
Integrated Transcriptome and Proteome Analysis Revealed the Regulatory Mechanism of Hypocotyl Elongation in Pakchoi
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Phenotypic Characterization of lhy7.1Mutant
2.2. Different Light Quality Affects Hypocotyl Growth
2.3. Transcriptome Analysis Results
2.3.1. Summary of Transcriptome Sequencing
2.3.2. GO Enrichment Analysis of DEGs
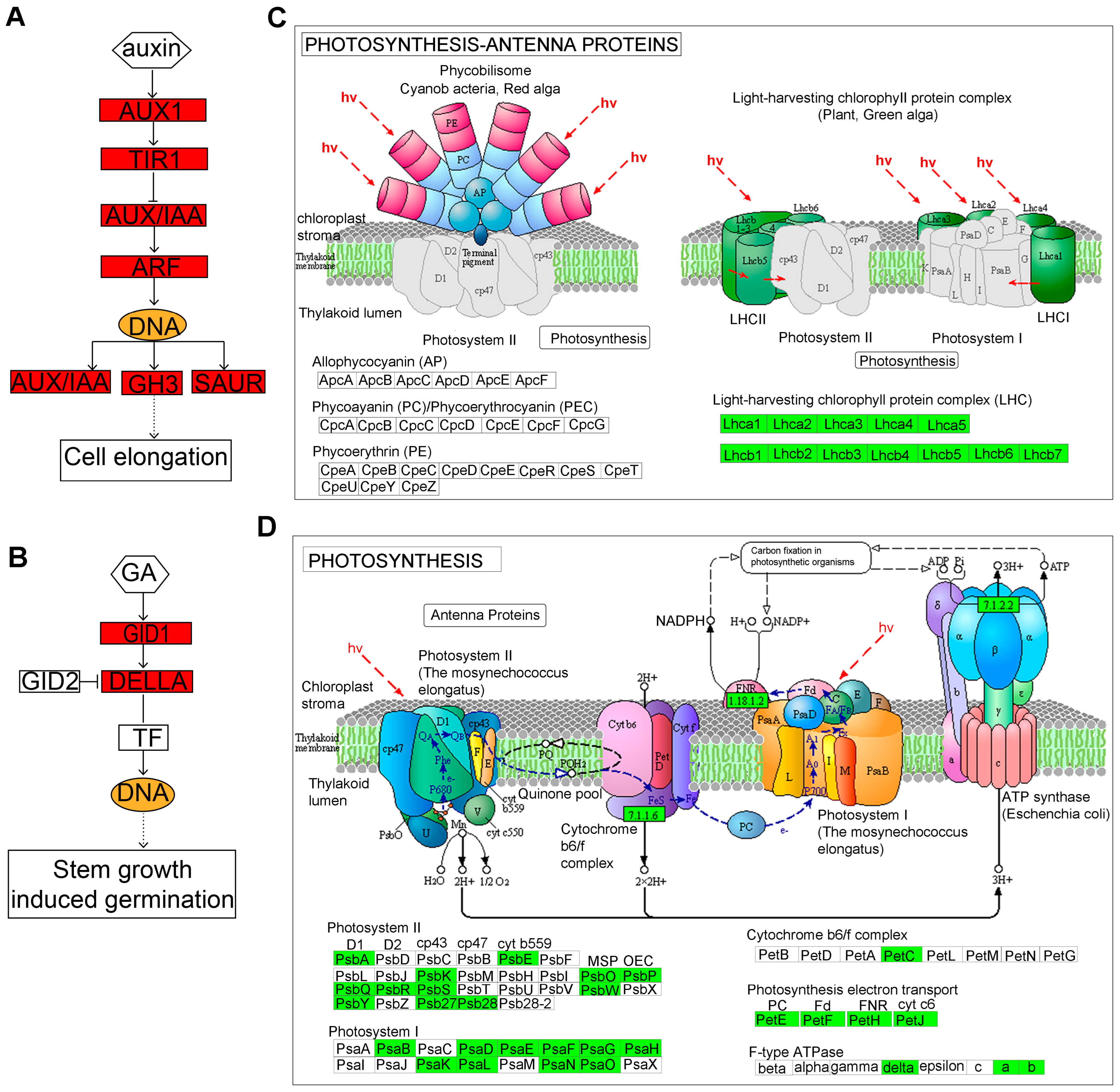
2.3.3. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis
2.3.4. Identification of Key Regulatory Genes Involved in Hypocotyl Elongation
2.3.5. qRT-PCR Validation
2.4. Proteome Analysis Results
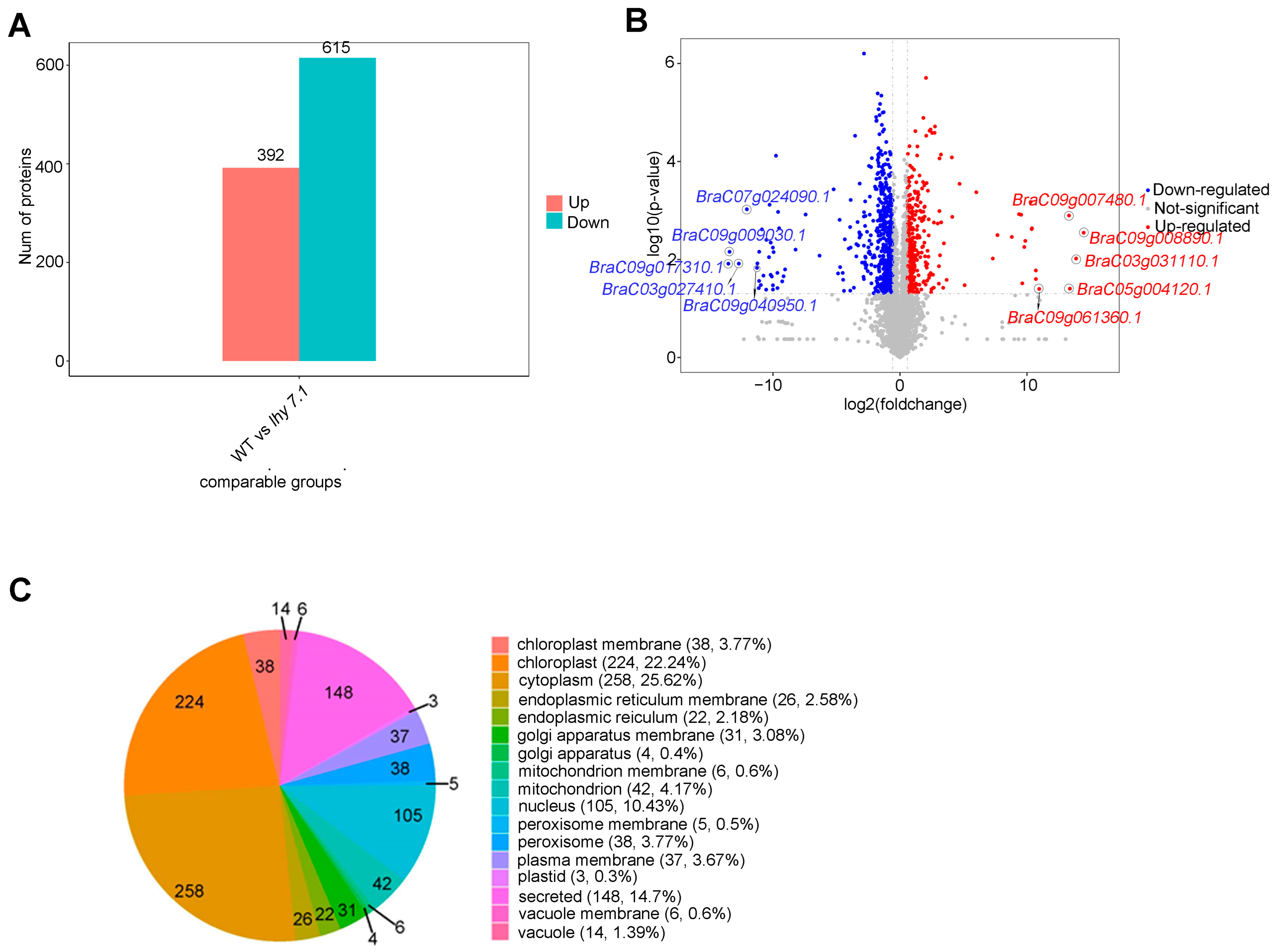
2.4.1. Differentially Expressed Protein (DEP) Analysis and Subcellular Localization
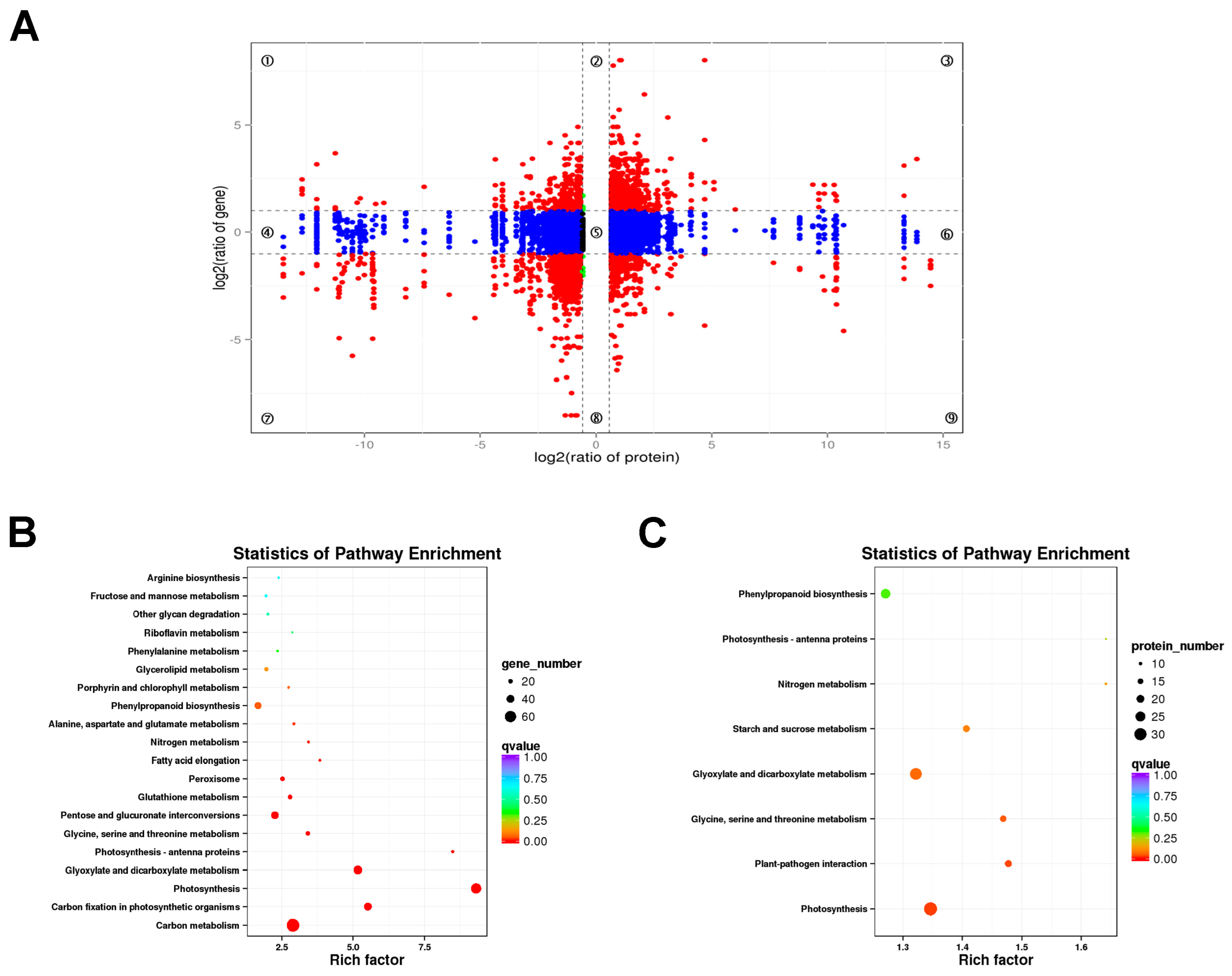
2.4.2. GO and KEGG Enrichment Functional Analyses of DEPs
2.5. Association Analysis of the Transcriptome and Proteome Results
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growing Conditions
4.2. Microscopic Observation of the Hypocotyl Cells
4.3. RNA Extraction, cDNA Library Construction and Sequencing
4.4. Proteome Analysis
4.5. Quantitative Real Time PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, R. The Brassica rapa Genome: Economic/Academic Importance of Brassica rapa; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–15. [Google Scholar]
- Li, Y.; Liu, G.; Ma, L.; Liu, T.; Zhang, C.; Xiao, D.; Zheng, H.; Chen, F.; Hou, X. A chromosome-level reference genome of non-heading Chinese cabbage [Brassica campestris (syn. Brassica rapa) ssp. chinensis]. Hortic. Res. 2020, 7, 212. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Su, T.; Zhao, X.; Wang, W.; Zhang, D.; Yu, Y.; Bayer, P.E.; Edwards, D.; Yu, S.; Zhang, F. Assembly of the non-heading pak choi genome and comparison with the genomes of heading Chinese cabbage and the oilseed yellow sarson. Plant Biotechnol. J. 2021, 19, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Xi, D.; Li, X.; Zhang, C.; Gao, L.; Li, Y.; Zhu, Y.; Zhu, H. Genetic diversity analysis of non-heading Chinese cabbage of resistance to clubroot disease based on SNP molecular markers. Agronomy 2023, 13, 245. [Google Scholar] [CrossRef]
- Zhang, K.; Pan, J.; Chen, Y.; Wei, Y.; Du, H.; Sun, J.; Lv, D.; Wen, H.; He, H.; Wang, G.; et al. Mapping and identification of CsSh5.1, a gene encoding a xyloglucan galactosyltransferase required for hypocotyl elongation in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2021, 134, 979–991. [Google Scholar] [CrossRef]
- Bo, K.; Wang, H.; Pan, Y.; Behera, T.K.; Pandey, S.; Wen, C.; Wang, Y.; Simon, P.W.; Li, Y.; Chen, J.; et al. SHORT HYPOCOTYL 1 encodes a SMARCA3-like chromatin remodeling factor regulating elongation. Plant Physiol. 2016, 172, 1273–1292. [Google Scholar] [CrossRef]
- Jiao, Y.; Lau, O.S.; Deng, X.W. Light-regulated transcriptional networks in higher plants. Nat. Rev. Genet. 2007, 8, 217–230. [Google Scholar] [CrossRef]
- Tavridou, E.; Pireyre, M.; Ulm, R. Degradation of the transcription factors PIF4 and PIF5 under UV-B promotes UVR8-mediated inhibition of hypocotyl growth in Arabidopsis. Plant J. 2019, 101, 507–517. [Google Scholar] [CrossRef]
- Kahle, N.; Sheerin, D.J.; Fischbach, P.; Koch, L.A.; Schwenk, P.; Lambert, D.; Rodriguez, R.; Kerner, K.; Hoecker, U.; Zurbriggen, M.D.; et al. COLD REGULATED 27 and 28 are targets of CONSTITUTIVELY PHOTOMORPHOGENIC 1 and negatively affect phytochrome B signalling. Plant J. 2020, 104, 1038–1053. [Google Scholar] [CrossRef]
- Oh, J.; Park, E.; Song, K.; Bae, G.; Choi, G. PHYTOCHROME INTERACTING FACTOR8 inhibits phytochrome A-mediated far-red light responses in Arabidopsis. Plant Cell 2020, 32, 186–205. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, L.; Chen, P.; Liang, T.; Li, X.; Liu, H. UV-B photoreceptor UVR8 interacts with MYB73/MYB77 to regulate auxin responses and lateral root development. EMBO J. 2020, 39, e101928. [Google Scholar] [CrossRef]
- Zhao, Y.; Shi, H.; Pan, Y.; Lyu, M.; Yang, Z.; Kou, X.; Deng, X.W.; Zhong, S. Sensory circuitry controls cytosolic calcium-mediated phytochrome B phototransduction. Cell 2023, 186, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Deng, X.W. Phytochrome signaling mechanism. Arab. Book 2011, 3, e71–e74. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; He, S.; Xu, F.; Wei, X.; Jiang, L.; Liu, Y.; Wang, W.; Li, T.; Xu, P.; Du, S.; et al. Photoexcited CRY1 and phyB interact directly with ARF6 and ARF8 to regulate their DNA-binding activity and auxin-induced hypocotyl elongation in Arabidopsis. New Phytol. 2020, 225, 848–865. [Google Scholar] [CrossRef]
- Xu, H.; Lantzouni, O.; Bruggink, T.; Benjamins, R.; Lanfermeijer, F.; Denby, K.; Schwechheimer, C.; Bassel, G.W. A molecular signal integration network underpinning Arabidopsis seed germination. Curr. Biol. 2020, 30, 3703–3712. [Google Scholar] [CrossRef]
- Vandenbussche, F.; Verbelen, J.; Van Der Straeten, D. Of light and length: Regulation of hypocotyl growth in Arabidopsis. Bioessays 2005, 27, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.W.; Wu, M.; Reeves, P.H.; Hodgens, C.; Yadav, V.; Hayes, S.; Pierik, R. Three auxin response factors promote hypocotyl elongation. Plant Physiol. 2018, 178, 864–875. [Google Scholar] [CrossRef]
- Smalle, J.; Haegman, M.; Kurepa, J.; Van Montagu, M.; Van Der Straeten, D. Ethylene can stimulate Arabidopsis hypocotyl elongation in the light. Proc. Natl. Acad. Sci. USA 1997, 94, 2756–2761. [Google Scholar] [CrossRef]
- Cary, A.J.C.U.; Liu, W.; Howell, S.H. Cytokinin action is coupled to ethylene in its effects on the inhibition of root and hypocotyl elongation in Arabidopsis thaliana seedlings. Plant Physiol. 1995, 107, 1075–1082. [Google Scholar] [CrossRef]
- Morillon, R.; Catterou, M.; Sangwan, R.S.; Sangwan, B.S.; Lassalles, J. Brassinolide may control aquaporin activities in Arabidopsis thaliana. Planta 2001, 212, 199–204. [Google Scholar] [CrossRef]
- Faurobert, M.; Mihr, C.; Bertin, N.; Pawlowski, T.; Negroni, L.; Sommerer, N.; Causse, M. Major proteome variations associated with cherry tomato pericarp development and ripening. Plant Physiol. 2007, 143, 1327–1346. [Google Scholar] [CrossRef]
- Lou, X.; Wang, H.; Ni, X.; Gao, Z.; Iqbal, S. Integrating proteomic and transcriptomic analyses of loquat (Eriobotrya japonica Lindl.) in response to cold stress. Gene 2018, 30, 57–65. [Google Scholar] [CrossRef]
- Wu, M.; Zou, Y.; Yu, Y.; Chen, B.; Zheng, Q.; Ye, Z.; Wei, T.; Ye, S.; Guo, L.; Lin, J. Comparative transcriptome and proteome provide new insights into the regulatory mechanisms of the postharvest deterioration of Pleurotus tuoliensis fruitbodies during storage. Food Res. Int. 2021, 147, 110540. [Google Scholar] [CrossRef]
- Li, L.; Zhang, H.; Chai, X.; Wei, S.; Luo, S.; Wang, H.; Lv, J.; Yu, J.; Liu, Z. Transcriptome and proteome conjoint analysis revealed that exogenous sulfur regulates glucosinolate synthesis in cabbage. Plants 2021, 10, 2104. [Google Scholar] [CrossRef]
- Lai, Y.; Zhang, D.; Wang, J.; Wang, J.; Ren, P.; Yao, L.; Si, E.; Kong, Y.; Wang, H. Integrative transcriptomic and proteomic analyses of molecular mechanism responding to salt stress during seed germination in hulless barley. Int. J. Mol. Sci. 2020, 21, 359. [Google Scholar] [CrossRef]
- Powell, A.E.; Lenhard, M. Control of organ size in plants. Curr. Biol. 2012, 22, R360–R367. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Yin, S.; Sun, J.; Linghu, Y.; Ma, J.; Pan, J.; Wang, C. Clathrin light chains regulate hypocotyl elongation by affecting the polarization of the auxin transporter PIN3 in Arabidopsis. J. Integr. Plant Biol. 2021, 63, 1922–1936. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liu, P.; Jin, Z.; Sun, J.; Weng, Y.; Chen, P.; Du, S.; Wei, A.; Li, Y. A mutation in CsHY2 encoding a phytochromobilin (PΦB) synthase leads to an elongated hypocotyl 1(elh1) phenotype in cucumber (Cucumis sativus L.). Theor. Appl. Genet. 2021, 134, 2639–2652. [Google Scholar] [CrossRef] [PubMed]
- Cendreau, E.; Traas, J.; Demos, T.; Crandjean, O.; Caboche, M.; Hofte, H. Cellular basis of hypocotyl growth in Arabidopsis thaliana. Plant Physiol. 1997, 114, 295–305. [Google Scholar] [CrossRef]
- Kutschera, U.; Niklas, K.J. The epidermal-growth-control theory of stem elongation: An old and a new perspective. J. Plant Physiol. 2007, 164, 1395–1409. [Google Scholar] [CrossRef]
- Song, X.; Liu, G.; Huang, Z.; Duan, W.; Tan, H.; Li, Y.; Hou, X. Temperature expression patterns of genes and their coexpression with LncRNAs revealed by RNA-Seq in non-heading Chinese cabbage. BMC Genom. 2016, 17, 297. [Google Scholar] [CrossRef]
- Song, L.; Li, X.; Zu, F.; Gao, C.; Wang, B.; Lin, C.; Tu, J.; Wang, A.; Zhou, G. Comparative transcript profiling and cytological observation of the newly bred recessive genic male sterility non-heading Chinese cabbage (Brassica rapa ssp. chinensis) line WS24-3A. Genes Genom. 2019, 41, 1475–1492. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Xiao, D.; Yin, Y.; Wang, H.; Wang, J.; Liu, T.; Hou, X.; Li, Y. Comparative transcriptome analysis of purple and green non-heading Chinese cabbage and function analyses of BcTT8 gene. Genes 2022, 13, 988. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Feng, X. Transcriptome and proteome profiling revealed molecular mechanism of selenium responses in bread wheat (Triticum Aestivum L.). BMC Plant Biol. 2021, 21, 584. [Google Scholar] [CrossRef]
- Hagen, G. Auxin signal transduction. Essays Biochem. 2015, 58, 1–12. [Google Scholar] [CrossRef]
- Kong, Y.; Wang, G.; Tang, H.; Yang, J.; Yang, Y.; Wang, J.; Li, G.; Li, Y.; Yuan, J. Multi-omics analysis provides insight into the phytotoxicity of chicken manure and cornstalk on seed germination. Sci. Total Environ. 2023, 861, 160611. [Google Scholar] [CrossRef]
- Li, L.; Hou, X.; Tsuge, T.; Ding, M.; Aoyama, T.; Oka, A.; Gu, H.; Zhao, Y.; Qu, L. The possible action mechanisms of indole-3-acetic acid methyl ester in Arabidopsis. Plant Cell Rep. 2008, 27, 575–584. [Google Scholar] [CrossRef]
- Xi, Y.; Yang, Y.; Yang, J.; Zhang, X.; Pan, Y.; Guo, H. IAA3-mediated repression of PIF proteins coordinates light and auxin signaling in Arabidopsis. PLoS Genet. 2021, 17, e1009384. [Google Scholar] [CrossRef]
- Takase, T.Y.C.U.; Nakazawa, M.; Ishikawa, A.; Manabe, K.; Matsui, M. DFL2, a new member of the Arabidopsis GH3 gene family, is involved in red light-specific hypocotyl elongation. Plant Cell Physiol. 2003, 44, 1071–1080. [Google Scholar] [CrossRef]
- Takase, T.; Nakazawa, M.; Ishikawa, A.; Kawashima, M.; Ichikawa, T.; Takahashi, N.; Shimada, H.; Manabe, K.; Matsui, M. ydk1-D, an auxin-responsive GH3 mutant that is involved in hypocotyl and root elongation. Plant J. 2004, 37, 471–483. [Google Scholar] [CrossRef]
- Sun, T. The molecular mechanism and evolution of the GA–GID1–DELLA signaling module in plants. Curr. Biol. 2011, 21, R338–R345. [Google Scholar] [CrossRef]
- Das, D.; St Onge, K.R.; Voesenek, L.A.C.J.; Pierik, R.; Sasidharan, R. Ethylene- and shade-induced hypocotyl elongation share transcriptome patterns and functional regulators. Plant Physiol. 2016, 172, 725–2016. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, Y.B.A.Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Lu, C.; Hwang, J. Prediction of protein subcellular localization. Proteins Struct. Funct. Bioinform. 2006, 64, 643–651. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Total Clean Reads | Clean Reads | Mapped Reads | Unique Mapped Reads (%) | GC Content (%) | Q30 (%) |
|---|---|---|---|---|---|---|
| WT-1 | 44,040,902 | 22,020,451 | 39,643,566 (90.02%) | 38,125,106 (86.57%) | 46.99 | 94.68 |
| WT-2 | 38,548,448 | 19,274,224 | 33,831,669 (87.76%) | 32,386,509 (84.02%) | 47.53 | 93.81 |
| WT-3 | 47,041,512 | 23,520,756 | 41,826,754 (88.91%) | 40,171,957 (85.40%) | 47.24 | 94.16 |
| lhy7.1-1 | 40,919,018 | 20,459,509 | 37,127,273 (90.73%) | 35,753,814 (87.38%) | 47.65 | 93.80 |
| lhy7.1-2 | 41,214,806 | 20,607,403 | 36,540,515 (88.66%) | 35,216,896 (85.45%) | 47.63 | 94.04 |
| lhy7.1-3 | 42,684,144 | 21,342,072 | 38,598,224 (90.43%) | 37,184,460 (87.12%) | 47.73 | 93.89 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Xi, D.; Gao, L.; Zhu, H.; Yang, X.; Song, X.; Zhang, C.; Miao, L.; Zhang, D.; Zhang, Z.; et al. Integrated Transcriptome and Proteome Analysis Revealed the Regulatory Mechanism of Hypocotyl Elongation in Pakchoi. Int. J. Mol. Sci. 2023, 24, 13808. https://doi.org/10.3390/ijms241813808
Li X, Xi D, Gao L, Zhu H, Yang X, Song X, Zhang C, Miao L, Zhang D, Zhang Z, et al. Integrated Transcriptome and Proteome Analysis Revealed the Regulatory Mechanism of Hypocotyl Elongation in Pakchoi. International Journal of Molecular Sciences. 2023; 24(18):13808. https://doi.org/10.3390/ijms241813808
Chicago/Turabian StyleLi, Xiaofeng, Dandan Xi, Lu Gao, Hongfang Zhu, Xiuke Yang, Xiaoming Song, Changwei Zhang, Liming Miao, Dingyu Zhang, Zhaohui Zhang, and et al. 2023. "Integrated Transcriptome and Proteome Analysis Revealed the Regulatory Mechanism of Hypocotyl Elongation in Pakchoi" International Journal of Molecular Sciences 24, no. 18: 13808. https://doi.org/10.3390/ijms241813808