
Pathway from Acute Kidney Injury to Chronic Kidney Disease: Molecules Involved in Renal Fibrosis
 , , ,
, , ,
Abstract
:1. Introduction
2. Epidemiology of AKI to CKD Transition
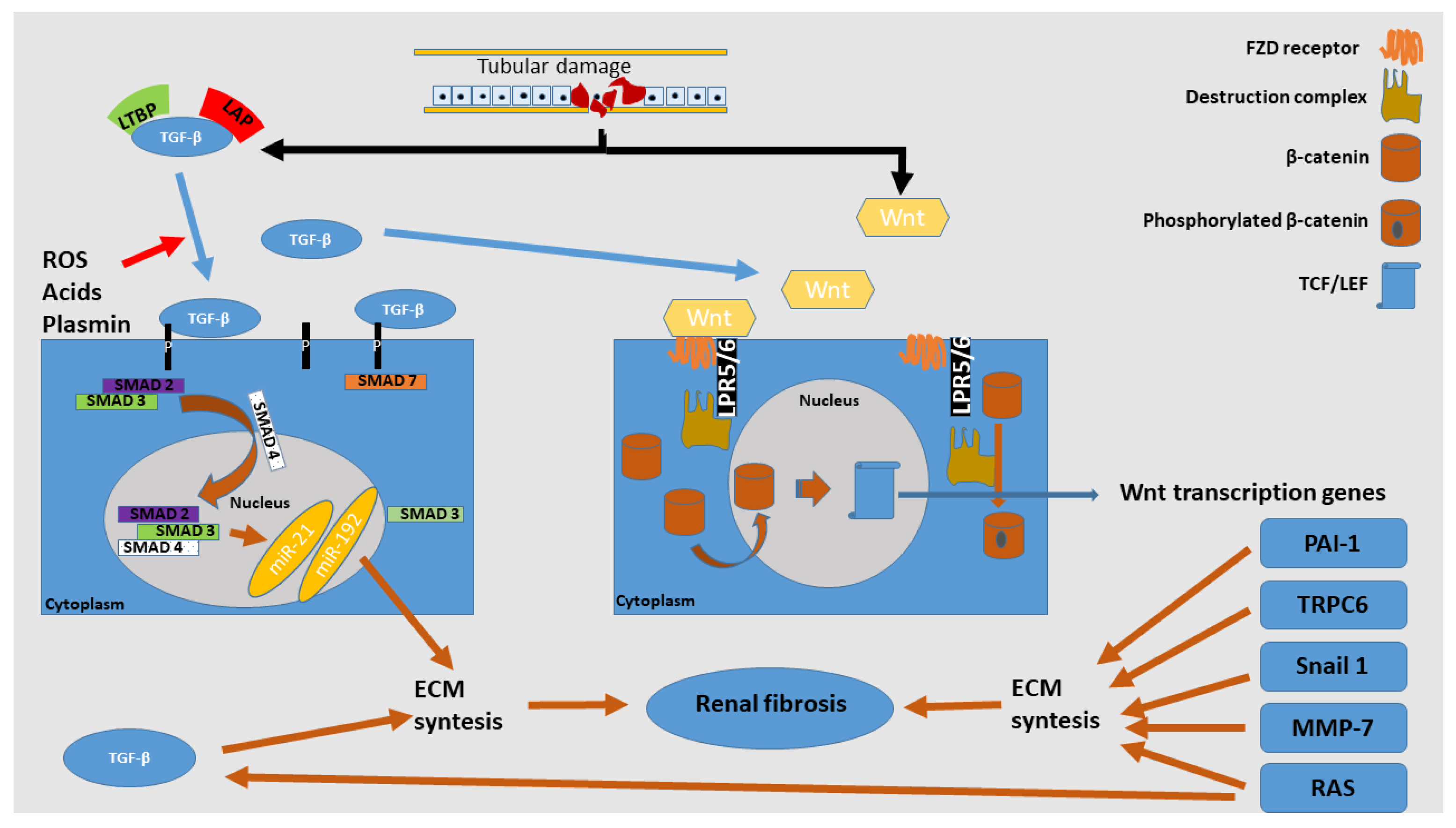
3. Pathway to Renal Fibrosis
4. Main Pro-Fibrotic Factors and Mechanisms of Action
4.1. Wnt/β-Catenin Signaling Pathway
4.2. Snail Pathway
4.3. MMP-7 Signaling
4.4. PAI-1 Signaling
4.5. TRPC6
4.6. RAS Activation
4.7. TGF-β1 and SMAD Signaling Pathway
4.8. Cell Death Pathways
4.9. Hypoxia-Inducible Factor Pathways
4.10. VEGF
4.11. Mitochondrial Dysfunction
4.12. G2/M Arrest Pathway
4.13. Innate and Adaptive Immunological Pathways
5. Therapeutic Potential in Renal Fibrosis
6. Discussion
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Takaori, K.; Nakamura, J.; Yamamoto, S.; Nakata, H.; Sato, Y.; Takase, M.; Nameta, M.; Yamamoto, T.; Economides, A.N.; Kohno, K.; et al. Severity and Frequency of Proximal Tubule Injury Determines Renal Prognosis. J. Am. Soc. Nephrol. 2016, 27, 2393–2406. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, T.D.; Boon, W.C.; Simpson, E.R.; Smith, E.R.; Samuel, C.S. Estrogens do not protect, but androgens exacerbate, collagen accumulation in the female mouse kidney after ureteric obstruction. Life Sci. 2016, 158, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, M.; Grandalino, G.; Gesualdo, L.; Castellano, G. Acute Kidney Injury to Chronic Kidney Disease Transition. Contrib. Nephrol. 2018, 193, 45–54. [Google Scholar]
- KDIGO Clinical Practice Guideline for Acute Kidney Injury. Kidney Int. Suppl. 2012, 2, 1–138.
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. Acute Kidney Injury Advisory Group of the American Society of Nephrology. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef]
- Mammen, C.; Al Abbas, A.; Skippen, P.; Nadel, H.; Levine, D.; Collet, J.P.; Matsell, D.G. Long-term risk of CKD in children surviving episodes of acute kidney injury in the intensive care unit: A prospective cohort study. Am. J. Kidney Dis. 2012, 59, 523–530. [Google Scholar] [CrossRef]
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute kidney injury increases risk of ESRD among elderly. J. Am. Soc. Nephrol. 2009, 20, 223–228. [Google Scholar] [CrossRef]
- Horne, K.L.; Packington, R.; Monaghan, J.; Reilly, T.; Selby, N.M. Three-year outcomes after acute kidney injury: Results of a prospective parallel group cohort study. BMJ Open 2017, 7, e015316. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 2014, 10, 493–503. [Google Scholar] [CrossRef]
- Hong, K.M.; Belperio, J.A.; Keane, M.P.; Burdick, M.D.; Strieter, R.M. Differentiation of human circulating fibrocytes as mediated by transforming growth factor-beta and peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2007, 282, 22910–22920. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.C.; Hung, L.Y.; Chen, Y.C.; Lo, W.C.; Lin, C.H.; Tam, K.W.; Wu, M.Y. Effects of febuxostat on renal function in patients with chronic kidney disease: A systematic review and meta-analysis. Medicine 2019, 98, e16311. [Google Scholar] [CrossRef] [PubMed]
- Allison, S.J. Fibrosis: The source of myofibroblasts in kidney fibrosis. Nat. Rev. Nephrol. 2013, 9, 494. [Google Scholar] [PubMed]
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. Suppl. 2014, 4, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Boor, P. EP4: A new piece in the fibrotic puzzle. Kidney Int. 2012, 82, 132–135. [Google Scholar] [CrossRef]
- Huang, P.; Yan, R.; Zhang, X.; Wang, L.; Ke, X.; Qu, Y. Activating Wnt/β-catenin signaling pathway for disease therapy: Challenges and opportunities. Pharmacol. Ther. 2019, 196, 79–90. [Google Scholar] [CrossRef]
- Xie, H.; Miao, N.; Xu, D.; Zhou, Z.; Ni, J.; Yin, F.; Wang, Y.; Cheng, Q.; Chen, P.; Li, J.; et al. FoxM1 promotes Wnt/β-catenin pathway activation and renal fibrosis via transcriptionally regulating multi-Wnts expressions. J. Cell. Mol. Med. 2021, 25, 1958–1971. [Google Scholar] [CrossRef]
- Schunk, S.J.; Floege, J.; Fliser, D.; Speer, T. WNT-β-catenin signalling—A versatile player in kidney injury and repair. Nat. Rev. Nephrol. 2021, 17, 172–184. [Google Scholar] [CrossRef]
- Hu, H.H.; Cao, G.; Wu, X.Q.; Vaziri, N.D.; Zhao, Y.Y. Wnt signaling pathway in aging-related tissue fibrosis and therapies. Ageing Res. Rev. 2020, 60, 101063. [Google Scholar] [CrossRef]
- Li, S.S.; Sun, Q.; Hua, M.R.; Suo, P.; Chen, J.R.; Yu, X.Y.; Zhao, Y.Y. Targeting the Wnt/β-Catenin Signaling Pathway as a Potential Therapeutic Strategy in Renal Tubulointerstitial Fibrosis. Front. Pharmacol. 2021, 12, 719880. [Google Scholar] [CrossRef]
- Kim, S.; Nie, H.; Nesin, V.; Tran, U.; Outeda, P.; Bai, C.X.; Keeling, J.; Maskey, D.; Watnick, T.; Wessely, O.; et al. The polycystin complex mediates Wnt/Ca(2+) signalling. Nat. Cell Biol. 2016, 18, 752–764. [Google Scholar] [CrossRef] [PubMed]
- Acebron, S.P.; Niehrs, C. β-Catenin-Independent Roles of Wnt/LRP6 Signaling. Trends Cell Biol. 2016, 26, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Tan, R.J.; Fu, H.; Liu, Y. Wnt/β-catenin signaling in kidney injury and repair: A double-edged sword. Lab. Investig. 2016, 96, 156–167. [Google Scholar] [CrossRef]
- Simon-Tillaux, N.; Hertig, A. Snail and kidney fibrosis. Nephrol. Dial. Transplant. 2017, 32, 224–233. [Google Scholar] [CrossRef]
- García de Herreros, A.; Baulida, J. Cooperation, amplification, and feed-back in epithelial-mesenchymal transition. Biochim. Biophys. Acta. 2012, 1825, 223–228. [Google Scholar] [CrossRef]
- Kim, D.Y.; Kang, M.K.; Park, S.H.; Lee, E.J.; Kim, Y.H.; Oh, H.; Choi, Y.J.; Kang, Y.H. Eucalyptol ameliorates Snail1/β-catenin-dependent diabetic disjunction of renal tubular epithelial cells and tubulointerstitial fibrosis. Oncotarget 2017, 8, 106190–106205. [Google Scholar] [CrossRef]
- Zhang, S.; Qian, G.; Zhang, Q.Q.; Yao, Y.; Wang, D.; Chen, Z.G.; Wang, L.J.; Chen, M.; Sun, S.Y. mTORC2 Suppresses GSK3-Dependent Snail Degradation to Positively Regulate Cancer Cell Invasion and Metastasis. Cancer Res. 2019, 79, 3725–3736. [Google Scholar] [CrossRef]
- Togawa, H.; Nakanishi, K.; Mukaiyama, H.; Hama, T.; Shima, Y.; Sako, M.; Miyajima, M.; Nozu, K.; Nishii, K.; Nagao, S.; et al. Epithelial-to-mesenchymal transition in cyst lining epithelial cells in an orthologous PCK rat model of autosomal-recessive polycystic kidney disease. Am. J. Physiol. Renal Physiol. 2011, 300, F511–F520. [Google Scholar] [CrossRef]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef]
- García-Sánchez, O.; López-Hernández, F.J.; López-Novoa, J.M. An integrative view on the role of TGF-beta in the progressive tubular deletion associated with chronic kidney disease. Kidney Int. 2010, 77, 950–955. [Google Scholar] [CrossRef]
- Wozniak, J.; Floege, J.; Ostendorf, T.; Ludwig, A. Key metalloproteinase-mediated pathways in the kidney. Nat. Rev. Nephrol. 2021, 17, 513–527. [Google Scholar] [CrossRef] [PubMed]
- Villa-Morales, M.; Fernández-Piqueras, J. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.J.; Li, Y.; Rush, B.M.; Cerqueira, D.M.; Zhou, D.; Fu, H.; Ho, J.; Beer Stolz, D.; Liu, Y. Tubular injury triggers podocyte dysfunction by β-catenin-driven release of MMP-7. JCI Insight 2019, 4, e122399. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tan, R.J.; Liu, Y. The Many Faces of Matrix Metalloproteinase-7 in Kidney Diseases. Biomolecules 2020, 10, 960. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Zhou, D.; Zhu, H.; Liao, J.; Lin, L.; Hong, X.; Hou, F.F.; Liu, Y. Matrix metalloproteinase-7 protects against acute kidney injury by priming renal tubules for survival and regeneration. Kidney Int. 2019, 95, 1167–1180. [Google Scholar] [CrossRef]
- Malik, S.A.; Modarage, K.; Goggolidou, P. The Role of Wnt Signalling in Chronic Kidney Disease (CKD). Genes 2020, 11, 496. [Google Scholar] [CrossRef]
- Flevaris, P.; Vaughan, D. The Role of Plasminogen Activator Inhibitor Type-1 in Fibrosis. Semin. Thromb. Hemost. 2017, 43, 169–177. [Google Scholar] [CrossRef]
- Rabieian, R.; Boshtam, M.; Zareei, M.; Kouhpayeh, S.; Masoudifar, A.; Mirzaei, H. Plasminogen Activator Inhibitor Type-1 as a Regulator of Fibrosis. J. Cell. Biochem. 2018, 119, 17–27. [Google Scholar] [CrossRef]
- Yao, L.; Wright, M.F.; Farmer, B.C.; Peterson, L.S.; Khan, A.M.; Zhong, J.; Gewin, L.; Hao, C.M.; Yang, H.C.; Fogo, A.B. Fibroblast-specific plasminogen activator inhibitor-1 depletion ameliorates renal interstitial fibrosis after unilateral ureteral obstruction. Nephrol. Dial. Transplant. 2019, 34, 2042–2050. [Google Scholar] [CrossRef]
- Gu, L.F.; Ge, H.T.; Zhao, L.; Wang, Y.J.; Zhang, F.; Tang, H.T.; Cao, Z.Y.; Yu, B.Y.; Chai, C.Z. Huangkui Capsule Ameliorates Renal Fibrosis in a Unilateral Ureteral Obstruction Mouse Model Through TRPC6 Dependent Signaling Pathways. Front. Pharmacol. 2020, 11, 996. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; Hao, S.; Zhou, D.; Tan, R.J.; Nie, J.; Hou, F.F.; Kahn, M.; Liu, Y. Multiple genes of the renin-angiotensin system are novel targets of Wnt/β-catenin signaling. J. Am. Soc. Nephrol. 2015, 26, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, T. Soluble (Pro)Renin Receptor in Hypertension. Nephron 2023, 147, 234–243. [Google Scholar] [CrossRef]
- Zhou, G.; Wu, J.; Gu, C.; Wang, B.; Abel, E.D.; Cheung, A.K.; Huang, Y. Prorenin independently causes hypertension and renal and cardiac fibrosis in cyp1a1-prorenin transgenic rats. Clin. Sci. 2018, 132, 1345–1363. [Google Scholar] [CrossRef] [PubMed]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91–92, 92–108. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, S.; Tao, L.; Gan, S.; Luo, H.; Xu, Y.; Shen, X. Inhibitory Effects of Oxymatrine on Transdifferentiation of Neonatal Rat Cardiac Fibroblasts to Myofibroblasts Induced by Aldosterone via Keap1/Nrf2 Signaling Pathways In Vitro. Med. Sci. Monit. 2019, 25, 5375–5388. [Google Scholar] [CrossRef]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-β/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Wu, W.; Wang, X.; Yu, X.; Lan, H.Y. Smad3 Signatures in Renal Inflammation and Fibrosis. Int. J. Biol. Sci. 2022, 18, 2795–2806. [Google Scholar] [CrossRef]
- Liu, G.X.; Li, Y.Q.; Huang, X.R.; Wei, L.; Chen, H.Y.; Shi, Y.J.; Heuchel, R.L.; Lan, H.Y. Disruption of Smad7 promotes ANG II-mediated renal inflammation and fibrosis via Sp1-TGF-β/Smad3-NF.κB-dependent mechanisms in mice. PLoS ONE 2013, 8, e53573. [Google Scholar] [CrossRef]
- Shuttleworth, V.G.; Gaughan, L.; Nawafa, L.; Mooney, C.A.; Cobb, S.L.; Sheerin, N.S.; Logan, I.R. The methyltransferase SET9 regulates TGFB1 activation of renal fibroblasts via interaction with SMAD3. J. Cell Sci. 2018, 131, jcs207761. [Google Scholar]
- Chen, B.; Wang, P.; Liang, X.; Jiang, C.; Ge, Y.; Dworkin, L.D.; Gong, R. Permissive effect of GSK3β on profibrogenic plasticity of renal tubular cells in progressive chronic kidney disease. Cell Death Dis. 2021, 12, 432. [Google Scholar] [CrossRef]
- Sato, M.; Muragaki, Y.; Saika, S.; Roberts, A.B.; Ooshima, A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J. Clin. Investig. 2003, 112, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Kriegel, A.J.; Liu, Y.; Fang, Y.; Ding, X.; Liang, M. The miR-29 family: Genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol. Genom. 2012, 44, 237–244. [Google Scholar] [CrossRef]
- Meng, X.M.; Huang, X.R.; Xiao, J.; Chung, A.C.; Qin, W.; Chen, H.Y.; Lan, H.Y. Disruption of Smad4 impairs TGF-β/Smad3 and Smad7 transcriptional regulation during renal inflammation and fibrosis in vivo and in vitro. Kidney Int. 2012, 81, 266–279. [Google Scholar] [CrossRef]
- Zhang, F.; Tsai, S.; Kato, K.; Yamanouchi, D.; Wang, C.; Rafii, S.; Liu, B.; Kent, K.C. Transforming growth factor-beta promotes recruitment of bone marrow cells and bone marrow-derived mesenchymal stem cells through stimulation of MCP-1 production in vascular smooth muscle cells. J. Biol. Chem. 2009, 284, 17564–17574. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Chung, A.C.; Chen, H.Y.; Meng, X.M.; Lan, H.Y. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J. Am. Soc. Nephrol. 2011, 22, 1668–1681. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, J.; Shi, J.; Liu, K.; Wang, X.; Jia, Y.; He, T.; Shen, K.; Wang, Y.; Liu, J.; et al. Exosomes derived from human adipose mesenchymal stem cells attenuate hypertrophic scar fibrosis by miR-192-5p/IL-17RA/Smad axis. Stem Cell Res. Ther. 2021, 12, 221. [Google Scholar] [CrossRef]
- Zhao, S.; Li, W.; Yu, W.; Rao, T.; Li, H.; Ruan, Y.; Yuan, R.; Li, C.; Ning, J.; Li, S.; et al. Exosomal miR-21 from tubular cells contributes to renal fibrosis by activating fibroblasts via targeting PTEN in obstructed kidneys. Theranostics 2021, 11, 8660–8673. [Google Scholar] [CrossRef]
- Fan, Q.; Lu, R.; Zhu, M.; Yan, Y.; Guo, X.; Qian, Y.; Zhang, L.; Dai, H.; Ni, Z.; Gu, L. Serum miR-192 Is Related to Tubulointerstitial Lesion and Short-Term Disease Progression in IgA Nephropathy. Nephron 2019, 142, 195–207. [Google Scholar] [CrossRef]
- Liu, Z.; Yang, Q.; Wei, Q.; Chang, Y.; Qu, M.; Yu, L. The protective effect of miR-377 inhibitor against renal ischemia-reperfusion injury through inhibition of inflammation and oxidative stress via a VEGF-dependent mechanism in mice. Mol. Immunol. 2019, 106, 153–158. [Google Scholar] [CrossRef]
- Wang, H.; Wang, B.; Zhang, A.; Hassounah, F.; Seow, Y.; Wood, M.; Ma, F.; Klein, J.D.; Price, S.R.; Wang, X.H. Exosome-Mediated miR-29 Transfer Reduces Muscle Atrophy and Kidney Fibrosis in Mice. Mol. Ther. 2019, 27, 571–583. [Google Scholar] [CrossRef]
- Huang, J.; Lai, W.; Li, M.; Li, C.; Lou, T.; Peng, H.; Ye, Z. SIS3 Alleviates Cisplatin-Induced Acute Kidney Injury by Regulating the LncRNA Arid2-IR-Transferrin Receptor Pathway. Kidney Blood Press. Res. 2022, 47, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Tan, R.Z.; Yu, Y.; Niu, Y.Y.; Yu, C. LncRNA GAS5 protects against TGF-β-induced renal fibrosis via the Smad3/miRNA-142-5p axis. Am. J. Physiol. Renal Physiol. 2021, 321, F517–F526. [Google Scholar] [CrossRef] [PubMed]
- Yang, L. How Acute Kidney Injury Contributes to Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 117–142. [Google Scholar] [PubMed]
- Nagata, S.; Segawa, K. Sensing and clearance of apoptotic cells. Curr. Opin. Immunol. 2021, 68, 1–8. [Google Scholar] [CrossRef]
- Maremonti, F.; Meyer, C.; Linkermann, A. Mechanisms and Models of Kidney Tubular Necrosis and Nephron Loss. J. Am. Soc. Nephrol. 2022, 33, 472–486. [Google Scholar] [CrossRef]
- Wiernicki, B.; Dubois, H.; Tyurina, Y.Y.; Hassannia, B.; Bayir, H.; Kagan, V.E.; Vandenabeele, P.; Wullaert, A.; Vanden, B.T. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 2020, 11, 922. [Google Scholar] [CrossRef]
- Jiang, L.; Liu, X.Q.; Ma, Q.; Yang, Q.; Gao, L.; Li, H.D.; Wang, J.N.; Wei, B.; Wen, J.; Li, J.; et al. hsa-miR-500a-3P alleviates kidney injury by targeting MLKL-mediated necroptosis in renal epithelial cells. FASEB J. 2019, 33, 3523–3535. [Google Scholar] [CrossRef]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales, P.J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. [Google Scholar] [CrossRef]
- Yang, Q.; Gao, L.; Hu, X.W.; Wang, J.N.; Zhang, Y.; Dong, Y.H.; Lan, H.Y.; Meng, X.M. Smad3-Targeted Therapy Protects against Cisplatin-Induced AKI by Attenuating Programmed Cell Death and Inflammation via a NOX4-Dependent Mechanism. Kidney Dis. 2021, 7, 372–390. [Google Scholar] [CrossRef]
- Tang, P.M.; Zhang, Y.Y.; Xiao, J.; Tang, P.C.; Chung, J.Y.; Li, J.; Xue, V.W.; Huang, X.R.; Chong, C.C.; Ng, C.F.; et al. Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage-myofibroblast transition. Proc. Natl. Acad. Sci. USA 2020, 117, 20741–20752. [Google Scholar] [CrossRef]
- Tang, P.M.; Zhou, S.; Li, C.J.; Liao, J.; Xiao, J.; Wang, Q.M.; Lian, G.Y.; Li, J.; Huang, X.R.; To, K.; et al. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 2018, 93, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Gong, A.Y.; Haller, S.T.; Dworkin, L.D.; Liu, Z.; Gong, R. The ageing kidney: Molecular mechanisms and clinical implications. Ageing Res. Rev. 2020, 63, 101–151. [Google Scholar] [CrossRef] [PubMed]
- Gifford, C.C.; Lian, F.; Tang, J.; Costello, A.; Goldschmeding, R.; Samarakoon, R.; Higgins, P.J. PAI-1 induction during kidney injury promotes fibrotic epithelial dysfunction via deregulation of klotho, p53, and TGF-β1-receptor signaling. FASEB J. 2021, 35, e21725. [Google Scholar] [CrossRef] [PubMed]
- You, Y.K.; Huang, X.R.; Chen, H.Y.; Lyu, X.F.; Liu, H.F.; Lan, H.Y. C-reactive protein Promotes Diabetic Kidney Disease in db/db Mice via the CD32b-Smad3-mTOR signaling Pathway. Sci. Rep. 2016, 6, 26740. [Google Scholar] [CrossRef]
- Sun, S.F.; Tang, P.M.K.; Feng, M.; Xiao, J.; Huang, X.R.; Li, P.; Ma, R.C.W.; Lan, H.Y. Novel lncRNA Erbb4-IR Promotes Diabetic Kidney Injury in db/db Mice by Targeting miR-29b. Diabetes 2018, 67, 731–744. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Fontecha-Barriuso, M.; Carrasco, S.; Sanchez-Niño, M.D.; Mässenhausen, A.V.; Linkermann, A.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; et al. TWEAK and RIPK1 mediate a second wave of cell death during AKI. Proc. Natl. Acad. Sci. USA 2018, 115, 4182–4187. [Google Scholar] [CrossRef]
- Su, L.; Jiang, X.; Yang, C.; Zhang, J.; Chen, B.; Li, Y.; Yao, S.; Xie, Q.; Gomez, H.; Murugan, R.; et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J. Biol. Chem. 2019, 294, 19395–19404. [Google Scholar] [CrossRef]
- Li, S.; Zheng, L.; Zhang, J.; Liu, X.; Wu, Z. Inhibition of ferroptosis by up-regulating Nrf2 delayed the progression of diabetic nephropathy. Free Radic. Biol. Med. 2021, 162, 435–449. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, H.; Yi, B.; Yang, S.; Liu, J.; Hu, J.; Wang, J.; Cao, K.; Zhang, W. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis. 2020, 11, 73. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, J.; Xu, H.; Zhou, C.; Han, B.; Zhu, H.; Hu, Z.; Ma, Z.; Ming, Z.; Yao, Y.; et al. XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis. 2020, 11, 629. [Google Scholar] [CrossRef]
- Yang, L.; Guo, J.; Yu, N.; Liu, Y.; Song, H.; Niu, J.; Gu, Y. Tocilizumab mimotope alleviates kidney injury and fibrosis by inhibiting IL-6 signaling and ferroptosis in UUO model. Life Sci. 2020, 261, 118487. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, D.; Yu, Y.; Zhao, T.; Min, N.; Wu, Y.; Kang, L.; Zhao, Y.; Du, L.; Zhang, M.; et al. Legumain promotes tubular ferroptosis by facilitating chaperone-mediated autophagy of GPX4 in AKI. Cell Death Dis. 2021, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Ding, X.; Zheng, J.; Wang, B.; Li, Y.; Xiang, H.; Dou, M.; Qiao, Y.; Tian, P.; Xue, W. miR-182-5p and miR-378a-3p regulate ferroptosis in I/R-induced renal injury. Cell Death Dis. 2020, 11, 929. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Mei, M.; Pu, Y.; Zhang, H.; Liu, H.; Tang, M.; Pan, Q.; He, Y.; Wu, X.; Zhao, H. Necrostatin-1 Attenuates Renal Ischemia and Reperfusion Injury via Meditation of HIF-1α/mir-26a/TRPC6/PARP1 Signaling. Mol. Ther. Nucleic Acids 2019, 17, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Song, N.; Zhang, X.; Jiao, X.; Hu, J.; Liang, M.; Teng, J.; Ding, X. Renal Protection Mediated by Hypoxia Inducible Factor-1α Depends on Proangiogenesis Function of miR-21 by Targeting Thrombospondin 1. Transplantation 2017, 101, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, L.; Liu, M.; Du, R.; Dang, Y.; Bai, M.; Zhang, L.; Ma, F.; Yang, X.; Ning, X.; et al. MicroRNA-493 targets STMN-1 and promotes hypoxia-induced epithelial cell cycle arrest in G2/M and renal fibrosis. FASEB J. 2019, 32, 1565–1577. [Google Scholar] [CrossRef] [PubMed]
- Jamadarkhana, P.; Chaudhary, A.; Chhipa, L.; Dubey, A.; Mohanan, A.; Gupta, R.; Deshpande, S. Treatment with a novel hypoxia-inducible factor hydroxylase inhibitor (TRC160334) ameliorates ischemic acute kidney injury. Am. J. Nephrol. 2012, 36, 208–218. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, L.; Zhang, Y.; Yu, X.; Sun, X.; Zhu, T.; Li, X.; Liang, W.; Han, Y.; Qin, C. PINK1 Deficiency Ameliorates Cisplatin-Induced Acute Kidney Injury in Rats. Front. Physiol. 2019, 10, 1225. [Google Scholar] [CrossRef]
- Livingston, M.J.; Wang, J.; Zhou, J.; Wu, G.; Ganley, I.G.; Hill, J.A.; Yin, X.M.; Dong, Z. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef]
- Li, N.; Wang, H.; Jiang, C.; Zhang, M. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp. Cell Res. 2018, 369, 27–33. [Google Scholar] [CrossRef]
- Feng, J.; Li, H.; Zhang, Y.; Wang, Q.; Zhao, S.; Meng, P.; Li, J. Mammalian STE20-Like Kinase 1 Deletion Alleviates Renal Ischaemia-Reperfusion Injury via Modulating Mitophagy and the AMPK-YAP Signalling Pathway. Cell. Physiol. Biochem. 2018, 1, 2359–2376. [Google Scholar] [CrossRef]
- Tang, C.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 2019, 10, 677. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Sun, H.; Song, S.; Liu, Y.; Liu, P.; Livingston, M.J.; Wang, J.; Liang, M.; Mi, Q.S.; Huo, Y.; et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J. Clin. Investig. 2018, 128, 5448–5464. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, Y.H.; Tang, C.; Xue, M.; Li, X.Y.; Chang, Y.P.; Cheng, Y.; Li, T.; Yu, X.C.; Sun, B.; et al. Calcium Dobesilate Restores Autophagy by Inhibiting the VEGF/PI3K/AKT/mTOR Signaling Pathway. Front. Pharmacol. 2019, 10, 886. [Google Scholar] [CrossRef] [PubMed]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef]
- Chen, L.; Lin, G.; Chen, K.; Liang, R.; Wan, F.; Zhang, C.; Tian, G.; Zhu, X. VEGF promotes migration and invasion by regulating EMT and MMPs in nasopharyngeal carcinoma. J. Cancer 2020, 11, 7291–7301. [Google Scholar] [CrossRef]
- Hwang, S.D.; Song, J.H.; Kim, Y.; Lim, J.H.; Kim, M.Y.; Kim, E.N.; Hong, Y.A.; Chung, S.; Choi, B.S.; Kim, Y.S.; et al. Inhibition of lymphatic proliferation by the selective VEGFR-3 inhibitor SAR131675 ameliorates diabetic nephropathy in db/db mice. Cell Death Dis. 2019, 10, 219. [Google Scholar] [CrossRef]
- Canaud, G.; Brooks, C.R.; Kishi, S.; Taguchi, K.; Nishimura, K.; Magassa, S.; Scott, A.; Hsiao, L.L.; Ichimura, T.; Terzi, F.; et al. Cyclin G1 and TASCC regulate kidney epithelial cell G2-M arrest and fibrotic maladaptive repair. Sci. Transl. Med. 2019, 11, 4754. [Google Scholar] [CrossRef]
- Lemos, D.R.; McMurdo, M.; Karaca, G.; Wilflingseder, J.; Leaf, I.A.; Gupta, N.; Miyoshi, T.; Susa, K.; Johnson, B.G.; Soliman, K.; et al. Interleukin-1β Activates a MYC-Dependent Metabolic Switch in Kidney Stromal Cells Necessary for Progressive Tubulointerstitial Fibrosis. J. Am. Soc. Nephrol. 2018, 29, 1690–1705. [Google Scholar] [CrossRef]
- Yiu, W.H.; Li, R.X.; Wong, D.W.L.; Wu, H.J.; Chan, K.W.; Chan, L.Y.Y.; Leung, J.C.K.; Lai, K.N.; Sacks, S.H.; Zhou, W.; et al. Complement C5a inhibition moderates lipid metabolism and reduces tubulointerstitial fibrosis in diabetic nephropathy. Nephrol. Dial. Transplant. 2018, 33, 1323–1332. [Google Scholar] [CrossRef]
- Mehrotra, P.; Sturek, M.; Neyra, J.A.; Basile, D.P. Calcium channel Orai1 promotes lymphocyte IL-17 expression and progressive kidney injury. J. Clin. Investig. 2019, 129, 4951–4961. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chen, T.; Wang, C.; Zhang, Z.; Wang, X.M.; Li, Q.; Lee, V.W.S.; Wang, Y.M.; Zheng, G.; Alexander, S.I.; et al. Flt3 inhibition alleviates chronic kidney disease by suppressing CD103+ dendritic cell-mediated T cell activation. Nephrol. Dial. Transplant. 2019, 34, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Zhang, X.; Liu, L.C.; Kim, A.Y.; Curley, S.P.; Chen, X.; Dworkin, L.D.; Cooper, C.J.; Gupta, R. Interleukin-10 attenuates renal injury after myocardial infarction in diabetes. J. Investig. Med. 2022, 70, 1233–1242. [Google Scholar] [CrossRef]
- Martín, M.B.T.; Satoh, M.; Hernández, P.R.; Martínez, G.E.A.; Petri, M.H.; Sandoval-García, F.; Pizano-Martinez, O.; García-Iglesias, T.; Corona-Meraz, F.I.; Vázquez, D.M.M. The DNA co-vaccination using Sm antigen and IL-10 as prophylactic experimental therapy ameliorates nephritis in a model of lupus induced by pristane. PLoS ONE 2021, 16, 0259114. [Google Scholar]
- Gong, J.; Noel, S.; Hsu, J.; Bush, E.L.; Arend, L.J.; Sadasivam, M.; Lee, S.A.; Kurzhagen, J.T.; Hamad, A.R.A.; Rabb, H. TCR+CD4−CD8− (double negative) T cells protect from cisplatin-induced renal epithelial cell apoptosis and acute kidney injury. Am. J. Physiol. Renal Physiol. 2020, 318, F1500–F1512. [Google Scholar] [CrossRef]
- Andrade, S.M.; Cenedeze, M.A.; Perandini, L.A.; Felizardo, R.J.F.; Watanabe, I.K.M.; Agudelo, J.S.H.; Castoldi, A.; Gonçalves, G.M.; Origassa, C.S.T.; Semedo, P.; et al. TLR2 and TLR4 play opposite role in autophagy associated with cisplatin-induced acute kidney injury. Clin. Sci. 2018, 132, 1725–1739. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, C. From AKI to CKD: Maladaptive Repair and the Underlying Mechanisms. Int. J. Mol. Sci. 2022, 23, 10880. [Google Scholar] [CrossRef]
- Shu, S.; Wang, Y.; Zheng, M.; Liu, Z.; Cai, J.; Tang, C.; Dong, Z. Hypoxia and Hypoxia-Inducible Factors in Kidney Injury and Repair. Cells 2019, 8, 207. [Google Scholar] [CrossRef]
- Liu, J.; Wei, Q.; Guo, C.; Dong, G.; Liu, Y.; Tang, C.; Dong, Z. Hypoxia, HIF, and Associated Signaling Networks in Chronic Kidney Disease. Int. J. Mol. Sci. 2017, 18, 950. [Google Scholar] [CrossRef]
- Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. [Google Scholar] [CrossRef] [PubMed]
- Koyano, T.; Namba, M.; Kobayashi, T.; Nakakuni, K.; Nakano, D.; Fukushima, M.; Nishiyama, A.; Matsuyama, M. The p21 dependent G2 arrest of the cell cycle in epithelial tubular cells links to the early stage of renal fibrosis. Sci. Rep. 2019, 9, 12059. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.M.; Bonventre, J.V. Acute kidney injury and maladaptive tubular repair leading to renal fibrosis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Li, K.; Smyth, L.A.; Xing, G.; Wang, N.; Meader, L.; Lu, B.; Sacks, S.H.; Zhou, W. C3a and C5a Promote Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2012, 23, 1474–1485. [Google Scholar] [CrossRef]
- Peng, Q.; Wu, W.; Wu, K.Y.; Cao, B.; Qiang, C.; Li, K.; Sacks, S.H.; Zhou, W. The C5a/C5aR1 axis promotes progression of renal tubulointerstitial fibrosis in a mouse model of renal ischemia/reperfusion injury. Kidney Int. 2019, 96, 117–128. [Google Scholar] [CrossRef]
- Tang, P.C.; Zhang, Y.Y.; Chan, M.K.; Lam, W.W.; Chung, J.Y.; Kang, W.; To, K.F.; Lan, H.Y.; Tang, P.M. The Emerging Role of Innate Immunity in Chronic Kidney Diseases. Int. J. Mol. Sci. 2020, 21, 4018. [Google Scholar] [CrossRef]
- Wei, W.; Zhao, Y.; Zhang, Y.; Jin, H.; Shou, S. The role of IL-10 in kidney disease. Int. Immunopharmacol. 2022, 108, 108917. [Google Scholar] [CrossRef]
- Vinuesa, E.; Hotter, G.; Jung, M.; Herrero, F.I.; Torras, J.; Sola, A. Macrophage involvement in the kidney repair phase after ischaemia/reperfusion injury. J. Pathol. 2008, 214, 104–113. [Google Scholar] [CrossRef]
- Lu, X.; Rudemiller, N.P.; Ren, J.; Wen, Y.; Yang, B.; Griffiths, R.; Privratsky, J.R.; Madan, B.; Virshup, D.M.; Crowley, S.D. Opposing actions of renal tubular- and myeloid-derived porcupine in obstruction-induced kidney fibrosis. Kidney Int. 2019, 96, 1308–1319. [Google Scholar] [CrossRef]
- Tang, P.M.; Nikolic, P.D.J.; Lan, H.Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef]
- Feng, Y.; Ren, J.; Gui, Y.; Wei, W.; Shu, B.; Lu, Q.; Xue, X.; Sun, X.; He, W.; Yang, J.; et al. Wnt/β-Catenin-Promoted Macrophage Alternative Activation Contributes to Kidney Fibrosis. J. Am. Soc. Nephrol. 2018, 29, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Feng, X.; Liu, X.; Wang, Y.; Hu, M.; Cao, Q.; Zhang, Z.; Zhao, L.; Zhang, J.; Guo, R.; et al. Fate alteration of bone marrow-derived macrophages ameliorates kidney fibrosis in murine model of unilateral ureteral obstruction. Nephrol. Dial. Transplant. 2019, 34, 1657–1668. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Ganguly, S.; Nespoux, J.; Freeman, B.; Zhang, H.; Brenner, D.; Dhar, D.; Vallon, V. Western diet promotes renal injury, inflammation, and fibrosis in a murine model of Alström syndrome. Nephron 2020, 144, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Chen, H.Y.; Wang, J.N.; Han, H.Q.; Jiang, L.; Wu, W.F.; Wei, B.; Gao, L.; Ma, Q.Y.; Liu, X.Q.; et al. Alcohol promotes renal fibrosis by activating Nox2/4-mediated DNA methylation of Smad7. Clin. Sci. 2020, 134, 103–122. [Google Scholar] [CrossRef]
- Sousa, M.V.; Amaral, A.G.; Freitas, J.A.; Murata, G.M.; Watanabe, E.H.; Balbo, B.E.; Tavares, M.D.; Hortegal, R.A.; Rocon, C.; Souza, L.E.; et al. Smoking accelerates renal cystic disease and worsens cardiac phenotype in Pkd1-deficient mice. Sci. Rep. 2021, 11, 14443. [Google Scholar] [CrossRef]
- Sandino, J.; Martín-Taboada, M.; Medina-Gómez, G.; Vila-Bedmar, R.; Morales, E. Novel Insights in the Physiopathology and Management of Obesity-Related Kidney Disease. Nutrients 2022, 14, 3937. [Google Scholar] [CrossRef]
- Duan, Y.C.; Shi, L.; Jin, Z.; Hu, M.; Huang, H.; Yan, T.; Zhang, K.R. Swimming Exercise Ameliorates Hypertension-Induced Kidney Dysfunction via Alleviating Renal Interstitial Fibrosis and Apoptosis. Kidney Blood Press. Res. 2021, 46, 219–228. [Google Scholar] [CrossRef]
- Cho, M.E.; Smith, D.C.; Branton, M.H.; Penzak, S.R.; Kopp, J.B. Pirfenidone slows renal function decline in patients with focal segmental glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2005, 2, 906–913. [Google Scholar] [CrossRef]
- Vincenti, F.; Fervenza, F.C.; Campbell, K.N.; Diaz, M.; Gesualdo, L.; Nelson, P.; Praga, M.; Radhakrishnan, J.; Sellin, L.; Singh, A.; et al. A Phase 2, Double-Blind, Placebo-Controlled, Randomized Study of Fresolimumab in Patients With Steroid-Resistant Primary Focal Segmental Glomerulosclerosis. Kidney Int. Rep. 2017, 2, 800–810. [Google Scholar] [CrossRef]
- Tan, R.Z.; Wang, C.; Deng, C.; Zhong, X.; Yan, Y.; Luo, Y.; Lan, H.Y.; He, T.; Wang, L. Quercetin protects against cisplatin-induced acute kidney injury by inhibiting Mincle/Syk/NF-κB signaling maintained macrophage inflammation. Phytother. Res. 2020, 34, 139–152. [Google Scholar] [CrossRef]
- Ai, J.; Nie, J.; He, J.; Guo, Q.; Li, M.; Lei, Y.; Liu, Y.; Zhou, Z.; Zhu, F.; Liang, M.; et al. GQ5 Hinders Renal Fibrosis in Obstructive Nephropathy by Selectively Inhibiting TGF-β-Induced Smad3 Phosphorylation. J. Am. Soc. Nephrol. 2015, 26, 1827–1838. [Google Scholar] [CrossRef]
- Sierra, M.E.; Rodríguez, R.R.; Namorado, T.C.; Molina, E.; Romero, T.D.; Pedraza, C.J.; Reyes, J.L. All-Trans Retinoic Acid Attenuates Fibrotic Processes by Downregulating TGF-β1/Smad3 in Early Diabetic Nephropathy. Biomolecules 2019, 9, 525. [Google Scholar] [CrossRef] [PubMed]
- Diao, W.; Chen, W.; Cao, W.; Yuan, H.; Ji, H.; Wang, T.; Chen, W.; Zhu, X.; Zhou, H.; Guo, H.; et al. Astaxanthin protects against renal fibrosis through inhibiting myofibroblast activation and promoting CD8+ T cell recruitment. Biochim. Biophys. Acta. Gen. Subj. 2019, 1863, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, Z.; Luo, J.; Jiang, Y.; Li, L.; Chen, Y.; Zhang, L.; Huang, Q.; Cao, Y.; Zhou, P.; et al. Apigenin ameliorates hyperuricemic nephropathy by inhibiting URAT1 and GLUT9 and relieving renal fibrosis via the Wnt/β-catenin pathway. Phytomedicine 2021, 87, 53585. [Google Scholar] [CrossRef]
- Pan, B.; Zhang, H.; Hong, Y.; Ma, M.; Wan, X.; Cao, C. Indoleamine-2,3-Dioxygenase Activates Wnt/β-Catenin Inducing Kidney Fibrosis after Acute Kidney Injury. Gerontology 2021, 67, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Luo, M.; Chen, N.; Deng, X.; He, J.; Zhang, L.; Luo, P.; Wu, J. Inhibition of PAI-1 attenuates perirenal fat inflammation and the associated nephropathy in high-fat diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E260–E267. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.Y.; Cho, M.Y.; Baik, S.K.; Jeong, P.H.; Suk, K.T.; Jang, Y.O.; Yea, C.J.; Kim, J.W.; Kim, H.S.; Kwon, S.O.; et al. Beneficial effects of candesartan, an angiotensin-blocking agent, on compensated alcoholic liver fibrosis—A randomized open-label controlled study. Liver Int. 2012, 32, 977–987. [Google Scholar] [CrossRef]
- Zhu, Q.; Li, N.; Li, F.; Zhou, Z.; Han, Q.; Lv, Y.; Sang, J.; Liu, Z. Therapeutic effect of renin angiotensin system inhibitors on liver fibrosis. J. Renin Angiotensin Aldosterone Syst. 2016, 17, 14703203–16628717. [Google Scholar] [CrossRef]
- Tan, W.Q.; Fang, Q.Q.; Shen, X.Z.; Giani, J.F.; Zhao, T.V.; Shi, P.; Zhang, L.Y.; Khan, Z.; Li, Y.; Li, L.; et al. Angiotensin-converting enzyme inhibitor works as a scar formation inhibitor by down-regulating Smad and TGF-β-activated kinase 1 (TAK1) pathways in mice. Br. J. Pharmacol. 2018, 175, 4239–4252. [Google Scholar] [CrossRef]
- Brilla, C.G.; Funck, R.C.; Rupp, H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 2000, 102, 1388–1393. [Google Scholar] [CrossRef]
- Bomback, A.S.; Kshirsagar, A.V.; Amamoo, M.A.; Klemmer, P.J. Change in proteinuria after adding aldosterone blockers to ACE inhibitors or angiotensin receptor blockers in CKD: A systematic review. Am. J. Kidney Dis. 2008, 51, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. FIDELIO-DKD Investigators. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Koch, G.; Ouyang, J.; McQuade, R.D.; Blais, J.D.; Czerwiec, F.S.; et al. Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2017, 377, 1930–1942. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, H.; Wong, M.G.; Jardine, M.J.; Hladunewich, M.; Jha, V.; Monaghan, H.; Zhao, M.; Barbour, S.; Reich, H.; et al. Effect of Oral Methylprednisolone on Clinical Outcomes in Patients With IgA Nephropathy: The TESTING Randomized Clinical Trial. JAMA 2017, 18, 432–442. [Google Scholar] [CrossRef]
- Gross, O.; Girgert, R.; Rubel, D.; Temme, J.; Theissen, S.; Müller, G.A. Renal protective effects of aliskiren beyond its antihypertensive property in a mouse model of progressive fibrosis. Am. J. Hypertens. 2011, 24, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Li, F.; Qi, C.; Mao, X.; Wang, F.; Zhao, Z.; Chen, J.K.; Zhang, Z.; Wu, H. Metformin effectively treats Tsc1 deletion-caused kidney pathology by upregulating AMPK phosphorylation. Cell Death Discov. 2020, 6, 52. [Google Scholar] [CrossRef]
- Nargesi, A.A.; Lerman, L.O.; Eirin, A. Mesenchymal Stem Cell-derived Extracellular Vesicles for Renal Repair. Curr. Gene Ther. 2017, 17, 29–42. [Google Scholar] [CrossRef]
- Birtwistle, L.; Chen, X.M.; Pollock, C. Mesenchymal Stem Cell-Derived Extracellular Vesicles to the Rescue of Renal Injury. Int. J. Mol. Sci. 2021, 22, 6596. [Google Scholar] [CrossRef]
- Li, J.; Gong, X. Tetramethylpyrazine: An Active Ingredient of Chinese Herbal Medicine With Therapeutic Potential in Acute Kidney Injury and Renal Fibrosis. Front. Pharmacol. 2022, 13, 820071. [Google Scholar] [CrossRef]
- Reubi, F. On the history of kidney disease. Schweiz. Med. Wochenschr. 1987, 117, 369–376. [Google Scholar]
- Beale, L.S. Arterio-Capillary Fibrosis. Br. Med. J. 1873, 1, 32–33. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Class | Name | Pathways | Action | Evidence |
|---|---|---|---|---|
| Wnt/β-catenin | TRPC6 knockout | Wnt/β-catenin downstream signaling | Ameliorates renal fibrosis and α-SMA expression | [40] |
| Snail1 inhibition by Eucalyptol | Loweres α-SMA expression in tubulointerstitial space | [26] | ||
| Snail upregulation | Seen in largest cysts from polycystic renal disease | [28] | ||
| PAI-1 knockout | Ameliorates tubulointerstitial fibrosis via FMT inhibition and lower collagen I deposition | [37] | ||
| MMP-7 knockout | Protects from podocyte destruction | [33] | ||
| MMP-7 upregulation | Promotes apoptosis and FMT | [32] | ||
| ANG II | ANG II upregulation | JAK and MAPK activation Increased intracellular Ca2+ influx Upregulation of TGF-β/SMAD2/SMAD3 signaling | Renal fibrosis | [44] |
| Oxymatrine | ANG II and aldosterone inhibition | Overproduction of fibronectin, collagen I, PAI-1 and PAI-2 | [45] | |
| TGF-β1/SMAD signaling | POU4F1 silencing | SMAD3 downstream signaling | Prevents MMT | [70] |
| Src inhibition | Prevents MMT | [71] | ||
| SET9 activation | Increases SMA expression | [49] | ||
| SMAD4 inhibition | Lowers renal fibrosis in UUO mouse models | [53] | ||
| SMAD3 inhibition | Lowers renal fibrosis in diabetic, obstructive and hypertensive nephropathy | [46,51,52] | ||
| GSK3β inhibition | Induces higher CREB activity with lower CBP, essential in SMAD3 activation | [50] | ||
| Snail1 knockout | Reduces renal fibrosis in mice obstructive nephropathy | [29] | ||
| CDKIs: p16, p21, p27, p38 | Induces tubular epithelial cell death, through G1 cell cycle arrest and are involved in kidney ageing | [72] | ||
| PAI-1 activation | Klotho inhibition with TGF-β and p53 upregulation | Promotes kidney fibrosis | [73] | |
| CRP | Smad activation via CD32b-ERK/p38 MAP kinase crosstalk pathway | Promotes kidney inflammation and fibrosis | [74] | |
| miR-21 knockout | SMAD3 downstream signaling Through PTEN/Akt pathway | In vivo studies—abolished FMT In vitro studies—alleviates renal fibrosis in obstructive nephropathy | [57] | |
| miR-192 | SMAD3 downstream signaling | High serum/intrarenal and urinary levels were associated with higher grades of tubulointerstitial fibrosis | [58] | |
| Exo/miR-29 | SMAD3 downstream signaling | MiR-29 exerts anti-fibrotic effects through TGF-β1 inhibition | [60] | |
| Erbb4-IR | SMAD dependent lncRNA | Promotes renal fibrosis in diabetic and obstructive nephropathy Erbb4-IR inhibition alleviates renal fibrosis via miR-29 upregulation | [75] | |
| Arid2-IR | Protective in cell cycle control | [61] | ||
| GAS5 | Inhibits TGF-β1 and is downregulated by SMAD signaling Evidences blocked renal fibrosis in vitro studies | [62] | ||
| Cell death pathways | Fn-14 knockout gene | TWEAK pathway | Reduces death of tubular epithelial cells | [76] |
| Pannexin-1 inhibition | MAPK/ERK pathway | Inhibits ferroptosis and evidences decreased serum creatinine, cell necrosis and melondialdehyde expression | [77] | |
| Ferrostatin-1 | Ferroptosis inhibitor | Lowers tissue renal damage in mouse models with cisplatin-induced AKI | [78] | |
| VDR | Inhibits GPX4—key control of ferroptosis | Knockout for VDR in mice evidenced worsened renal injury Paricalcitol activates VDR and evidenced lower AKI stage after cisplatin treatment | [79] | |
| XJB-5-131 | Inhibits ferroptosis | Decreases AKI stage, attenuated inflammation and promoted tubular epithelial cell proliferation | [80] | |
| Tocilizumab | IL-6 and ferroptosis inhibition | Alleviates renal injuries in obstructive nephropathy | [81] | |
| Legumain | Downregulate GPX4—key control of ferroptosis | Alleviates AKI stage in rats | [82] | |
| miR-387a-3p knockout | Ferroptosis transcription factor | Downregulates IRI on mouse models | [83] | |
| miR-182-5p knockout | Ferroptosis transcription factor | Downregulates IRI on mouse models | [83] | |
| Necrostatin-1 | RIPK1 inhibition | Ameliorates IRI via HIF-1α/mir-26a/TRPC6/PARP1 inhibition | [84] | |
| hsa-miR-500a-3P knockout (mRNAs for MLKL) | SMAD3 downstream signaling | Alleviates kidney injury by necroptosis inhibition | [67] | |
| RIPK upregulation | SMAD3 downstream signaling | Promotes necroptosis | [69] | |
| Hypoxia pathways | HIF-1/2α stimulation | miR-21 upregulation and VEGF upregulation | Renoprotective due to increased angiogenesis | [85] |
| miR-493 overexpression | STMN-1 inhibition | Is stimulated by hypoxia and induces renal fibrosis through G2/M cell cycle arrest | [86] | |
| TRC160334 | PHD inhibitor | Induce HIF-1α upregulation with reduced IRI | [87] | |
| PINK1/PARK2 | Upregulated mitophagy | Inhibits Drp1, renal inflammation and tubular epithelial cell apoptosis | [88,89] | |
| Drp1 knockout | Mitochondrial fragmentation | Ameliorates renal inflammation, renal injury and renal fibrosis | [90] | |
| Mst1 knockout | Upregulates mitophagy | Renoprotective via AMPK and OPA1 downregulation | [91] | |
| BNIP3 knockout | Downregulates mitophagy | Increased ROS, damaged mitochondria and inflammatory renal response | [92] | |
| miR-668 | HIF-1α downstream signaling | Renoprotective due to reduced apoptosis and mitochondrial fragmentation | [93] | |
| VEGF inhibition | Induces thrombotic microangiopathy | [94,95] | ||
| VEGF upregulation | Induces collapsing glomerulopathies | [96] | ||
| VEGF upregulation | Increases EMT via upregulation of matrix metalloproteinases | [97] | ||
| SAR131675 | VEGF receptor—tyrosine kinase inhibition | Reduces apoptosis, lymphangiogenesis, inflammation and renal fibrosis | [98] | |
| G2/M cell cycle arrest | GC1 inhibition | TASCC inhibition | Ameliorates renal fibrosis | [99] |
| MYD88 knockout | TLR/IL-1R downstream signaling and NF-κB upstream signaling | Ameliorates renal fibrosis | [100] | |
| Immunological pathways | NOX-D21 | C5a/C5aR inhibition | Attenuates tubulointerstitial fibrosis in diabetic nephropathy | [101] |
| Orai1 knockout | Th17 inhibition | Alleviates renal fibrosis | [102] | |
| Flt3 inhibitor | DCs downstream signaling | Reduces proinflammatory cytokines and chemokines, TNF-α, IL-6 and IL-1β | [103] | |
| IL-10 | Cd4+ T cell activation | Reduces renal fibrosis in diabetic nephropathy | [104] | |
| IL-10 co-vaccination | Reduces tubular damage in SLE nephropathy on mouse models | [105] | ||
| Double negative T cells upregulation | PTECs-reduced apoptosis | [106] | ||
| TLR4 depletion | Diminishes renal damage after Cisplatin induced AKI | [107] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niculae, A.; Gherghina, M.-E.; Peride, I.; Tiglis, M.; Nechita, A.-M.; Checherita, I.A. Pathway from Acute Kidney Injury to Chronic Kidney Disease: Molecules Involved in Renal Fibrosis. Int. J. Mol. Sci. 2023, 24, 14019. https://doi.org/10.3390/ijms241814019
Niculae A, Gherghina M-E, Peride I, Tiglis M, Nechita A-M, Checherita IA. Pathway from Acute Kidney Injury to Chronic Kidney Disease: Molecules Involved in Renal Fibrosis. International Journal of Molecular Sciences. 2023; 24(18):14019. https://doi.org/10.3390/ijms241814019
Chicago/Turabian StyleNiculae, Andrei, Mihai-Emil Gherghina, Ileana Peride, Mirela Tiglis, Ana-Maria Nechita, and Ionel Alexandru Checherita. 2023. "Pathway from Acute Kidney Injury to Chronic Kidney Disease: Molecules Involved in Renal Fibrosis" International Journal of Molecular Sciences 24, no. 18: 14019. https://doi.org/10.3390/ijms241814019