain Monte Carlo with convergence diagnosis. BMC Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Zhang, H.; Yin, L.; Wang, M.; Yuan, X.; Liu, X. Factors affecting the accuracy of genomic selection for agricultural economic traits in maize, cattle, and pig populations. Front. Genet. 2019, 10, 189. [Google Scholar] [CrossRef]
- Shi, S.; Li, X.; Fang, L.; Liu, A.; Su, G.; Zhang, Y.; Luobu, B.; Ding, X.; Zhang, S. Genomic prediction using Bayesian regression models with global–local prior. Front. Genet. 2021, 12, 628205. [Google Scholar] [CrossRef]
- Sandhu, K.S.; Lozada, D.N.; Zhang, Z.; Pumphrey, M.O.; Carter, A.H. Deep learning for predicting complex traits in spring wheat breeding program. Front. Plant Sci. 2021, 11, 613325. [Google Scholar] [CrossRef] [PubMed]
- Morota, G.; Koyama, M.; M Rosa, G.J.; Weigel, K.A.; Gianola, D. Predicting complex traits using a diffusion kernel on genetic markers with an application to dairy cattle and wheat data. Genet. Sel. Evol. 2013, 45, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Reinoso-Peláez, E.L.; Gianola, D.; González-Recio, O. Genome-enabled prediction methods based on machine learning. Methods Mol. Biol. 2022, 2467, 189–218. [Google Scholar] [CrossRef] [PubMed]
- Pal, R. Feature selection and extraction from heterogeneous genomic characterizations. In Predictive Modeling of Drug Sensitivity; Academic Press: Cambridge, MA, USA, 2017; pp. 45–81. [Google Scholar] [CrossRef]
- Yu, L. Feature selection for Genomic Data Analysis. In Computational Methods of Feature Selection, 1st ed.; Liu, H., Motoda, H., Eds.; CRC: New York, NY, USA, 2007; pp. 337–353. [Google Scholar] [CrossRef]
- Hastie, T.; Friedman, J.; Tisbshirani, R. The Elements of Statistical Learning: Data Mining, Inference, and Prediction; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Shi, L.; Westerhuis, J.A.; Rosén, J.; Landberg, R.; Brunius, C. Variable selection and validation in multivariate modelling. Bioinformatics 2018, 35, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Morillo-Salas, J.L.; Bolón-Canedo, V.; Alonso-Betanzos, A. Dealing with heterogeneity in the context of distributed feature selection for classification. Knowl. Inf. Syst. 2020, 63, 233–276. [Google Scholar] [CrossRef]
- Paul, J. Feature Selection from Heterogeneous Biomedical Data: Semantic Scholar. Undefined. 2015. Available online: https://www.semanticscholar.org/paper/Feature-selection-from-heterogeneous-biomedical-Paul/47054794c57a8c57665d83bed606fd40b7ef011f (accessed on 29 October 2022).
- Rustam, Z.; Kharis, S.A. Multiclass classification on brain cancer with multiple support Vector Machine and feature selection based on kernel function. AIP Conf. Proc. 2018, 2023, 020233. [Google Scholar] [CrossRef]
- Efron, B.; Hastie, T. Computer Age Statistical Inference; Cambridge University Press: Cambridge UK, 2021. [Google Scholar]
- Cuevas, J.; Crossa, J.; Soberanis, V.; Pérez-Elizalde, S.; Pérez-Rodríguez, P.; Campos, G.d.; Montesinos-López, O.A.; Burgueño, J. Genomic prediction of genotype × environment interaction kernel regression models. Plant Genome 2016, 9. [Google Scholar] [CrossRef]
- Li, B.; Zhang, N.; Wang, Y.-G.; George, A.W.; Reverter, A.; Li, Y. Genomic prediction of breeding values using a subset of SNPs identified by three machine learning methods. Front. Genet. 2018, 9, 237. [Google Scholar] [CrossRef]
- Asafu-Adjei, J.K.; Sampson, A.R. Covariate adjusted classification trees. Biostatistics 2017, 19, 42–53. [Google Scholar] [CrossRef]
- Sillanpää, M.J. Overview of techniques to account for confounding due to population stratification and cryptic relatedness in genomic data association analyses. Heredity 2010, 106, 511–519. [Google Scholar] [CrossRef]
- Thavamanikumar, S.; Dolferus, R.; Thumma, B.R. Comparison of genomic selection models to predict flowering time and spike grain number in two hexaploid wheat doubled haploid populations. G3 Genes Genomes Genet. 2015, 5, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Martini, J.W.; Hearne, S.J.; Gardunia, B.; Wimmer, V.; Toledo, F.H. Editorial: Genomic selection: Lessons learned and Perspectives. Front. Plant Sci. 2022, 13, 890434. [Google Scholar] [CrossRef] [PubMed]
- Montesinos-López, O.A.; Montesinos-López, A.; Pérez-Rodríguez, P.; Barrón-López, J.A.; Martini, J.W.; Fajardo-Flores, S.B.; Gaytan-Lugo, L.S.; Santana-Mancilla, P.C.; Crossa, J. A review of deep learning applications for Genomic Selection. BMC Genom. 2021, 22, 1–23. [Google Scholar] [CrossRef]
- Shen, X.; De Jonge, J.; Forsberg, S.K.; Pettersson, M.E.; Sheng, Z.; Hennig, L.; Carlborg, Ö. Natural CMT2 variation is associated with genome-wide methylation changes and temperature seasonality. PLoS Genet. 2014, 10, e1004842. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y. Kernel regularization for low-frequency decay systems. In Proceedings of the 60th IEEE Conference on Decision and Control (CDC), Austin, TX, USA, 13–17 December 2021. [Google Scholar] [CrossRef]
- Piles, M.; Bergsma, R.; Gianola, D.; Gilbert, H.; Tusell, L. Feature selection stability and accuracy of prediction models for genomic prediction of residual feed intake in pigs using machine learning. Front. Genet. 2021, 12, 611506. [Google Scholar] [CrossRef]
- Liang, M.; An, B.; Li, K.; Du, L.; Deng, T.; Cao, S.; Du, Y.; Xu, L.; Gao, X.; Zhang, L.; et al. Improving genomic prediction with machine learning incorporating TPE for hyperparameters optimization. Biology 2022, 11, 1647. [Google Scholar] [CrossRef]
- Antonio, M.L.O.; López, A.M.; Crossa, J. Multivariate Statistical Machine Learning Methods for Genomic Prediction; Springer International Publishing AG: Berlin/Heidelberg, Germany, 2022. [Google Scholar] [CrossRef]
- Ho, D.S.; Schierding, W.; Wake, M.; Saffery, R.; O’Sullivan, J. Machine learning SNP based prediction for Precision Medicine. Front. Genet. 2019, 10, 267. [Google Scholar] [CrossRef]
- Mathew, B.; Sillanpää, M.J.; Léon, J. Advances in statistical methods to handle large data sets for genome-wide association mapping in crop breeding. In Advances in Breeding Techniques for Cereal Crops; Burleigh Dodds Science Publishing: Cambridge, UK, 2019; pp. 437–450. [Google Scholar] [CrossRef]
- Chollet, F. Deep Learning with Python; Manning Publications Co.: London, UK, 2018. [Google Scholar]
- Liang, M.; Chang, T.; An, B.; Duan, X.; Du, L.; Wang, X.; Miao, J.; Xu, L.; Gao, X.; Zhang, L.; et al. A Stacking Ensemble Learning Framework for genomic prediction. Front. Genet. 2021, 12, 600040. [Google Scholar] [CrossRef]
- Moll, R.H.; Kamprath, E.J.; Jackson, W.A. Analysis and interpretation of factors which contribute to efficiency of nitrogen utilization 1. Agron. J. 1982, 74, 562–564. [Google Scholar] [CrossRef]
- Mathew, B.; Léon, J.; Sillanpää, M.J. A novel linkage-disequilibrium corrected genomic relationship matrix for SNP-heritability estimation and genomic prediction. Heredity 2017, 120, 356–368. [Google Scholar] [CrossRef]
- Usai, M.G.; Goddard, M.E.; Hayes, B.J. Lasso with cross-validation for Genomic Selection. Genet. Res. 2009, 91, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.D.; Verbyla, A.P.; Pitchford, W.S. Incorporating lasso effects into a mixed model for quantitative trait loci detection. J. Agric. Biol. Environ. Stat. 2007, 12, 300–314. [Google Scholar] [CrossRef]
- Chen, C.; Steibel, J.P.; Tempelman, R.J. Genome wide association analyses based on broadly different specifications for prior distributions, genomic windows, and Estimation Methods. Genetics 2017, 206, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Pérez, P.; de los Campos, G. Genome-wide regression and prediction with the BGLR statistical package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Meuwissen, T.H.; Hayes, B.J.; Goddard, M.E. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Habier, D.; Tetens, J.; Seefried, F.-R.; Lichtner, P.; Thaller, G. The impact of genetic relationship information on genomic breeding values in German Holstein cattle. Genet. Sel. Evol. 2010, 42, 1–12. [Google Scholar] [CrossRef]
- Morota, G.; Gianola, D. Kernel-based whole-genome prediction of complex traits: A Review. Front. Genet. 2014, 5, 363. [Google Scholar] [CrossRef]
- Campos, G.D.L.; Gianola, D.; Rosa, G.J.M.; Weigel, K.A.; Crossa, J. Semi-parametric genomic-enabled prediction of genetic values using reproducing kernel Hilbert spaces methods. Genet. Res. 2010, 92, 295–308. [Google Scholar] [CrossRef]
- Gianola, D.; van Kaam, J.B. Reproducing kernel Hilbert spaces regression methods for genomic assisted prediction of Quantitative Traits. Genetics 2008, 178, 2289–2303. [Google Scholar] [CrossRef]
- Zhao, W.; Lai, X.; Liu, D.; Zhang, Z.; Ma, P.; Wang, Q.; Zhang, Z.; Pan, Y. Applications of support vector machine in genomic prediction in pig and maize populations. Front. Genet. 2020, 11, 598318. [Google Scholar] [CrossRef] [PubMed]
- González-Recio, O.; Jiménez-Montero, J.A.; Alenda, R. The gradient boosting algorithm and random boosting for genome-assisted evaluation in large data sets. J. Dairy Sci. 2013, 96, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Grinberg, N.F.; Orhobor, O.I.; King, R.D. An evaluation of machine-learning for predicting phenotype: Studies in yeast, Rice, and wheat. Mach. Learn. 2019, 109, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Perez, B.C.; Bink MC, A.M.; Churchill, G.A.; Svenson, K.L.; Calus MP, L. Prediction Performance of Linear Models and Gradient Boosting Machine on Complex Phenotypes in Outbred Mice. bioRxiv 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nazzicari, N.; Biscarini, F. Stacked kinship CNN vs. GBLUP for genomic predictions of additive and complex continuous phenotypes. Sci. Rep. 2022, 12, 19889. [Google Scholar] [CrossRef] [PubMed]
- Franchini, G.; Porta, F.; Ruggiero, V.; Trombini, I.; Zanni, L. Learning rate selection in stochastic gradient methods based on line search strategies. Appl. Math. Sci. Eng. 2023, 31, 2164000. [Google Scholar] [CrossRef]
- Na, G.S. Efficient learning rate adaptation based on hierarchical optimization approach. Neural Netw. 2022, 150, 326–335. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inference | Model | N Level | a | Training Set (CVK_fold = 10) | Test Set (CVK_fold = 5) | p-Value f | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GEBVs Mean b | Boot. GEBVs Mean c | Vg d | Ve e | GEBVs Mean | Boot. GEBVs Mean | Vg | Ve | |||||
| Frequentist | rrBLUP | LN | 0.30 | 0.65 | 0.66 | 27.49 | 64.15 | 0.64 | 0.66 | 28.77 | 67.13 | 0.05 * |
| HN | 0.33 | 0.73 | 0.70 | 9.79 | 19.88 | 0.73 | 0.70 | 8.77 | 17.81 | |||
| gBLUP | LN | 0.28 | 0.64 | 0.61 | 26.78 | 68.87 | 0.64 | 0.61 | 22.61 | 54.14 | 0.044 * | |
| HN | 0.30 | 0.71 | 0.69 | 4.93 | 11.51 | 0.71 | 0.68 | 4.57 | 10.61 | |||
| Bayesian | LASSO | LN | 0.31 | 0.62 | 0.61 | 32.40 | 72.12 | 0.62 | 0.60 | 32.03 | 71.12 | 0.144 ns |
| HN | 0.31 | 0.70 | 0.68 | 7.94 | 17.69 | 0.69 | 0.65 | 8.39 | 18.69 | |||
| BGLR | LN | 0.32 | 0.63 | 0.68 | 35.49 | 75.43 | 0.65 | 0.65 | 35.35 | 75.12 | 0.042 * | |
| HN | 0.30 | 0.72 | 0.69 | 9.70 | 22.65 | 0.74 | 0.65 | 26.24 | 61.24 | |||
| Kernel | RKHS | LN | 0.45 | 0.64 | 0.64 | 57.11 | 69.81 | 0.64 | 0.64 | 53.29 | 65.14 | 0.031 * |
| HN | 0.61 | 0.72 | 0.72 | 28.16 | 18.01 | 0.72 | 0.71 | 42.71 | 27.21 | |||
| SVM | LN | 0.38 | 0.13 | 0.22 | 24.30 | 39.66 | 0.18 | 0.32 | 24.64 | 40.21 | 0.048 * | |
| HN | 0.57 | 0.18 | 0.29 | 73.09 | 55.14 | 0.18 | 0.33 | 59.66 | 45.01 | |||
| Ensemble | BOOST | LN | 0.48 | 0.61 | 0.61 | 62.84 | 67.08 | 0.61 | 0.65 | 61.92 | 67.08 | 0.164 ns |
| HN | 0.62 | 0.68 | 0.69 | 28.73 | 17.61 | 0.68 | 0.72 | 27.76 | 17.02 | |||
| BAGG | LN | 0.55 | 0.60 | 0.68 | 71.03 | 58.12 | 0.61 | 0.71 | 69.82 | 57.13 | 0.679 ns | |
| HN | 0.55 | 0.64 | 0.71 | 39.80 | 32.57 | 0.64 | 0.74 | 38.12 | 31.19 | |||
| STACK | LN | 0.62 | 0.69 | 0.78 | 49.33 | 30.24 | 0.69 | 0.79 | 50.85 | 31.17 | 0.0924 ns | |
| HN | 0.71 | 0.76 | 0.78 | 72.98 | 29.81 | 0.76 | 0.79 | 73.49 | 30.02 | |||
| Inference | Model | N Level | Training Set (CVK_fold = 10) | Test Set (CVK_fold = 5) | p-Value b | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LR a | No. of Iteration | No. of Batch Size | Accuracy (%) | LR | No. of Iteration | No. of Batch Size | Accuracy (%) | ||||
| Frequentist | rrBLUP | LN | 0.01 | 27 | 10 | 81.12 | 0.01 | 27 | 5 | 92.22 | 0.007 ** |
| HN | 0.01 | 27 | 100 | 81.09 | 0.01 | 27 | 25 | 92.21 | |||
| gBLUP | LN | 0.001 | 27 | 10 | 80.88 | 0.001 | 27 | 5 | 92.21 | 0.001 ** | |
| HN | 0.001 | 27 | 100 | 80.41 | 0.001 | 27 | 25 | 92.22 | |||
| Bayesian | LASSO | LN | 0.01 | 468 | 10 | 85.51 | 0.01 | 468 | 5 | 92.43 | 0.0024 * |
| HN | 0.01 | 468 | 100 | 85.12 | 0.01 | 468 | 25 | 92.44 | |||
| BGLR | LN | 0.001 | 468 | 10 | 84.47 | 0.001 | 468 | 5 | 92.74 | 0.0031 * | |
| HN | 0.001 | 468 | 100 | 85.77 | 0.001 | 468 | 25 | 92.76 | |||
| Kernel | RKHS | LN | 0.01 | 2050 | 100 | 85.11 | 0.01 | 2050 | 25 | 95.64 | 0.001 ** |
| HN | 0.01 | 2050 | 1000 | 85.33 | 0.01 | 2050 | 250 | 95.37 | |||
| SVM | LN | 0.001 | 2050 | 100 | 84.04 | 0.001 | 2050 | 25 | 95.33 | 0.0014 ** | |
| HN | 0.001 | 2050 | 1000 | 85.77 | 0.001 | 2050 | 250 | 95.18 | |||
| Ensemble | BOOST | LN | 0.01 | 5520 | 100 | 91.12 | 0.01 | 5520 | 25 | 96.01 | 0.098 ns |
| HN | 0.01 | 5520 | 1000 | 92.11 | 0.01 | 5520 | 250 | 96.12 | |||
| BAGG | LN | 0.001 | 5520 | 100 | 92.31 | 0.001 | 5520 | 25 | 96.48 | 0.1445 ns | |
| HN | 0.001 | 5520 | 1000 | 92.16 | 0.001 | 5520 | 250 | 97.22 | |||
| STACK | LN | 0.001 | 5520 | 100 | 93.58 | 0.001 | 5520 | 25 | 97.54 | 0.0905 ns | |
| HN | 0.001 | 5520 | 1000 | 93.79 | 0.001 | 5520 | 250 | 97.84 | |||
| Inference | Model | Low N | High N | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Frequentist | rrBLUP | 9.1 × 105 | 6.1 × 107 | 6.2 × 107 | 0.0009 | 0.2 × 106 | 0.9 × 106 | 1.1 × 106 | 0.0009 |
| gBLUP | 3.1 × 106 | 0.8 × 106 | 1.2 × 106 | 0.0009 | 0.22 × 106 | 3.7 × 106 | 4.0 × 106 | 0.0009 | |
| Bayesian | LASSO | 3.1 × 106 | 0.82 × 106 | 1.2 × 106 | 0.0009 | 0.2 × 106 | 0.8 × 106 | 1.2 × 106 | 0.0009 |
| BGLR | 3.1 × 106 | 5.57 × 106 | 6.4 × 106 | 0.0009 | 0.2 × 106 | 5.2 × 106 | 5.5 × 106 | 0.0009 | |
| Kernel | RKHS | 25 × 104 | 49 × 104 | 55 × 104 | 0.00081 | 0.3 × 106 | 1.1 × 106 | 1.5 × 106 | 0.00081 |
| SVM | 0.1 × 1012 | 2.5 ×1 012 | 2.7 × 1012 | 0.00080 | 0.15 × 1010 | 1.5 × 1010 | 1.6 × 1010 | 0.00080 | |
| Ensemble | BOOST | 4.1 × 106 | 4.2 × 106 | 5.1 × 106 | 0.00007 | 0.09 × 107 | 1.1 × 107 | 1.19 × 107 | 0.00007 |
| BAGG | 0.75 × 107 | 0.5 × 107 | 0.8 × 107 | 0.00007 | 0.11 × 107 | 1.6 × 107 | 1.75 × 107 | 0.00007 | |
| STACK | 0.41 × 106 | 0.6 × 106 | 1.2 × 106 | 0.00002 | 0.5 × 106 | 5.1 × 106 | 5.8 × 106 | 0.00002 | |
| Mean | 12.3 × 109 | 30.0 × 109 | 33 × 109 | 0.00059 | 1.67 × 108 | 1.68 × 109 | 1.78 × 109 | 0.00059 | |
| LSD (0.05) | 0.92 × 109 | 0.48 × 109 | 1.25 × 109 | 2.11 | 0.34 × 108 | 0.70 × 109 | 1.03 × 109 | 2.11 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadeqi, M.B.; Ballvora, A.; Dadshani, S.; Léon, J. Genetic Parameter and Hyper-Parameter Estimation Underlie Nitrogen Use Efficiency in Bread Wheat. Int. J. Mol. Sci. 2023, 24, 14275. https://doi.org/10.3390/ijms241814275
Sadeqi MB, Ballvora A, Dadshani S, Léon J. Genetic Parameter and Hyper-Parameter Estimation Underlie Nitrogen Use Efficiency in Bread Wheat. International Journal of Molecular Sciences. 2023; 24(18):14275. https://doi.org/10.3390/ijms241814275
Chicago/Turabian StyleSadeqi, Mohammad Bahman, Agim Ballvora, Said Dadshani, and Jens Léon. 2023. "Genetic Parameter and Hyper-Parameter Estimation Underlie Nitrogen Use Efficiency in Bread Wheat" International Journal of Molecular Sciences 24, no. 18: 14275. https://doi.org/10.3390/ijms241814275