1. Introduction
The latest figures released by the International Diabetes Federation in 2021 show that 537 million (1 in 10) adults now live with diabetes worldwide; a rise of 16% (74 million) since the previous estimates in 2019 [
1]. Due to the microangiopathy caused by hyperglycemia, up to 30~50% of diabetic patients have diabetic kidney disease (DKD) [
2]. DKD has been recognized as the uppermost cause of end stage kidney disease, which brings tremendous health and property deficiency to patients and medical finance.
Mitochondria exhibit a semi-autonomous nature due to their double-membrane structure. They possess their own genome and function as the cellular energy-generating factories, providing over 90% of the cell’s ATP supply. Mitochondria can oxidize small molecular organic acids, fatty acids and ketones to produce energy, extensively participating in the regulation of cell metabolism, energy supply, cell cycle and other intracellular activities. Numerous studies have demonstrated that the mitochondrial disorder caused by metabolic changes of diabetes is one of the important causes of DKD [
3]. In DKD, mitochondrial dysfunction has a definite relation with DKD because of the reduced quantity, impaired oxidative function, kinetic imbalance and fuel transition [
4]. Furthermore, the oxygen contents vary across different regions of the kidney, leading to diverse metabolic characteristics of mitochondria in various types of renal cells. Consequently, the changes of mitochondria in distinct cell types at different stages of DKD may exhibit variations as well. Therefore, it is imperative to carry out a comprehensive analysis of mitochondria considering heterogeneity among different cell types in kidney of DKD at single-cell resolution.
Bulk RNA-Seq, known for its high-throughput capabilities, has been a powerful technology for screening biomarkers or therapeutic targets for diverse tissues and organs [
5]. Zhao et al. utilized this technology to investigate the biomarkers of DKD in db/db mice and found the mitochondrial dysfunction, abnormal lipid metabolism and oxidative stress [
6]. Bulk RNA-Seq, however, has a limitation owning to the internal heterogeneity within tissues and organs. In recent years, single-cell RNA sequencing (scRNA-seq) has been widely used in the research of diseases such as hepatology [
7], cardiovascular disease [
8], and neurodegenerative disease [
9]. Compared to bulk RNA-Seq, scRNA-Seq advantageously offers the ability to comprehensively consider the analysis of heterogeneity among the types of cells. Leveraging scRNA-Seq, we recently identified the cell-specific targets involved in the aetiology of DKD in db/db mice [
10].
In the current study, we performed an extensive analysis of the mitochondrial landscape in DKD based upon the data of specific cell types of scRNA renal transcription to reveal the regulatory network and its heterogeneity of renal mitochondrial damages in distinct renal cell types of DKD. There are universal transitions of material transportation, metabolism, cell cycle in the descending loop of Henle (DLH), collecting duct principal cells (CD-PC), endothelial cells (EnCs), B cells and macrophages (Mac). Especially notable is the fluctuation in the CD-PC and B cells, whose cell state can be reversed by angiotensin receptor blockers (ARB) and Huangkui capsule (HKC).
3. Discussion
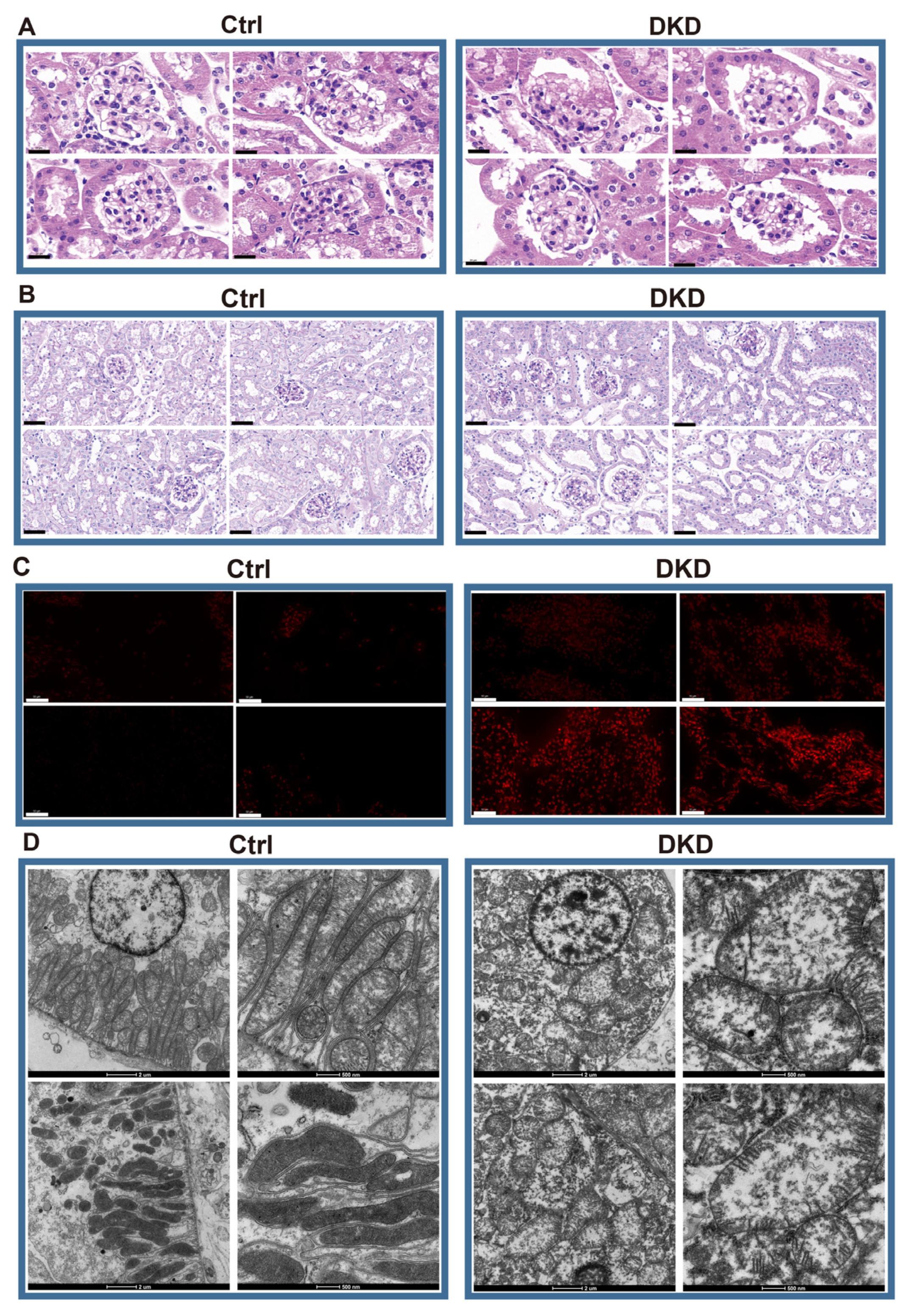
In the current study, we have uncovered that the mitochondrial contents in the kidney were not consistent within the cell types. Instead, the mitochondrial contents showed the distinct trends in different parts of the nephron and infiltrative IC, and the fold changes of mitochondrial content were significantly decreased in DSRC and IC of DKD than what in PTCs (
Figure 3D). Subsequently, DRSC demonstrated the most differences in mitochondrial transcriptome. Therefore, our study has provided evidence that the different cell types of kidney may have the different degrees of mitochondrial stress.
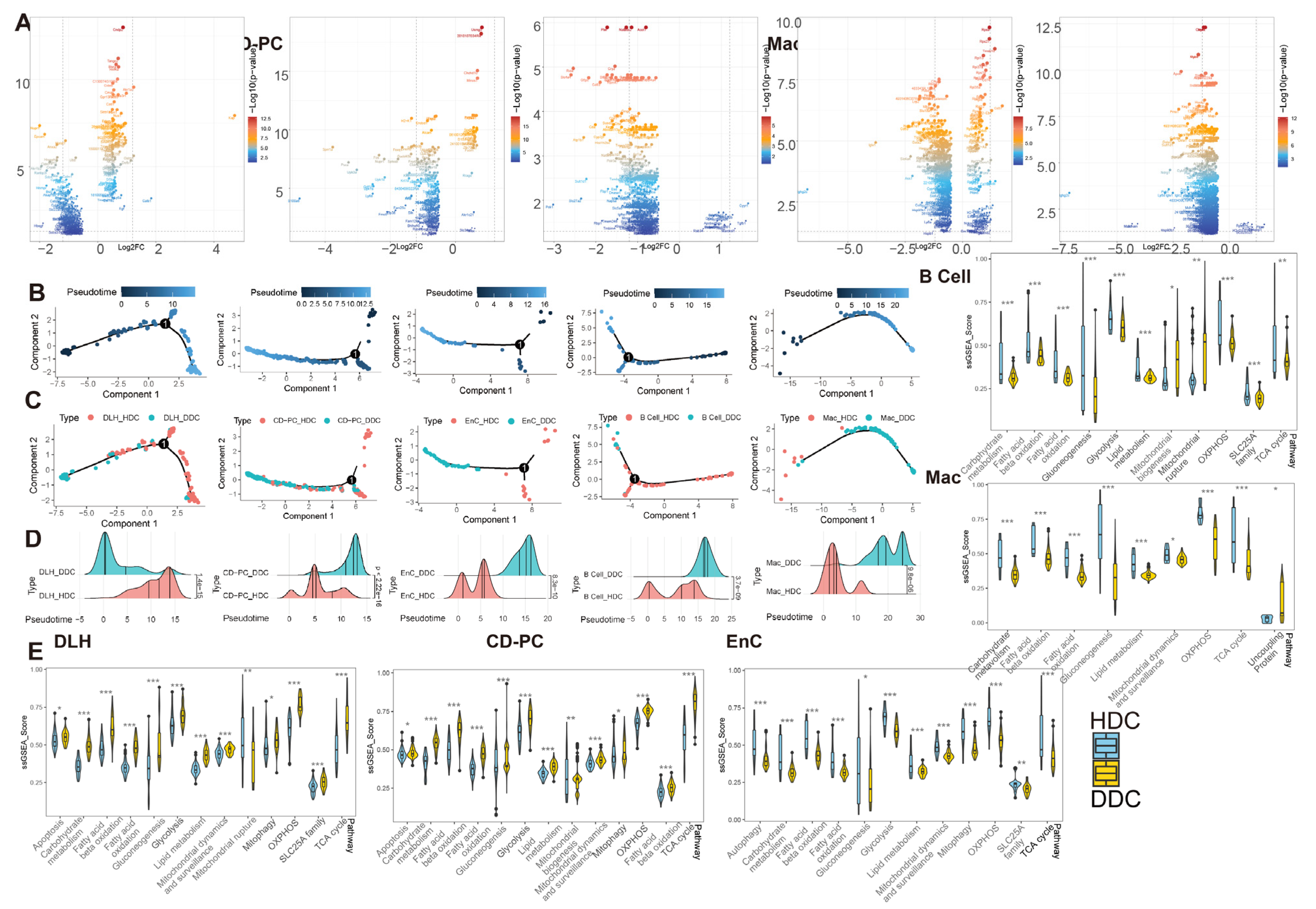
The heterogeneity among various renal cell types in DKD may not only be inherent but also pathological. To verify the occurrence of normal-like subpopulation among the renal cell of DKD, we have assuredly identified two cell subsets (HDC and DDC) in DLH, CD-PC, EnC, B cells and Mac by scoring 151 mitochondria-related pathways. The differential genes between DDCs and HDCs overlap with the differences between DKD and Ctrl groups to a certain extent, and these overlaps have an intensively positive correlation (
Figure 4C). Furthermore, the enrichment results showed that the overlap genes can be divided into DCP (
Figure 4F). Although MANGs were excluded when the intersective genes were enriched to DCP, we have demonstrated that several pathways, especially oxidation and metabolism pathways, may be indirectly related to mitochondrial function in current research. For instance, both ROS elimination and ROS GSEA pathways were observed in the five cell types, suggesting that the excessive production of ROS in DKD would lead to oxidative stress in the microenvironment to stimulate the expression of TGF-
β [
15], and further cause the mitochondrial morphological deterioration, apoptosis, extracellular matrix degradation, mesangial dilation, glomerular basement membrane thickening, renal tubule fibrosis and other characteristics (
Figure 1). Indeed, we have detected the emergence of apoptosis, TGF-
β, NF
κB, epithelial-mesenchymal transition (EMT) and other related pathways (
Figure 4F). In addition, the metabolic pathways related to amino acids, glucose, fatty acids and their derivatives were also enriched. As we have previously reported, there are indeed metabolite differences in the peripheral blood of db/db mice with DKD [
20]. These disorders in carbohydrates, amino acids and fats could be further observed in the kidneys, which is not just limited in the peripheral blood. Carbohydrates and lipids, as the principal energy sources, may have varying availabilities in different types and phases of cells. In healthy renal tubulointerstitium, ATP is produced by the oxidation of free fatty acids and ketones, except for glomerulus [
3]. Under the induction of diabetic high glucose environment; however, the damaged renal tubules may undergo metabolic transformation from oxidative phosphorylation of lipid metabolism to glycolysis, which will further lead to lipid accumulation. This will cause further oxidative impairment in renal stromal cells, and the accumulation of lipid in Mac will inhibit the autophagy of macrophages, thus inhibiting their ability to transform from M1 (immune activation) to M2 (immunosuppression), and ultimately leading to the increase of local inflammation activity [
21]. Based upon the ssGSEA score of mitochondria-related DKD pathways, we have demonstrated that the glucose and lipid metabolism-related pathways are regulated differently across distinct cell types. Metabolism carbohydrate, fatty acid
β oxidation, glycolysis, OXPHOS, and TCA cycle exhibited a consistent trend between HDCs and DDCs, and all of them were up-regulated in the DDCs of DLH and CD-PC, while down-regulated in the DDCs of EnC, B cells, and Mac. Thereby, DLH and CD-PC but not EnC, B cells, and Mac may attribute to the role of substance transport within the kidney. These findings prove that the metabolism consisted with renal cell function and both of them showed a heterogeneity trend in DKD progress.
DEGs in the members of SLC superfamilies may explain the metabolic disorder in kidney (
Figure S6). In the current study, we found that the expression levels of SLCs in mitochondria, including
Slc25a3,
Slc25a4,
Slc25a5 and
Slc25a30, were significantly up-regulated in the DDCs of CD-PC (
Figure S6C) and might lead to an increase in ROS and enable the latter to act as a signaling molecule to activate tubular cell apoptosis in DKD [
22]. The increase of ROS content and up-regulation of apoptosis could be found in both ROS staining (
Figure 1C) and ssGSEA score (
Figure 5E). In addition to mitochondrial SLCs, we found that professional carbohydrate and lipid SLCs were mostly up-regulated in the DDCs of DLH and CD-PC (
Figure S6C). The kidney has the highest resting metabolic rate in the body. The elevation of these SLCs is consistent with the observed elevation of ssGSEA score in the carbohydrate metabolism, gluconeogenesis, glycolysis, fatty acid oxidation, lipid metabolism, and fatty acid oxidation pathways in the DDCs of DLH and CD-PC (
Figure 5E). A previous study has demonstrated that, even though the renal system intakes a large amount of glucose in DKD, the proportion utilized for aerobic oxidation is reduced instead. This shift towards the overactivated glycolysis breaks the metabolic balance, leading to oxidative stress and subsequent kidney damage [
21]. Herein, we speculate that the oxidative stress damage of mitochondria may be caused by abnormal transport from the cytoplasm to mitochondria.
The potential of SLCs as targets in treatment of DKD has been concerned by researchers. Invokana (inhibitor of Slc5a2) has been used for blood glucose management to alleviate kidney injury in type 2 diabetes patients. Slc5a2, encoding the SGLT2, is localized in the early proximal tubule and response for 90% glucose reuptake, while high glucose conditions increase SGLT2 levels and enhance glucose recovery in the proximal tubule of diabetic patients. By inhibiting the activity of SGLT2, blood glucose levels can be reduced, inflammation, fibrosis, and damage to the glomerular interstitium improved, and proteinuria reduced by 30% to 50% [
23]. In the current study, we have observed that
Slc5a2 is upregulated in the DDCs of DLH compared to HDCs, while upregulation of
Slc2a4 in DDCs and CD-PC is also seen (
Figure S6).
Slc2a4 is another SLC member that can transport glucose, and the knockout of this gene in podocytes can prevent glomerular hypertrophy, mesangial dilatation, albuminuria and other symptoms in mice induced by DKD, thus reducing the risk of DKD [
24]. These members of the SLC superfamily could be potential treatment targets of DKD.
To reveal the characteristic genes associated with DDCs outside mitochondria, we have defined ESDEG with deleting MANGs. There are differences in RNA dynamics between DDCs and HDCs (
Figure 5A–D). A massive single nuclear RNA-Seq (snRNA-Seq) study has documented that CD-PC, DLH, and EnCs exhibit the highest count of differentially expressed gene in DKD progression [
25]. Furthermore, GO enrichment analysis of ESDEGs in these five cells revealed that they were belong to heterogeneous regulatory modes (
Figure 6A). ESDEGs of DLH were enriched to wound healing, regulation of supramolecular fiber organization, cell-substrate adhesion, regulation of actin filament-based process etc., suggesting that it may have undergone cyto-dynamics changes. CD-PC have enriched abundant gene sets related to epithelial cell development or differentiation. B cells and Mac exhibited metabolism-related or immune-related pathways similar to the MHC pathways. Cell communication and TF prediction allow us to understand how these intricate pathways regulate complex biological network by controlling downstream TF and TG with a very small number of receptors (
Figure 7). For instance,
Fzd7 codes a surface labeled molecule, and is expressed as a marker of progenitor cells or stem cells in mammalian renal mesenchymal [
17]. As a receptor of ncWNT pathway,
Fzd7 was found to be significantly decreased in DDCs of CD-PC. It controlled the cell polarity of CD-PC through a total of 25 transcription factors such as EGR1, EP300, ESR1, ETV1, etc. (
Figure 7B), resulting in the decreased developmental ability in the DDCs of CD-PC and showed a decreased proportion of G2M phase cells in the cell cycle. Consistent with this phenomenon, inhibition of Fzd7 tends to imply the decreased epithelialization and increased cell death and apoptosis [
26].
F11r encodes junction adhesion molecular and plays a key role in the adhesion of endothelial and epithelial cells and is relevant to angiogenesis, immune response, and maintenance of vascular permeability. This gene ontology and TF regulatory network showed a similar trend in HDCs and the DDCs of CD-PC (
Figure 7 and
Figure 8). In the lesions of chronic diseases, irreversible cell proliferation stagnation, increased protein production, decreased anti-apoptotic ability and metabolic ability disorder often occur. For instance, impairment of cell cycle and proliferation can be found in renal tubular epithelial cells in CKD, HIV-associated nephropathy and polycystic nephropathy [
27]. Evidence has demonstrated that hyperglycemia and lipotoxicity lead to tubular senescence in DKD in relation to cell death pathways such as apoptosis, autophagy, necrosis and ferroptosis [
28]. In the current study, data have showed that in the DDCs of CD-PC, there was a state of Epi development and cell cycle accompanied by the decreased expression of
Fzd, which not only corresponded to the impaired normal function of CD-PC, but also duplicated the impaired metal ion transport ability of CD-PC in the process of DKD [
10]. What excites us is the proportion shift of DDCs and HDCs in CD-PC can not only be testified between Bulk RNA-Seq deconvolution of DM and DKD (
Figure 8G), but also be reversed by treating with ARB and HKC. However, since SGLT2i acts on S1 PTCs, we did not find that it has a reverse effect on the composition of DDCs and HDCs in CD-PC (
Figure 8D).
The metabolic and cell cycle changes described above may further cause fibrosis and inflammation of renal tubules. The previous study has demonstrated that M1-type Mac play a leading role in DKD, and T cells may have an extensive synergistic effect with Mac in DKD inflammation [
10]. In the current study, we have alternately provided evidence suggesting that B cells, except T cells, may also be driven by mitochondria and participate in the process of DKD with
Cd74 in the APP pathway (
Figure 7A,B) and its interaction with other renal cells through MHC-II, TGF-
β, in the SELL pathway (
Table 2 and
Figure S7).
Cd74 encodes a class of MHC-Ⅱ transmembrane glycoprotein molecules and plays a certain role in the regulation of macrophage migration and T and B cell development.
Cd74 receptor activity was first discovered in B cells, in which, this gene regulates cell cycle and immune activation of B cells [
29]. As an additional survival receptor for B cells, the activation of Cd74 can induce B cells to enter the S-phase to improve their anti-apoptotic ability and thus enhance their survival ability, which leads to the formation of B cell libraries and the activation of immune response [
19]. Therefore, the activation of Cd74 is associated with plentiful inflammatory diseases, such as fibrosis, T1D and systemic lupus erythematosus [
30]. In the kidney, the absence of this gene is usually a significant obstacle to glomerular injury, nephritis and renal tubule fibrosis [
30]. Intriguingly, the up-regulation of B cell DDCs caused by DKD can be reversed by SLGT2i, ARB and HKC.
There are limitations in the current study. The cells in glomerulus, including podocytes and mesangial cell cannot be captured by scRNA-Seq, but these cells are important in renal injury and cause of proteinuria in DKD. This is due to a combination of the peculiarity of 10X Genomics and the small number of glomerular cells in total renal cell number. Another disadvantage of scRNA-seq is that the spatial coordinate information is lost during the process of tissue dissociation into single-cells suspension. Further investigation of the transcriptome map in DKD with spatial information has been taken into our consideration.
In conclusion, the current study has provided experimental evidence revealing the heterogeneity of mitochondrial disorders in distinct cell types, particularly in the CD-PC and B cells during the process of DKD and administration of ARB, SGLT2i and HKC. The information is useful for the discovery of new biomarkers in DKD and the proposal of diagnosis and treatment schemes for the disease.
4. Methods and Materials
4.1. Animal Management
BKS.Cg-Dock7m
+/+ Lepr
db/J (db/db) mice (DKD group) and heterozygous db/m mice (Ctrl group) aged 10 weeks were acquired from Huachang Xinnuo Medical Technology Co., LTD (Taizhou, China). All experiments with mice were carried out in accordance with the guidelines of, and were approved by, the Institutional Animal Care and Use Committee at China Pharmaceutical University (CPU). All male mice were housed in cages with a barrier environment (22–25 °C; 40–50% humidity; and 12-h light/dark cycle) and received regular chow and water at liberty. At 16 weeks, UACR and BG were measured as we described before [
10]. Based on blood glucose and UACR results, mice in the DM (BG > 16.7 mmol/L) and DKD (UACR > 30 ng/μg) groups were identified and four left kidneys of each group were used for bulk RNA-Seq. At 20 weeks, the mice were sacrificed by cervical dislocation method and the peripheral blood was removed by cardiac perfusion with 1x phosphate buffered solution and the kidneys of both sides were collected. The left kidney was placed in a frozen tube and stored in liquid nitrogen, and the right kidney was uniformly cut lengthwise and placed in 4% paraformaldehyde solution and 4% glutaraldehyde solution, respectively.
4.2. Reactive Oxygen Species, Hematoxylin-Eosin, and Periodic Acid-Schiff Stain
After the kidney was removed from the cryostorage tube, the frozen section was placed in the Cryotome E freezing microtome (Thermo, Shanghai China). The fluorescence staining and sealing were then performed in accordance with the specification of the D7008 ROS kit (Servicebio, Wuhan, China). Images were collected by Fluorescent Microscopy (NIKON ECLIPSE C1, NIKON). DAPI glows blue at a UV excitation wavelength of 330–380 nm and emission wavelength of 420 nm; FITC glows green at an excitation wavelength of 465–495 nm and emission wavelength of 515–555 nm; CY3 glows red at an excitation wavelength 510–560 nm and emission wavelength of 590 nm. In the Hematoxylin-Eosin (HE)-staining process, half of the right kidneys were embedded in paraffin wax, and then dehydration, staining, and elution were performed in accordance with the specification of the HE-staining kit (Hunan Aifang Biotechnology CO., Ltd., Changsha, China). Finally, the slices were sealed with neutral gum and the images were collected by AE41 optical microscope (Motic, Shenyang, China). In the PAS-staining procedure, the section and dewatering process after paraffin embedding was consistent with the HE procedure. The actual use of the above PAS process was included in the G1008 PAS dye kit (Hunan Aifang Biotechnology CO., Ltd., Changsha, China).
4.3. Transmission Electron Microscopy
The upper part of the kidney was taken from the glutaraldehyde and rinsed with 0.1 M phosphoric acid buffer 3 times for 15 min each time. The sample was infiltrated with 1% osmic acid solution configured with 0.1 M phosphoric acid buffer to avoid light for 7 h, and then the phosphoric acid rinsing operation was repeated. Alcohol gradient dehydration for 1 h each: 30%, 50%, 70%, 80%, 95%, 100%, 100%. Different proportions of the mixture of anhydrous ethanol and acetone were used for dehydration: 3:1, 0.5 h; 1:1, 0.5 h; 1:3, 1.5 h; 0:1,1 h. The samples were treated at 37 °C for 12 h with 812 embedding agent and then treated at 60 °C for 48 h to complete polymerization. The ultrathin sectioning mechanism was used to make sections ranging from 60 to 80 nm. After the dyeing step, the images were collected: 2% uranium acetate saturated alcohol solution away from light, 8 min; 70% alcohol rinse, 3 times; ultra-pure water rinse, 3 times; 2.6% lead citrate solution away from carbon dioxide, 8 min; ultra-pure water rinse, 3 times; the filter paper absorbs excess water and dries at room temperature for 12 h.
4.4. ScRNA-Seq Data Preprocessing, Dimension Reduction, Clustering, Cell Annotation and Quality Control
Renal scRNA-seq data of 4 Ctrl mice were adopted from the National Center for Biotechnology Information (NCBI) GEO database (GSE107585) and scRNA-Seq data of DKD kidneys were previously registered in Sequence Read Archive (PRJNA749372). The steps of quantitative processing, quality control, filtering, clustering, and annotation after obtaining FASTQ files of scRNA-Seq were described in our previous publications [
10]. Seurat container with cell annotation information was generated, and a total of 58,259 individual cells were obtained. After the cell annotation was completed, the DoubletFinder (Version 2.0.3) package was used to classify the doublets and singlets by iterating the optimal parameter of proportion of artificial nearest neighbors (pANN) based on principal component (PC) domain size as 1 to 30. We used the find.pK() function of DoubleFinder package to determine the optimal pANN value of proximal tubular cells (PTC) and rest cell types as 0.1 and 0.18 respectively, and relied on the pANN value to identify double or multiple droplets (doublets) and single droplets (singlets) under Poisson distribution.
As described in the methods above, the uniform manifold approximation and projection (UMAP) of single-cell droplet identification of PTC and other cell types are shown in
Figure S1A,B, respectively. The proportion of doublets and singlets within all PTCs and the remaining cell types are visualized in
Figure S1C,D and the count data are shown in
Supplemental Table S1. We noticed that PTCs contained abundant doublets (21.7%, 9804/45,205). Among the PTC types, S2 (2.86%, 696/24,347) had the least proportion of doublets and S3 (50.8%, 6287/12,385) had the greatest proportion of doublets (
Figure S1C). The Double-finder was less sensitive when used for unsupervised cell clusters with similar expression patterns [
31]. We thus realized that the false positive heterotypic doublets in S1 and S3 might be incorrectly identified. Among the DRSC, we found that Mac (62.7%, 260/415) had the highest percentage of doublets, while the ALH (0.0341%, 1/2932) had the lowest proportion of doublets (
Figure S1D). Considering these factors, we refused to discard doublets misidentified in PTCs but did not reserve doublets in other cells for downstream advanced analysis. Consequently, we used the Subset function in the Seurat (Version 4.0.3) package to take out all cells in PTC and singlets of other cells for subsequent analysis. The RunUMAP function in Seurat was re-executed and UMAP score were visualized by adding confidence intervals via ggplot2 (Version 3.3.5).
4.5. Gene Set Scoring and Enrichment Analysis
The calculation of mitochondrial coding gene set content was performed by the PercentageFeatureSet() function in Seurat. After the percentage was obtained, the wilcox.test() in R platform was used to calculate the difference. The normalized expression quantity of each cell was used as GSVA (Version 1.44.4) input matrix and Kernel estimation of the cumulative density function (kcdf) was calculated according to Gaussian distribution to obtain GSVA and single sample ssGSEA scores. The inter-group difference analysis of GSVA and ssGSEA scores was implemented by the limma (Version 3.48.3) package in R based on empirical linear models of Bayesian methods. The p value of both differential analysis is corrected by false discovery rate (FDR) to get the adjusted p value. Then, GSVA significant pathways (p adjustment < 0.05 and |Log2FC| > 0.5) were selected. Linear regressions between DEGs were calculated by lm() function based on the qr method and visualized by ggplot2.
In the current study, DCP, GO and KEGG enrichment analyses were implemented in ClusterProfiler (Version 4.4.4) [
32] through the principle of hypergeometric distribution. In DCP enrichment analysis, among 518 intersection genes belonging to DLH, 297 were enriched in 33 DCP (9 pathways met the significant enrichment condition of
p < 0.05), such as the Glutathione metabolism, Taurine and hypotaurine metabolism, ROS elimination, Biosynthesis of unsaturated fatty acids, Alanine aspartate and glutamate metabolism and so on. While among 324 exclusive genes of DA2 in DLH, 140 of them were enriched in 27 DCP and only one pathway, named Collagen formation, was significantly enriched. Among 664 intersection genes belonging to CD-PC, 112 were enriched in 28 DCP (2 of them met the significant condition), such as Glutathione metabolism, ROS elimination, EMT, Glycolysis Gluconeogenesis and Apoptosis. While among 218 exclusive genes of DA2 in CD-PC, 80 of them were enriched in 16 DCP and only TGF-
β1 pathway was significantly enriched. Among 115 intersection genes belonging to EnCs, 26 were enriched in 13 DCP (ROS-GSEA and Glutathione metabolism met the significant condition). Among 510 exclusive genes of DA2 in EnCs, 145 of them were enriched in 29 DCP; however, only the Glycerolipid metabolism and Glutathione metabolism pathways were significantly enriched. Considering B cells, 76 of 297 intersection genes were enriched in 19 DCP (ROS GSEA and TGF-β pathways were significantly enriched), while 245 of 563 exclusive genes of DA2 were enriched in 25 DCP (Glutathione metabolism, Alanine, aspartate and glutamate metabolism, ROS elimination, Nitrogen metabolism and Arginine and proline metabolism pathways were enriched). Regarding Mac, 80 of 146 intersection genes were enriched in 20 DCP (Apoptosis and ROS GSEA pathways were significant). While 146 of 835 exclusive genes of DA2 were enriched in 39 DCP, however, only Glycine, serine and threonine metabolism pathway were significantly enriched (
Table S5).
In the pseudotime analysis, we first took out the UMI count in the Seurat object that conforms to the negative binomial distribution to create the CellDataSet object of monocle (Version 2.18.0). DDRTree dimensionality reduction determined the topological structure of data, and we then used exclusive significant difference genes (ESDEG) of corresponding cells to guide the semi-supervised trajectory analysis while the orderCells() function was used to determine the coordinates of the cells in pseudotime [
33]. Trajectory diagram and its color rendering was performed by the plot_cell_trajectory() function in monocle, while at the same time, the cumulative density distribution curve generated by ggridges (0.5.3) and
t test was executed by ggsignif (0.6.3).
4.6. Cell Communication Prediction
After the normalized matrix of the DKD group in the Seurat object container is obtained, the input RNA matrix was converted into a reliable protein network with STRINGdb support using a random propagation technique through projectData() function. Then, by referring to the ligand–receptor interactions database containing 229 signaling pathways including secreted signaling, cell–cell contact, ECM–Receptor three types in CellChat (Version 1.5.0), the obtained protein matrix was calculated by computeCommunProbPathway() function to obtain the intercellular interaction intensity to represent the possibility of manifold-leaning inferred intercellular communication network. In order to focus on the heterogeneity of cell subsets within DKD, five cell types including DLH, CD-PC, EnCs, B cells and Mac, were divided according to HDC and DDC, and the rest of the cells were not. Considering that some subsets contained only a tiny number of cells, we did not set the minimum number of cells involved in the pathway for filtration. Consequently, 46 pathways including 90 ligand receptor pairs verified by permutation test (p-value < 0.05) were preserved. The downstream visualization of cell communication was performed through netVisual_chord_cell (), netAnalysis_signalingRole_scatter (), netAnalysis_signalingRole_heatmap (), netVisual _bubble () and so on provided by CellChat.
4.7. Transcription Factor Prediction
The single-cell transcription factor regulatory network is mainly completed by SCENIC [
16] (Version 1.3.1) and its dependent GENIE3 [
34] (Version 1.18.0), RcisTarget (Version 1.3.1) and AUCell [
16] (Version 1.1.6) packages in R. To avoid artificial co-variation, summarized counts (UMI count) were used as input data of GENIE3 (R version) to calculate the co-expression between TF and potential target gene through random forest models. By using motif framework of iRegulon information recorded by i-cisTarget (mm9-500bp-upstream-7species.mc9nr.feather and mm9-tss-centered-10kb-7species.mc9nr.feather), TF-motif enrichment analysis was carried out on sequences of 10 kb around and 500 bp upstream from the transcriptional start site (TSS). In the results, a normalized enrichment score (NES) greater than 3.0 was regarded as a qualified regulon and preserved. The genetic matrix of regulon was scored by AUCell packages, a method for area under the curve (AUC) calculation. According to the correspondence between TF and TG in RcisTarget and hypotaxis of receptors and TF in scMLnet [
35], we constructed the regulatory network of receptor, TF and TG. The continuous AUCell score of regulon was analyzed for difference through the limma package, Then circlize [
36] (Version 0.4.15) and plotly (Version 4.10.0) were performed, respectively, to display top50 (if sufficient) significantly different regulon (
p value < 0.01) in annular heat map and sankey diagram.
4.8. Bulk RNA-Seq Was Deconvolved by Cibersort Using the scRNA-Seq Data
Fastq files of bulk RNA-Seq were trimmed and aligned to the reference index (mm10 Version). Then gene counts were obtained by featureCount (Version 2.0.1) to calculate the transcripts per million (TPM). After figuring out the top 10 marker genes of 10 sub cell cluster, deconvolution was implemented by Cibersort.
4.9. scRNA-Seq Labels Transformation
In addition to the Ctrl and DKD groups, scRNA data from the HKC treatment group came from SRA (PRJNA991651), data of ARB and Sodium-glucose cotransporter-2 inhibitors (SGLT2i) treatment groups came from GEO (GSE181382). The data of all the groups underwent the same preprocessing process and were annotated with the same cell type as the DKD group. Finally, CD-PC and B cells of the Ctrl, HKC, ARB and SGLT2i groups were severally transformed with labels of HDC and DDC through singleR (Version 1.10.0).

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}