
Mitochondrial Dysfunction in PCOS: Insights into Reproductive Organ Pathophysiology

Abstract
:1. Introduction
2. Methods
3. Animal and Cellular Models of PCOS
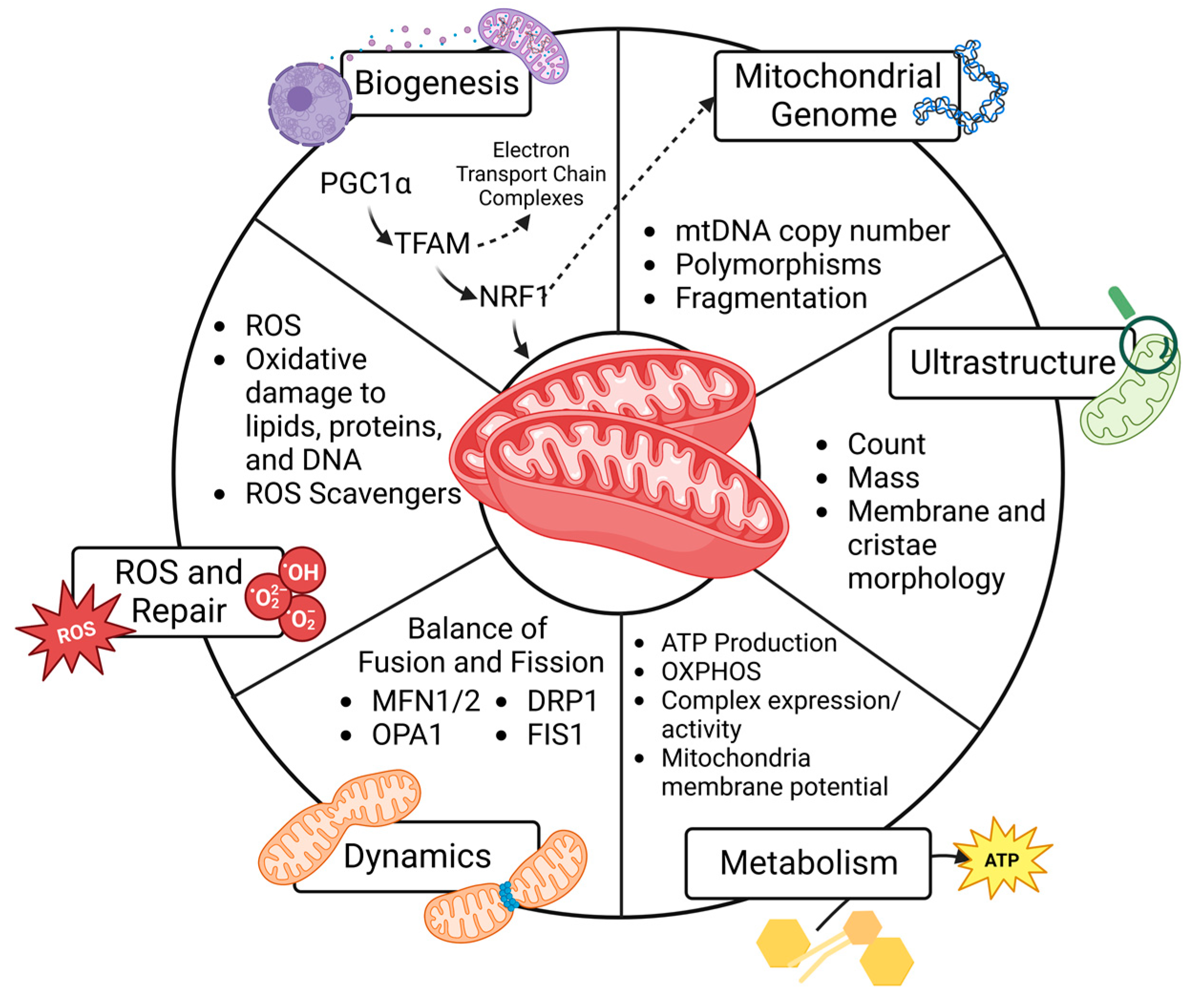
4. Mitochondrial Function and Cell Fate
4.1. Mitochondrial Biogenesis
4.2. Mitochondrial Genome
4.3. Ultrastructure
4.4. Metabolism
4.5. Dynamics
4.6. Reactive Oxygen Species and Repair
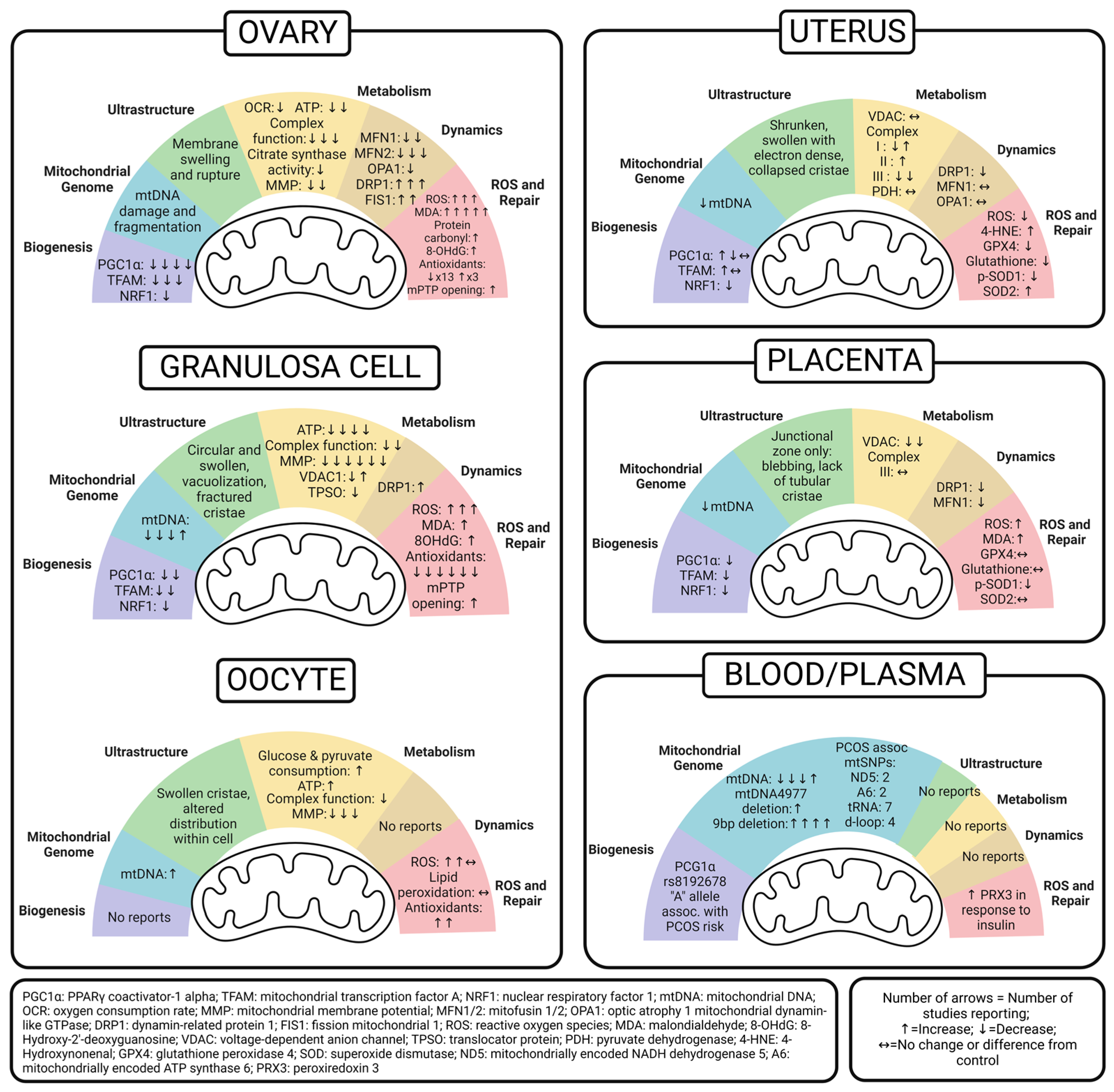
5. Ovary Mitochondria in PCOS
5.1. Whole Ovary

{kind=link}
{kind=link}
| Mitochondrial Function | Effect of PCOS Condition on Mitochondrial Function | Model—Species | PCOS Model/Diagnosis | Treatment Timeframe | Method | Therapeutic Intervention | Reference |
|---|---|---|---|---|---|---|---|
| Biogenesis | Decreased PGC1 | Rat | IP Letrozole + HFD | 21 days, 21 days, 35 days, 12 weeks | qPCR, WB | Cangfudaotan (IG) and metformin (IG) increased PGC1 to control levels | [52] |
| Rat | IG Letrozole | 21 days | WB | [53] | |||
| Mouse | SQ DHT | 35 days | WB | Overexpressing SIRT3 in vivo increased PGC1 back to control levels | [56] | ||
| Mouse | HF/HGD (58% kcal fat + sucrose) | 12 weeks | qPCR | Neurokinin-B antagonist increased PGC1 back to control levels | [54] | ||
| Decreased TFAM | Mouse | SQ DHEA | 20 days | WB | (1) L-carnitine (LC) + acetyl-L-carnitine (ACL) (2) LC and ACL plus propionyl-L-carnitine Both formulations increased TFAM compared to DHEA alone and controls | [55] | |
| Rat | IG Letrozole | 21 days | WB | [53] | |||
| Mouse | HF/HGD (58% kcal fat + sucrose) | 12 weeks | qPCR | Neurokinin-B antagonist increased TFAM back to control levels | [54] | ||
| Decreased NRF1 | Mouse | HF/HGD (58% kcal fat + sucrose) | 12 weeks | qPCR | Neurokinin-B antagonist increased NRF1 back to control levels | [54] | |
| Mitochondrial Genome | Increased mtDNA fragmentation | Rat | On day 22 of HFD (46% fat), OG letrozole | 21 days | qPCR | Cangfudaotan (IG) and metformin (IG) decreased mtDNA damage and fragmentation | [52] |
| Ultrastructure | Membrane swelling and ruptures | Rat | IP Letrozole + HFD | 21 days | EM | % of total damaged mitochondria decreased with either metformin (IG) or cangfudaotan (IG) but were still higher than control levels | [52] |
| Metabolism | Increased basal, maximal and ATP-linked OCR, proton leak | Mice—offspring | DHT injection in dams post-coitus, assessed pup neonatal ovaries | GD 16.5, 17.5, 18.5 | XF (Agilent) of whole neonatal ovaries | [58] | |
| Decreased OCR, RCR | Rat | IP letrozole + HFD | 21 days | Oxytherm Clark-type electrode on isolated mitochondria | Cangfudaotan (IG) increased OCR, RCR | [52] | |
| Decreased ATP | Rat | On day 22 of HFD (46% fat), OG letrozole | 21 days | Colorimetric ATP assay | SeNP alone and in combination with metformin increased ATP (most increase in combination) | [57] | |
| Rat | IP letrozole + HFD | 21 days | ATP assay | Cangfudaotan (IG) increased ATP levels | [52] | ||
| No difference in ATP | Mice—offspring | DHT injection in dams post-coitus, assessed pup neonatal ovaries | GD 16.5, 17.5, 18.5 | XF (Agilent) of whole neonatal ovaries | [58] | ||
| Decreased activity of mitochondrial complex enzymes | Rat | IP letrozole + HFD | 21 days | Complex enzyme activity assays | Cangfudaotan (IG) increased mitochondrial complex activity | [52] | |
| Decreased Complex I activity | Rat | On day 22 of HFD (46% fat), OG letrozole | 21 days | Complex I enzyme activity assay | SeNP alone and in combination with metformin increased Complex 1 activity (most increase in combination) | [57] | |
| Rat | SQ DHEA | 20 days | Bushen Huatan Granules (OG) increased activity of Complex I | [51] | |||
| Decreased Complexes III, IV activity | SQ DHEA | 20 days | Complexes III, IV enzyme activity assays | Bushen Huatan Granules (OG) increased activity of Complexes III and IV | [51] | ||
| Decreased Complex IV (Cox6a2 subunit) | Mice—offspring | DHT injection in dams post-coitus, assessed pup neonatal ovaries | GD 16.5, 17.5, 18.5 | RNAseq | [58] | ||
| Decreased citrate synthase activity | Rat | OG letrozole | 21 days | Citrate synthase activity assay | Metformin (OG) and sodium selenite (OG) increased mitochondrial citrate synthase activity but still lower than control group | [59] | |
| Decreased MMP | Rat | On day 22 of HFD (46% fat), OG letrozole | 21 days | JC-1 staining | SeNP alone and in combination with metformin increased MMP (most increase in combination) | [57] | |
| Rat | IP letrozole + HFD | Cangfudaotan (IG) or metformin (IG) increased MMP | [52] | ||||
| Dynamics | Decreased MFN1 | Rat | IP letrozole + HFD | 21 days | qPCR/WB | Cangfudaotan (IG) or metformin (IG) increased MFN1 | [52] |
| Rat | IG Letrozole | 21 days | WB | [53] | |||
| Decreased MFN2 | Rat | IP letrozole + HFD | 21 days | qPCR/WB | Cangfudaotan (IG) or metformin (IG) increased MFN2 | [52] | |
| Rat | OG letrozole (OG) | 21 days | qPCR/ELISA kit | Metformin (OG) and sodium selenite (OG) increased MFN2 but still lower than control group | [59] | ||
| Rat | IG Letrozole | 21 days | WB | [53] | |||
| Decreased OPA1 | Rat | IP letrozole + HFD | 21 days | qPCR/WB | Cangfudaotan (IG) or metformin (IG) increased OPA1 | [52] | |
| Increased DRP1 | Rat | IP letrozole + HFD | 21 days | qPCR/WB | Cangfudaotan (IG) or metformin (IG) decreased DRP1 | [52] | |
| Rat | OG letrozole | 21 days | qPCR/ELISA kit | Metformin (OG) and sodium selenite (OG) decreased DRP1 but still higher than control group | [59] | ||
| Rat | IG Letrozole | 21 days | WB | [53] | |||
| Increased FIS1 | Rat | IP letrozole + HFD | 21 days | qPCR/WB | Cangfudaotan (IG) or metformin (IG) decreased FIS1 | [52] | |
| Rat | IG Letrozole | 21 days | WB | [53] | |||
| ROS and Repair | Increased ROS | Rat | IP letrozole + HFD | 21 days | DCF staining | Cangfudaotan (IG) or metformin (IG) decreased ROS | [52] |
| Rat | IG Letrozole | 21 days | Activity to produce superoxide anion assay | [53] | |||
| Increased mitochondrial superoxide | Rat | SQ DHEA | 20 days | MitoSOX staining | Bushen Huatan Granules (OG) decreased mitochondrial superoxide | [51] | |
| Increased lipid peroxidation | Rat | On day 22 of HFD (46% fat), OG letrozole | 21 days | MDA assay | SeNP alone or in combination with metformin decreased lipid peroxidation | [57] | |
| Rat | OG letrozole | 21 days | Metformin (OG) and sodium selenite (OG) decreased lipid peroxidation but still higher than control group | [59] | |||
| Rat | SQ DHEA | 21 days | [60] | ||||
| Rat | IG Letrozole | 21 days | [53] | ||||
| Mouse | SQ DHEA | 20 days | Genistein decreased lipid peroxidation | [61] | |||
| Increased protein oxidation | Rat | OG letrozole | 21 days | DNPH reaction assay | Metformin (OG) and sodium selenite (OG) decreased protein oxidation but still higher than control group | [59] | |
| Increased DNA oxidation | Mouse | SQ DHEA | 20 days | 8-OHdG ELISA | Genistein decreased DNA oxidation levels | [61] | |
| Decreased antioxidant capacity | Rat | OG letrozole | 21 days | Ferric-reducing antioxidant power assay | Metformin (OG) and sodium selenite (OG) increased antioxidant capacity but still lower than control group | [59] | |
| Decreased SOD activity | Rat | On day 22 of HFD (46% fat), OG letrozole | 21 days | SOD enzyme activity assay | SeNP alone or in combination with metformin increased SOD levels | [57] | |
| Rat | SQ DHEA | 21 days | [60] | ||||
| Mouse | SQ DHEA | 20 days | Genistein increased SOD | [61] | |||
| Decreased SOD1 | Mouse | HF/HGD (58% kcal fat + sucrose) | 12 weeks | qPCR | Neurokinin-B antagonist increased SOD1 | [54] | |
| Increased SOD2 (MnSOD) | Mouse | SQ DHEA | 20 days | WB | (1) LC + ACL and (2) LC, ACL + propionyl-L-carnitine both decreased SOD2 | [55] | |
| Rat | IG Letrozole | 21 days | WB | [53] | |||
| Decreased GSH | Rat | On day 22 of HFD (46% fat), OG letrozole | 21 days | GSH level | SeNP alone or in combination with metformin increased GSH levels | [57] | |
| Decreased GSH-Px (GPx) | Rat | OG letrozole | 21 days | GPx enzyme activity assay | Metformin (OG) and sodium selenite (OG) increased GPx activity but still lower than control group | [59] | |
| Mouse | SQ DHEA | 20 days | GSH-Px level | Genistein increased GSH-Px | [61] | ||
| Increased GSH-Px | Rat | IG Letrozole | 21 days | GSH-Px enzyme activity assay | [53] | ||
| Decreased GR | Mouse | SQ DHEA | 20 days | GR enzyme activity assay | [61] | ||
| Decreased GSH:GSSG ratio | Mouse | SQ DHEA | 20 days | GSH and GSSG levels | Genistein increased GSH:GSSG ratio | [61] | |
| Decreased CAT activity | Mouse | SQ DHEA | 20 days | CAT enzyme activity assay | Genistein increased CAT activity | [61] | |
| Rat | SQ DHEA | 21 days | [60] | ||||
| Mouse | HF/HGD (58% kcal fat + sucrose) | 12 weeks | qPCR | Neurokinin-B antagonist increased CAT expression | [54] | ||
| Increased opening of mPTP | Rat | IP letrozole + HFD | 21 days | Mitochondrial Membrane Pore-Channel Colorimetric Assay | Canfudaton (IG) or metformin (IG) decreased opening of mPTP | [52] | |
| Increased levels of Cytochrome C in cytosol than in mitochondria | Rat | SQ DHEA | 20 days | WB | Bushen Huatan Granules (OG) decreased levels of Cytochrome C in cytosol fraction compared to mitochondrial fraction | [51] |
5.2. Granulosa Cell
| Mitochondrial Function | Effect of PCOS on Mitochondrial Function | Model—Species | PCOS Model/Diagnosis | Treatment Timeframe | Method | Therapeutic Intervention | Reference |
|---|---|---|---|---|---|---|---|
| Biogenesis | Decreased PGC1 | Cell line KGN | 500 mM DHT | 24 h | WB | Overexpression of SIRT3 (cell transfection) increased PGC1 levels comparable to controls | [56] |
| Cell line KGN | Palmitic Acid + DHT | Various | qPCR | si-NK3R increased PGC1 | [54] | ||
| Decreased TFAM | Mouse | SQ DHEA | 20 days prior to puberty | qPCR | Vitamin D3 (100 mM for 24 h) increased TFAM | [62] | |
| Cell line KGN | Palmitic Acid + DHT | Various | qPCR | si-NK3R increased TFAM | [54] | ||
| Decreased NRF1 | Cell line KGN | Palmitic Acid + DHT | Various | qPCR | si-NK3R increased NRF1 | [54] | |
| Mitochondrial Genome | Decreased mtDNA copy number | Mouse | SQ DHT | 20 days prior to puberty | qPCR | Vitamin D3 (100 mM for 24 h) increased mtDNA copy number | [62] |
| Rat | SQ DHEA | 20 days | qPCR (mtND1:beta-globin) | [24] | |||
| Human | Rotterdam | qPCR (mtND1:beta-actin) | [63] | ||||
| Increased mtDNA copy number | Cell line KGN | 500 nM DHT | 24 h | qPCR | Overexpression of SIRT3 decreased mtDNA copy number, which increased back to DHT-exposed levels with PGC1a inhibitor | [56] | |
| Ultrastructure | Disorganized cristae, vacuoles, less electron-dense | Mouse—offspring | DHT injection post-coitus, assessed pup neonatal GCs | GD 16.5, 17.5, 18.5 | TEM | [58] | |
| Mitochondrial aggregated distribution, cristae dissolution and fracture, presence of vacuoles (66.66% abnormal mitochondria compared to 0% in controls) | Rat | SQ DHEA | 20 days | TEM | [24] | ||
| Mitochondrial swelling and membrane defects | Mouse | SQ DHT | 35 days | TEM | Melatonin reduced mitochondrial swelling and membrane defects | [64] | |
| Mitochondrial swelling and membrane defects | Human | Rotterdam | TEM | [64] | |||
| Less rod-shaped mitochondria, more circular/constricted mitochondria | Rat | Continuous-release DHT pellet implant | 1 month (83 μg/day) | TEM | eCG increased rod-shaped mitochondria and decreased circular/constricted mitochondria | [65] | |
| Decreased number of mitochondria | Human | Rotterdam | Nonyl acridine orange (NAC) flow cytometry | [66] | |||
| Decreased mitochondrial mass, increased mitochondrial fragmentation, constrained cell expansion | Human | Rotterdam | pLV-mitoDsRed plasmid transfection (tags ATP synthase) and imaging | [66] | |||
| Metabolism | Decreased ATP levels | Mouse | 100 μM DHEA | 12 h | ATP assay | Genistein increased ATP levels, but still lower than controls. Additional treatment with NRF2 inhibitor ML385 decreased ATP levels compared to genistein alone but still higher than DHEA group | [61] |
| Rat | SQ DHEA | 20 days | [24] | ||||
| Cell line KGN | 500 nM DHT | 24 h | Overexpression of SIRT3 increased ATP but addition of PGC1a inhibitor reversed this effect | [56] | |||
| Human | Rotterdam | [63] | |||||
| Decreased activity of Complexes I, III, IV | Rat | Testosterone (10−5 M) | 24 h | Complex I/III/IV activity assay | 6 h incubation with serum from rats receiving Bushen Huatan Granules treatment led to increased activity of Complexes I, III, IV | [51] | |
| Decreased NDUFB8 (Complex I subunit) and ATP5j (ATP synthase subunit) | Rat | SQ DHEA | 20 days | qPCR/WB | [24] | ||
| Decreased MMP | Mouse | SQ DHEA | 20 days | JC-1 flow cytometry | Genistein increased MMP but still lower than controls. Additional treatment with NRF2 inhibitor ML385 had similar MMP to DHEA group | [61] | |
| Cell line KGN | 500 nM DHT | 24 h | JC-1 staining | Overexpression of SIRT3 increased MMP but addition of PGC1a inhibitor reversed this effect | [56] | ||
| Cell line KGN | 500 nM DHT | 24 h | Melatonin (1000 pM for 24 h) increased MMP | [64] | |||
| Human | Rotterdam | TMRE flow cytometry | [66] | ||||
| Human | Rotterdam | Mitotracker Red flow cytometry | [66] | ||||
| Human | Rotterdam | JC-1 Mitochondrial Membrane Potential Kit | [63] | ||||
| Increased VDAC1 | Rat | IG letrozole | 21 days | WB | [53] | ||
| Decreased VDAC1 | Human | Rotterdam | qPCR/ICC | [67] | |||
| Decreased TSPO | Human | Rotterdam | qPCR/ICC | [67] | |||
| Dynamics | Increased DRP1 | Rat | Continuous-release DHT pellet implant | 1 month (83 μg/day) | WB | [65] | |
| ROS and Repair | Increased ROS | Mouse | 100 μM DHEA | 12 h | DCFH-DA flow cytometry | Genistein decreased ROS | [61] |
| Human | Rotterdam | DCFH-DA | [63] | ||||
| Increased mitochondrial superoxide | Cell line KGN | 500 nM DHT | 24 h | MitoSOX staining | Overexpression of SIRT3 decreased mitochondrial superoxide but addition of PGC1-a inhibitor reversed this outcome | [56] | |
| Increased lipid peroxidation | Mouse | SQ DHEA | 20 days | MDA assay | Genistein decreased lipid peroxidation | [61] | |
| Increased DNA oxidation | Mouse | SQ DHEA | 20 days | 8-OHdG ELISA | Genistein decreased 8-OhdG levels | [61] | |
| Decreased SOD, GSH-Px, GR, GSH:GSSG ratio | Mouse | SQ DHEA | 20 days | SOD, GR enzyme activity assays/GSH-Px, GSH:GSSG content assays | Genistein increased SOD, CAT, GSH-Px, GSH:GSSG ratio | [61] | |
| Decreased SOD1 | Cell line KGN | Palmitic Acid + DHT | Various | qPCR | si-NK3R increased SOD1 | [54] | |
| Decreased CAT | Mouse | SQ DHEA | 20 days | CAT enzyme activity assay | Genistein increased CAT | [61] | |
| Cell line KGN | Palmitic Acid + DHT | Various | qPCR | si-NK3R increased CAT | [54] | ||
| Upregulation of UPR-MT (mitochondrial unfolded protein response) proteins | Human | Rotterdam | qPCR | [68] | |||
| Increased opening of mPTP | Cell line KGN | 500 nM DHT | 24 h | Mitochondrial permeability transition pore assay | Melatonin (1000 pM for 24 h) decreased opening of mPTP | [64] |
5.3. Oocyte
| Mitochondrial Function | Effect of PCOS on Mitochondrial Function | Model—Species | PCOS Model/Diagnosis | Treatment Timeframe | Method | Therapeutic Intervention | Reference |
|---|---|---|---|---|---|---|---|
| Biogenesis | No Reports | ||||||
| Mitochondrial Genome | Increased mtDNA copy number | Mouse | Controlled-release DHT pellet implant | 90 days (2.75 μg/day) | qPCR (mtCO1:tubulin) | [69] | |
| No difference in mtDNA copy number | Mouse—offspring | DHT injection in dam post-coitus, assessed post-pubertal pup oocytes | GD 16.5, 17.5, 18.5 | qPCR (mtCO1:tubulin) | [58] | ||
| Ultrastructure | Disorganized cristae, vacuoles, less electron-dense | Mouse—offspring | DHT injection in dam post-coitus, assessed post-pubertal pup oocytes | GD 16.5, 17.5, 18.5 | TEM | [58] | |
| Mitochondria with malformed cristae with concentric circles, swollen or loss of cristae | Mouse | Controlled-release DHT pellet implant | 90 days (2.75 μg/day) | SEM | [69] | ||
| Mitochondria have swollen cristae, no electron-dense contents, and are vacuolated | Mouse—offspring | DHT injection in dams post-coitus, assessed post-pubertal pup oocytes | GD 16.5, 17.5, 18.5 | TEM | [70] | ||
| Abnormal mitochondria distribution | Human/Mouse | EVs isolated from PCOS patients with non-hyperandrogenic phenotype were co-cultured with control murine oocytes | Mitochondrial Red Fluorescent Probe | [72] | |||
| Metabolism | Increased glucose, pyruvate consumption | Human | Rotterdam | Ultra-microfluorometric assay | [73] | ||
| Increased ATP levels | Mice | Controlled-release DHT pellet implant | 90 days (2.75 μg/day) | ATP assay | [69] | ||
| No difference in ATP levels | Mouse—offspring | DHT injection in dams post-coitus, assessed post-pubertal pup oocytes | GD 16.5, 17.5, 18.5 | [70] | |||
| Decreased mitochondrial Complex I genes (ND1, ND2, ND5) | Mice | Controlled-release DHT pellet implant | 90 days (2.75 μg/day) | qPCR | [69] | ||
| Increased mitochondrial Complexes I and IV genes (ND1, ND6 and CO1, CO2, CO3) | Mouse—offspring | DHT injection in dams post-coitus, assessed post-pubertal pup oocytes | GD 16.5, 17.5, 18.5 | [70] | |||
| Decreased MMP | Mice | Controlled-release DHT pellet implant | 90 days (2.75 μg/day) | JC-1 staining | [69] | ||
| Rat | DHEA injection (interscapular region) | 20 days | Rat-to-mouse BAT xenotransplant increased MMP | [74] | |||
| Mice—offspring | DHT injection in dams post-coitus, assessed post-pubertal pup oocytes | GD 16.5, 17.5, 18.5 | [70] | ||||
| ROS and Repair | Increased ROS | Rat | DHEA injection (interscapular region) | 20 days | ROS assay using DCFH-DA | Rat-to-mouse BAT xenotransplant decreased ROS level | [74] |
| Human | EVs isolated from PCOS patients with non-hyperandrogenic phenotype were co-cultured with control murine oocytes | DCHF-DA staining | [72] | ||||
| Mice—offspring | DHT injection in dams post-coitus, assessed post-pubertal pup oocytes | GD 16.5, 17.5, 18.5 | CellROX staining | [70] | |||
| No differences in ROS | Mice | Controlled-release DHT pellet implant | 90 days (2.75 μg/day) | [69] | |||
| No differences in lipid peroxidation | Mice | Controlled-release DHT pellet implant | 90 days (2.75 μg/day) | BODIPYC11® 581/591 staining | [69] | ||
| Increased CAT | Human | EVs isolated from PCOS patients with non-hyperandrogenic phenotype were co-cultured with control murine oocytes | qPCR | [72] | |||
| Increased GSS | Human | EVs isolated from PCOS patients with non-hyperandrogenic phenotype were co-cultured with control murine oocytes | qPCR | [72] |
6. Uterus Mitochondria in PCOS
| Mitochondrial Function | Effect of PCOS on Mitochondrial Function | Model/Species | PCOS Model/Diagnosis | Treatment Timeframe | Method | References | Therapeutic Intervention | References |
|---|---|---|---|---|---|---|---|---|
| Biogenesis | Increased PGC-1α | Mouse | SQ DHEA | 20 days | WB | [77] | ||
| No change in PGC-1α | Rat | IP DHT + INS | GD 0.5–GD 13.5 | qPCR | [78] | |||
| Decreased PGC-1α | Rat | IP DHT + INS | GD 7.5–GD 13.5 | qPCR | [79] | |||
| Increased TFAM | Human | Rotterdam Criteria | WB | [76] | ||||
| No change in TFAM | Rat | IP DHT + INS | GD 7.5–GD 13.5 or GD 14.5 | qPCR | [78,79] | |||
| Decreased NRF1 | Rat | IP DHT + INS | GD 7.5–GD 13.5 | qPCR | [79] | |||
| Mitochondrial Genome | Decreased mtDNA copy number | Rat | IP DHT + INS | GD 7.5–GD 13.5 | qPCR | [79] | ||
| Ultrastructure | Decreased TOMM20 | Mouse | SQ DHEA | 20 days | IHC | [77] | L-carnitine/acetyl-L-carnitine returned levels closer to control | [77] |
| Increased prohibitin I | Rat | IP DHT + INS | GD 7.5–GD 14.5 | WB | [81] | |||
| Shrunken mitochondria | Rat | IP DHT + INS | GD 7.5–GD 13.5 | TEM | [82] | |||
| Swollen mitochondria | Rat | IP DHT + INS | GD 7.5–GD 13.5 or GD 14.5 | TEM | [78,81] | N-acetyl-cysteine improved but did not fully rescue morphology and also impaired mitochondria in controls; flutamide decreased number of small swollen mitochondria but cristae remained disorganized | [78,81] | |
| Electron-dense and collapsed cristae | Rat | IP DHT + INS | GD 7.5–GD 13.5 or GD 14.5 | TEM | [78,81,82] | |||
| Metabolism | No difference in VDAC | Rat | IP DHT + INS | GD 7.5–GD 14.5 | WB | [81] | N-acetyl-cysteine did not change VDAC but did decrease it in controls | [81] |
| Decreased Complex I | Rat | IP DHT + INS | GD 7.5–GD 14.5 | WB | [81] | N-acetyl-cysteine normalized | [81] | |
| Increased Complex I | Rat | IP DHT + INS | GD 0.5–GD 13.5 | WB | [78] | Flutamide normalized | [78] | |
| Increased Complex II | Rat | IP DHT + INS | GD 0.5–GD 13.5 | WB | [78] | Flutamide normalized | [78] | |
| Decreased Complex III | Rat | IP DHT + INS | GD 7.5–GD 13.5 | WB | [79,81] | N-acetyl-cysteine normalized | [81] | |
| No difference in PDH | Human | Rotterdam Criteria | WB | [76] | ||||
| Dynamics | Decreased DRP1 (Fission) | Rat | IP DHT + INS | GD 7.5–GD 13.5 | qPCR | [79] | ||
| No change in MFN1 (Fusion) | Rat | IP DHT + INS | GD 7.5–GD 13.5 | qPCR | [79] | |||
| No change in OPA1 (Fusion) | Rat | IP DHT + INS | GD 7.5–GD 13.5 | qPCR | [79] | |||
| ROS and Repair | Increased 4-HNE adducts | Mouse | SQ DHEA | 20 days | IHC | [77] | L-carnitine/acetyl-L-carnitine returned levels closer to control | [77] |
| Reduced ROS levels | Rat | IP DHT + INS | GD 7.5–GD 13.5 | OxiSelect In Vitro ROS/RNS assay | [79] | |||
| Reduced GPX4 | Rat | IP DHT + INS | GD 7.5–GD 13.5 | WB, IHC | [82] | |||
| Reduced glutathione | Rat | IP DHT + INS | GD 7.5–GD 13.5 | Glutathione/glutathione + glutathione disulfide assay | [82] | |||
| Reduced phosphorylated SOD1 | Rat | IP DHT + INS | GD 7.5–GD 13.5 | WB | [79] | |||
| Increased SOD2 | Mouse | SQ DHEA | 20 days | WB | [77] | Propionyl-L-carnitine altered levels | [77] |
7. Placenta Mitochondria in PCOS
8. Peripheral Markers of Mitochondrial Dysfunction in PCOS
| Mitochondrial Function | Effect of PCOS on Mitochondrial Function | Method | Therapeutic Intervention | References |
|---|---|---|---|---|
| Biogenesis | Reduced “GG”(WT) frequency of PGC-1α rs8192678 polymorphism | PCR, RFLP Analysis | [90] | |
| No difference in TFAM genotype or allele frequency | PCR, RFLP Analysis | [90] | ||
| Mitochondrial Genome | Lower mtDNA copy number | qPCR | [92,93,94] | |
| Higher mtDNA copy number | qPCR | [91] | ||
| Negative association between mtDNA copy number and fasting insulin, HOMA-IR, waist circumference, and triglycerides | Pearson correlation coefficient | [94] | ||
| Positive association between mtDNA copy number and quantitative insulin-sensitivity check index (QUICKI) and sex hormone-binding globulin (SHBG) | Pearson correlation coefficient | [94] | ||
| No correlation between mtDNA copy number and anthropometric measure or 8-OHdG | qPCR, 8OH-dG ELISA kit | Metformin decreased mtDNA copy number at 6 and 12 months of treatment | [95] | |
| Higher mtDNA4977 deletion rate | qPCR | [91] | ||
| Higher frequency of a 9 bp deletion | qPCR | [49,96,97,98,99] | ||
| ND5 gene polymorphisms: T12811C, T12338C | qPCR | [97,100,101] | ||
| A6 gene polymorphisms: G8584A, C8684T | qPCR | [97,98,100] | ||
| Unique tRNA variants and higher frequency of variants for Cys and Leu tRNAs | qPCR | [92,97,100] | ||
| Greater frequency of D-loop SNPs C150T, T146C, A189G, and D310 | PCR, Mitomap, and mtDB mitochondria databases | [49,93] | ||
| Carriers of AA genotype of PGC1a polymorphism rs8192678 and D-loop SNPs A189G and D310 had lower mtDNA | PCR, Mitomap, and mtDB mitochondria databases | [90,93] | ||
| Ultrastructure | No Reports | |||
| Metabolism | No Reports | |||
| Dynamics | No Reports | |||
| ROS and Repair | Decreased PRX3 2–3 h post-OGTT | ELISA | [103] | |
| Positive correlation between PRX3 at 2 h post-OGTT and insulin at 1 h post-OGTT | Spearman correlation analysis | [103] |
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Genazzani, A.D.; Genazzani, A.R. Polycystic Ovary Syndrome as Metabolic Disease: New Insights on Insulin Resistance. Eur. Endocrinol. 2023, 19, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Joham, A.E.; Norman, R.J.; Stener-Victorin, E.; Legro, R.S.; Franks, S.; Moran, L.J.; Boyle, J.; Teede, H.J. Polycystic ovary syndrome. Lancet Diabetes Endocrinol. 2022, 10, 668–680. [Google Scholar] [CrossRef]
- The Lancet Regional Health—Europe. Polycystic ovary syndrome: What more can be done for patients? Lancet Reg. Health-Eur. 2022, 21, 100524. [Google Scholar] [CrossRef]
- CDC. PCOS (Polycystic Ovary Syndrome) and Diabetes. Available online: https://www.cdc.gov/diabetes/basics/pcos.html (accessed on 12 July 2023).
- Deswal, R.; Narwal, V.; Dang, A.; Pundir, C.S. The Prevalence of Polycystic Ovary Syndrome: A Brief Systematic Review. J. Hum. Reprod. Sci. 2020, 13, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, E.A.; Kauffman, A.S. The Role of the Brain in the Pathogenesis and Physiology of Polycystic Ovary Syndrome (PCOS). Med. Sci. 2019, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Crespo, R.P.; Bachega, T.; Mendonça, B.B.; Gomes, L.G. An update of genetic basis of PCOS pathogenesis. Arch. Endocrinol. Metab. 2018, 62, 352–361. [Google Scholar] [CrossRef]
- Ajmal, N.; Khan, S.Z.; Shaikh, R. Polycystic ovary syndrome (PCOS) and genetic predisposition: A review article. Eur. J. Obstet. Gynecol. Reprod. Biol. X 2019, 3, 100060. [Google Scholar] [CrossRef]
- Hoeger, K.M.; Dokras, A.; Piltonen, T. Update on PCOS: Consequences, Challenges, and Guiding Treatment. J. Clin. Endocrinol. Metab. 2020, 106, e1071–e1083. [Google Scholar] [CrossRef]
- Khan, M.J.; Ullah, A.; Basit, S. Genetic Basis of Polycystic Ovary Syndrome (PCOS): Current Perspectives. Appl. Clin. Genet. 2019, 12, 249–260. [Google Scholar] [CrossRef]
- Teede, H.J.; Misso, M.L.; Costello, M.F.; Dokras, A.; Laven, J.; Moran, L.; Piltonen, T.; Norman, R.J. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Fertil. Steril. 2018, 110, 364–379. [Google Scholar] [CrossRef]
- Naderpoor, N.; Shorakae, S.; de Courten, B.; Misso, M.L.; Moran, L.J.; Teede, H.J. Metformin and lifestyle modification in polycystic ovary syndrome: Systematic review and meta-analysis. Hum. Reprod. Update 2016, 22, 408–409. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.F.; Wu, Y.; Zhu, Y.H.; Ding, T.; Batterham, R.L.; Qu, F.; Hardiman, P.J. Pharmacologic therapy to induce weight loss in women who have obesity/overweight with polycystic ovary syndrome: A systematic review and network meta-analysis. Obes. Rev. 2018, 19, 1424–1445. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, W.; Bordewijk, E.M.; Legro, R.S.; Zhang, H.; Wu, X.; Gao, J.; Morin-Papunen, L.; Homburg, R.; König, T.E.; et al. First-line ovulation induction for polycystic ovary syndrome: An individual participant data meta-analysis. Hum. Reprod. Update 2019, 25, 717–732. [Google Scholar] [CrossRef]
- Legro, R.S.; Driscoll, D.; Strauss, J.F., 3rd; Fox, J.; Dunaif, A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc. Natl. Acad. Sci. USA 1998, 95, 14956–14960. [Google Scholar] [CrossRef] [PubMed]
- Stepto, N.K.; Cassar, S.; Joham, A.E.; Hutchison, S.K.; Harrison, C.L.; Goldstein, R.F.; Teede, H.J. Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic-hyperinsulaemic clamp. Hum. Reprod. 2013, 28, 777–784. [Google Scholar] [CrossRef]
- Oestreich, A.K.; Moley, K.H. Developmental and Transmittable Origins of Obesity-Associated Health Disorders. Trends Genet. 2017, 33, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Franks, S.; Berga, S.L. Does PCOS have developmental origins? Fertil. Steril. 2012, 97, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Risal, S.; Pei, Y.; Lu, H.; Manti, M.; Fornes, R.; Pui, H.P.; Zhao, Z.; Massart, J.; Ohlsson, C.; Lindgren, E.; et al. Prenatal androgen exposure and transgenerational susceptibility to polycystic ovary syndrome. Nat. Med. 2019, 25, 1894–1904. [Google Scholar] [CrossRef]
- Insler, V.; Lunenfeld, B. Polycystic ovarian disease: A challenge and controversy. Gynecol. Endocrinol. 1990, 4, 51–70. [Google Scholar] [CrossRef]
- Azziz, R.; Adashi, E.Y. Stein and Leventhal: 80 years on. Am. J. Obstet. Gynecol. 2016, 214, 247.e241–247.e211. [Google Scholar] [CrossRef]
- EvidenceHunt. Available online: https://evidencehunt.com/ (accessed on 5 June 2023).
- International Protein Nomenclature Guidelines. Available online: https://www.ncbi.nlm.nih.gov/genome/doc/internatprot_nomenguide/ (accessed on 19 July 2023).
- Song, L.; Yu, J.; Zhang, D.; Li, X.; Chen, L.; Cai, Z.; Yu, C. Androgen Excess Induced Mitochondrial Abnormality in Ovarian Granulosa Cells in a Rat Model of Polycystic Ovary Syndrome. Front. Endocrinol. 2022, 13, 789008. [Google Scholar] [CrossRef] [PubMed]
- Popov, L.D. Mitochondrial biogenesis: An update. J. Cell. Mol. Med. 2020, 24, 4892–4899. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.F.; Hamilton, K.L. A perspective on the determination of mitochondrial biogenesis. Am. J. Physiol.-Endocrinol. Metab. 2012, 302, E496–E499. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Weigl, S.; Paradiso, A.; Tommasi, S. Mitochondria and familial predisposition to breast cancer. Curr. Genom. 2013, 14, 195–203. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, K.; Guo, S.; Wang, Y.; Ji, X.; Yuan, Q.; Su, L.; Guo, X.; Gu, X.; Xing, J. NGS-based accurate and efficient detection of circulating cell-free mitochondrial DNA in cancer patients. Mol. Ther.-Nucleic Acids 2021, 23, 657–666. [Google Scholar] [CrossRef]
- Bisserier, M.; Shanmughapriya, S.; Rai, A.K.; Gonzalez, C.; Brojakowska, A.; Garikipati, V.N.S.; Madesh, M.; Mills, P.J.; Walsh, K.; Arakelyan, A.; et al. Cell-Free Mitochondrial DNA as a Potential Biomarker for Astronauts’ Health. J. Am. Heart Assoc. 2021, 10, e022055. [Google Scholar] [CrossRef]
- Trecarichi, A.; Duggett, N.A.; Granat, L.; Lo, S.; Malik, A.N.; Zuliani-Álvarez, L.; Flatters, S.J.L. Preclinical evidence for mitochondrial DNA as a potential blood biomarker for chemotherapy-induced peripheral neuropathy. PLoS ONE 2022, 17, e0262544. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef]
- Rasmussen, N. Mitochondrial Structure and the Practice of Cell Biology in the 1950s. J. Hist. Biol. 1995, 28, 381–429. [Google Scholar] [CrossRef]
- Joshua, R.C.B. The Structure of Mitochondria. Trans. Am. Microsc. Soc. 1975, 94, 303–304. [Google Scholar] [CrossRef]
- Zick, M.; Rabl, R.; Reichert, A.S. Cristae formation—Linking ultrastructure and function of mitochondria. Biochim. Et Biophys. Acta (BBA)-Mol. Cell Res. 2009, 1793, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef]
- Perkins, G.A.; Frey, T.G. Recent structural insight into mitochondria gained by microscopy. Micron 2000, 31, 97–111. [Google Scholar] [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Else, A.J.; Barnes, S.J.; Danson, M.J.; Weitzman, P.D. A new spectrophotometric assay for citrate synthase and its use to assess the inhibitory effects of palmitoyl thioesters. Biochem. J. 1988, 251, 803–807. [Google Scholar] [CrossRef]
- Benard, G.; Bellance, N.; James, D.; Parrone, P.; Fernandez, H.; Letellier, T.; Rossignol, R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007, 120, 838–848. [Google Scholar] [CrossRef]
- Muller, B.; Lewis, N.; Adeniyi, T.; Leese, H.J.; Brison, D.R.; Sturmey, R.G. Application of extracellular flux analysis for determining mitochondrial function in mammalian oocytes and early embryos. Sci. Rep. 2019, 9, 16778. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Zemirli, N.; Morel, E.; Molino, D. Mitochondrial Dynamics in Basal and Stressful Conditions. Int. J. Mol. Sci. 2018, 19, 564. [Google Scholar] [CrossRef]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef] [PubMed]
- Kraus, F.; Ryan, M.T. The constriction and scission machineries involved in mitochondrial fission. J. Cell Sci. 2017, 130, 2953–2960. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Moosa, A.; Ghani, M.; O’Neill, H.C. Genetic associations with polycystic ovary syndrome: The role of the mitochondrial genome; a systematic review and meta-analysis. J. Clin. Pathol. 2022, 75, 815–824. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Xu, Y.; Pan, C.S.; Li, Q.; Zhang, H.L.; Yan, L.; Anwaier, G.; Wang, X.Y.; Yan, L.L.; Fan, J.Y.; Li, D.; et al. The Ameliorating Effects of Bushen Huatan Granules and Kunling Wan on Polycystic Ovary Syndrome Induced by Dehydroepiandrosterone in Rats. Front. Physiol. 2021, 12, 525145. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.L.; Tai, H.; Xiao, X.S.; Zhang, S.Y.; Cui, S.C.; Qi, S.B.; Hu, D.D.; Zhang, L.N.; Kuang, J.S.; Meng, X.S.; et al. Cangfudaotan decoction inhibits mitochondria-dependent apoptosis of granulosa cells in rats with polycystic ovarian syndrome. Front. Endocrinol. 2022, 13, 962154. [Google Scholar] [CrossRef]
- Wang, F.; Han, J.; Wang, X.; Liu, Y.; Zhang, Z. Roles of HIF-1α/BNIP3 mediated mitophagy in mitochondrial dysfunction of letrozole-induced PCOS rats. J. Mol. Histol. 2022, 53, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Fernando, T.; Zhu, X.; Shi, Y. The overexpression of neurokinin B-neurokinin 3 receptor system exerts direct effects on the ovary under PCOS-like conditions to interfere with mitochondrial function. Am. J. Reprod. Immunol. 2023, 89, e13663. [Google Scholar] [CrossRef]
- Di Emidio, G.; Rea, F.; Placidi, M.; Rossi, G.; Cocciolone, D.; Virmani, A.; Macchiarelli, G.; Palmerini, M.G.; D’Alessandro, A.M.; Artini, P.G.; et al. Regulatory Functions of L-Carnitine, Acetyl, and Propionyl L-Carnitine in a PCOS Mouse Model: Focus on Antioxidant/Antiglycative Molecular Pathways in the Ovarian Microenvironment. Antioxidants 2020, 9, 867. [Google Scholar] [CrossRef]
- Pang, X.; Cheng, J.; Wu, T.; Sun, L. SIRT3 ameliorates polycystic ovary syndrome through FOXO1/PGC-1α signaling pathway. Endocrine 2023, 80, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Rabah, H.M.; Mohamed, D.A.; Mariah, R.A.; Abd El-Khalik, S.R.; Khattab, H.A.; AbuoHashish, N.A.; Abdelsattar, A.M.; Raslan, M.A.; Farghal, E.E.; Eltokhy, A.K. Novel insights into the synergistic effects of selenium nanoparticles and metformin treatment of letrozole—induced polycystic ovarian syndrome: Targeting PI3K/Akt signalling pathway, redox status and mitochondrial dysfunction in ovarian tissue. Redox Rep. 2023, 28, 2160569. [Google Scholar] [CrossRef] [PubMed]
- Barsky, M.; Merkison, J.; Hosseinzadeh, P.; Yang, L.; Bruno-Gaston, J.; Dunn, J.; Gibbons, W.; Blesson, C.S. Fetal programming of polycystic ovary syndrome: Effects of androgen exposure on prenatal ovarian development. J. Steroid Biochem. Mol. Biol. 2021, 207, 105830. [Google Scholar] [CrossRef] [PubMed]
- Atef, M.M.; Abd-Ellatif, R.N.; Emam, M.N.; Abo El Gheit, R.E.; Amer, A.I.; Hafez, Y.M. Therapeutic potential of sodium selenite in letrozole induced polycystic ovary syndrome rat model: Targeting mitochondrial approach (selenium in PCOS). Arch. Biochem. Biophys. 2019, 671, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, N.; Zeng, Z.; Tang, L.; Zhao, S.; Zhou, F.; Zhou, L.; Xia, W.; Zhu, C.; Rao, M. Humanin regulates oxidative stress in the ovaries of polycystic ovary syndrome patients via the Keap1/Nrf2 pathway. Mol. Hum. Reprod. 2021, 27, gaaa081. [Google Scholar] [CrossRef]
- Luo, M.; Zheng, L.W.; Wang, Y.S.; Huang, J.C.; Yang, Z.Q.; Yue, Z.P.; Guo, B. Genistein exhibits therapeutic potential for PCOS mice via the ER-Nrf2-Foxo1-ROS pathway. Food Funct. 2021, 12, 8800–8811. [Google Scholar] [CrossRef]
- Safaei, Z.; Bakhshalizadeh, S.H.; Nasr Esfahani, M.H.; Akbari Sene, A.; Najafzadeh, V.; Soleimani, M.; Shirazi, R. Effect of Vitamin D3 on Mitochondrial Biogenesis in Granulosa Cells Derived from Polycystic Ovary Syndrome. Int. J. Fertil. Steril. 2020, 14, 143–149. [Google Scholar] [CrossRef]
- Wang, J.; Wu, X. The effects of mitochondrial dysfunction on energy metabolism switch by HIF-1α signalling in granulosa cells of polycystic ovary syndrome. Endokrynol. Pol. 2020, 71, 134–145. [Google Scholar] [CrossRef]
- Zheng, B.; Meng, J.; Zhu, Y.; Ding, M.; Zhang, Y.; Zhou, J. Melatonin enhances SIRT1 to ameliorate mitochondrial membrane damage by activating PDK1/Akt in granulosa cells of PCOS. J. Ovarian Res. 2021, 14, 152. [Google Scholar] [CrossRef] [PubMed]
- Salehi, R.; Mazier, H.L.; Nivet, A.L.; Reunov, A.A.; Lima, P.; Wang, Q.; Fiocco, A.; Isidoro, C.; Tsang, B.K. Ovarian mitochondrial dynamics and cell fate regulation in an androgen-induced rat model of polycystic ovarian syndrome. Sci. Rep. 2020, 10, 1021. [Google Scholar] [CrossRef]
- Urs, D.B.S.; Wu, W.H.; Komrskova, K.; Postlerova, P.; Lin, Y.F.; Tzeng, C.R.; Kao, S.H. Mitochondrial Function in Modulating Human Granulosa Cell Steroidogenesis and Female Fertility. Int. J. Mol. Sci. 2020, 21, 3592. [Google Scholar] [CrossRef]
- Mazloomi, S.; Farimani, M.S.; Tayebinia, H.; Karimi, J.; Amiri, I.; Abbasi, E.; Khodadadi, I. The Association of Mitochondrial Translocator Protein and Voltage-Dependent Anion Channel-1 in Granulosa Cells with Estradiol Levels and Presence of Immature Follicles in Polycystic Ovary Syndrome. J. Reprod. Infertil. 2022, 23, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, M.; Herraiz, S.; Cakiroglu, Y.; Garcia-Velasco, J.A.; Tiras, B.; Pacheco, A.; Rabadan, S.; Kohls, G.; Barrio, A.I.; Pellicer, A.; et al. Distress response in granulosa cells of women affected by PCOS with or without insulin resistance. Endocrine 2023, 79, 200–207. [Google Scholar] [CrossRef]
- Chappell, N.R.; Zhou, B.; Hosseinzadeh, P.; Schutt, A.; Gibbons, W.E.; Blesson, C.S. Hyperandrogenemia alters mitochondrial structure and function in the oocytes of obese mouse with polycystic ovary syndrome. F S Sci. 2021, 2, 101–112. [Google Scholar] [CrossRef]
- Chappell, N.R.; Zhou, B.; Schutt, A.K.; Gibbons, W.E.; Blesson, C.S. Prenatal androgen induced lean PCOS impairs mitochondria and mRNA profiles in oocytes. Endocr. Connect. 2020, 9, 261–270. [Google Scholar] [CrossRef]
- Babayev, E.; Seli, E. Oocyte mitochondrial function and reproduction. Curr. Opin. Obstet. Gynecol. 2015, 27, 175–181. [Google Scholar] [CrossRef]
- Liu, C.; Wang, M.; Yao, H.; Cui, M.; Gong, X.; Wang, L.; Sui, C.; Zhang, H. Inhibition of Oocyte Maturation by Follicular Extracellular Vesicles of Nonhyperandrogenic PCOS Patients Requiring IVF. J. Clin. Endocrinol. Metab. 2023, 108, 1394–1404. [Google Scholar] [CrossRef]
- Harris, S.E.; Maruthini, D.; Tang, T.; Balen, A.H.; Picton, H.M. Metabolism and karyotype analysis of oocytes from patients with polycystic ovary syndrome. Hum. Reprod. 2010, 25, 2305–2315. [Google Scholar] [CrossRef]
- Du, L.; Wang, Y.; Li, C.R.; Chen, L.J.; Cai, J.Y.; Xia, Z.R.; Zeng, W.T.; Wang, Z.B.; Chen, X.C.; Hu, F.; et al. Rat BAT xenotransplantation recovers the fertility and metabolic health of PCOS mice. J. Endocrinol. 2021, 248, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Lopes, I.M.; Baracat, M.C.; Simões Mde, J.; Simões, R.S.; Baracat, E.C.; Soares, J.M., Jr. Endometrium in women with polycystic ovary syndrome during the window of implantation. Rev. Assoc. Med. Bras. 2011, 57, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, J.; Hu, M.; Zhang, Y.; Cui, P.; Li, X.; Li, J.; Vestin, E.; Brännström, M.; Shao, L.R.; et al. Differential Expression Patterns of Glycolytic Enzymes and Mitochondria-Dependent Apoptosis in PCOS Patients with Endometrial Hyperplasia, an Early Hallmark of Endometrial Cancer, In Vivo and the Impact of Metformin In Vitro. Int. J. Biol. Sci. 2019, 15, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Palmerini, M.G.; Macchiarelli, G.; Cocciolone, D.; Mascitti, I.A.; Placidi, M.; Vergara, T.; Di Emidio, G.; Tatone, C. Modulating Morphological and Redox/Glycative Alterations in the PCOS Uterus: Effects of Carnitines in PCOS Mice. Biomedicines 2023, 11, 374. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, M.; Yang, F.; Zhang, Y.; Ma, S.; Zhang, D.; Wang, X.; Sferruzzi-Perri, A.N.; Wu, X.; Brännström, M.; et al. Increased uterine androgen receptor protein abundance results in implantation and mitochondrial defects in pregnant rats with hyperandrogenism and insulin resistance. J. Mol. Med. 2021, 99, 1427–1446. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhang, Y.; Guo, X.; Jia, W.; Liu, G.; Zhang, J.; Li, J.; Cui, P.; Sferruzzi-Perri, A.N.; Han, Y.; et al. Hyperandrogenism and insulin resistance induce gravid uterine defects in association with mitochondrial dysfunction and aberrant reactive oxygen species production. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E794–E809. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, W.; Xu, H.; Hu, M.; Guo, X.; Jia, W.; Liu, G.; Li, J.; Cui, P.; Lager, S.; et al. Hyperandrogenism and insulin resistance-induced fetal loss: Evidence for placental mitochondrial abnormalities and elevated reactive oxygen species production in pregnant rats that mimic the clinical features of polycystic ovary syndrome. J. Physiol. 2019, 597, 3927–3950. [Google Scholar] [CrossRef]
- Hu, M.; Zhang, Y.; Ma, S.; Li, J.; Wang, X.; Liang, M.; Sferruzzi-Perri, A.N.; Wu, X.; Ma, H.; Brännström, M.; et al. Suppression of uterine and placental ferroptosis by N-acetylcysteine in a rat model of polycystic ovary syndrome. Mol. Hum. Reprod. 2021, 27, gaab067. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, M.; Jia, W.; Liu, G.; Zhang, J.; Wang, B.; Li, J.; Cui, P.; Li, X.; Lager, S.; et al. Hyperandrogenism and insulin resistance modulate gravid uterine and placental ferroptosis in PCOS-like rats. J. Endocrinol. 2020, 246, 247–263. [Google Scholar] [CrossRef]
- Siemers, K.M.; Baack, M.L. The importance of placental lipid metabolism across gestation in obese and non-obese pregnancies. Clin. Sci. 2023, 137, 31–34. [Google Scholar] [CrossRef]
- Kirtana, A.; Seetharaman, B. Comprehending the Role of Endocrine Disruptors in Inducing Epigenetic Toxicity. Endocr. Metab. Immune Disord. Drug Targets 2022, 22, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Szczuko, M.; Kikut, J.; Komorniak, N.; Bilicki, J.; Celewicz, Z.; Ziętek, M. The Role of Arachidonic and Linoleic Acid Derivatives in Pathological Pregnancies and the Human Reproduction Process. Int. J. Mol. Sci. 2020, 21, 9628. [Google Scholar] [CrossRef]
- Sartori, C.; Lazzeroni, P.; Merli, S.; Patianna, V.D.; Viaroli, F.; Cirillo, F.; Amarri, S.; Street, M.E. From Placenta to Polycystic Ovarian Syndrome: The Role of Adipokines. Mediat. Inflamm. 2016, 2016, 4981916. [Google Scholar] [CrossRef]
- Sun, M.; Sun, B.; Qiao, S.; Feng, X.; Li, Y.; Zhang, S.; Lin, Y.; Hou, L. Elevated maternal androgen is associated with dysfunctional placenta and lipid disorder in newborns of mothers with polycystic ovary syndrome. Fertil. Steril. 2020, 113, 1275–1285.e1272. [Google Scholar] [CrossRef] [PubMed]
- Medina, G.; Maliqueo, M.; Crisosto, N.; Echiburú, B.; Sir-Petermann, T.; Lara, H.E. Metformin increases norepinephrine transporter expression in placenta of patients with polycystic ovary syndrome. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3482–3489. [Google Scholar] [PubMed]
- Kelley, A.S.; Smith, Y.R.; Padmanabhan, V. A Narrative Review of Placental Contribution to Adverse Pregnancy Outcomes in Women With Polycystic Ovary Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 5299–5315. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.V.; Govatati, S.; Deenadayal, M.; Shivaji, S.; Bhanoori, M. Polymorphisms in the TFAM and PGC1-α genes and their association with polycystic ovary syndrome among South Indian women. Gene 2018, 641, 129–136. [Google Scholar] [CrossRef]
- Ye, M.; Hu, B.; Shi, W.; Guo, F.; Xu, C.; Li, S. Mitochondrial DNA 4977 bp Deletion in Peripheral Blood Is Associated With Polycystic Ovary Syndrome. Front. Endocrinol. 2021, 12, 675581. [Google Scholar] [CrossRef]
- Saeed, N.; Hamzah, I.H.; Al-Gharrawi, S.A.R. Polycystic ovary syndrome dependency on mtDNA mutation; copy Number and its association with insulin resistance. BMC Res. Notes 2019, 12, 455. [Google Scholar] [CrossRef]
- Reddy, T.V.; Govatati, S.; Deenadayal, M.; Sisinthy, S.; Bhanoori, M. Impact of mitochondrial DNA copy number and displacement loop alterations on polycystic ovary syndrome risk in south Indian women. Mitochondrion 2019, 44, 35–40. [Google Scholar] [CrossRef]
- Lee, S.H.; Chung, D.J.; Lee, H.S.; Kim, T.J.; Kim, M.H.; Jeong, H.J.; Im, J.A.; Lee, D.C.; Lee, J.W. Mitochondrial DNA copy number in peripheral blood in polycystic ovary syndrome. Metabolism 2011, 60, 1677–1682. [Google Scholar] [CrossRef]
- Yang, P.K.; Chou, C.H.; Chang, C.H.; Chen, S.U.; Ho, H.N.; Chen, M.J. Changes in peripheral mitochondrial DNA copy number in metformin-treated women with polycystic ovary syndrome: A longitudinal study. Reprod. Biol. Endocrinol. 2020, 18, 69. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-P.; Wang, Y.; Mao, W.-W.; Zhang, X.-W.; Qiao, J.; Zhang, Q.-F. Single Nucleotide Polymorphisms (SNPs) and Variable Number Tandem Repeats (VNTRs) in mtDNA D-loop and CO II- tRNALys Intergenic Region with PCOS. J. Reprod. Contracept. 2011, 22, 129–138. [Google Scholar] [CrossRef]
- Zhuo, G.; Ding, Y.; Feng, G.; Yu, L.; Jiang, Y. Analysis of mitochondrial DNA sequence variants in patients with polycystic ovary syndrome. Arch. Gynecol. Obstet. 2012, 286, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, G.; Feng, G.; Leng, J.; Yu, L.; Jiang, Y. 9-bp Deletion Homoplasmy in Women with Polycystic Ovary Syndrome Revealed by Mitochondrial Genome-Mutation Screen. Biochem. Genet. 2010, 48, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Xia, B.-H.; Zhang, C.-J.; Zhuo, G.-C. Mitochondrial tRNALeu(UUR) C3275T, tRNAGln T4363C and tRNALys A8343G mutations may be associated with PCOS and metabolic syndrome. Gene 2018, 642, 299–306. [Google Scholar] [CrossRef]
- Ding, Y.; Xia, B.-H.; Zhang, C.-J.; Zhuo, G.-C. Mutations in mitochondrial tRNA genes may be related to insulin resistance in women with polycystic ovary syndrome. Am. J. Transl. Res. 2017, 9, 2984–2996. [Google Scholar] [PubMed]
- Ding, Y.U.; Zhuo, G.; Zhang, C.; Leng, J. Point mutation in mitochondrial tRNA gene is associated with polycystic ovary syndrome and insulin resistance. Mol. Med. Rep. 2016, 13, 3169–3172. [Google Scholar] [CrossRef]
- He, S.; Ji, D.; Liu, Y.; Deng, X.; Zou, W.; Liang, D.; Du, Y.; Zong, K.; Jiang, T.; Li, M.; et al. Polymorphisms of mtDNA in the D-loop region moderate the associations of BMI with HOMA-IR and HOMA-β among women with polycystic ovary syndrome: A cross-sectional study. J. Assist. Reprod. Genet. 2023, 40, 1983–1993. [Google Scholar] [CrossRef]
- Liu, H.L.; Li, T.T.; Yu, A.Q.; Li, J.; Li, X.; Li, L.; Kaifu, T. Plasma level of peroxiredoxin 3 in patients with polycystic ovarian syndrome. BMC Endocr. Disord. 2019, 19, 32. [Google Scholar] [CrossRef]
- Ding, Y.; Jiang, Z.; Xia, B.; Zhang, L.; Zhang, C.; Leng, J. Mitochondria-targeted antioxidant therapy for an animal model of PCOS-IR. Int. J. Mol. Med. 2019, 43, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Merviel, P.; James, P.; Bouée, S.; Le Guillou, M.; Rince, C.; Nachtergaele, C.; Kerlan, V. Impact of myo-inositol treatment in women with polycystic ovary syndrome in assisted reproductive technologies. Reprod. Health 2021, 18, 13. [Google Scholar] [CrossRef] [PubMed]


| Mitochondrial Function | Effect of PCOS Condition on Mitochondrial Function | Treatment Timeframe | Method | Therapeutic Intervention | References |
|---|---|---|---|---|---|
| Biogenesis | Decreased PGC-1α | GD 7.5–GD 13.5 | qPCR | [80] | |
| Decreased TFAM | GD 7.5–GD 13.5 | qPCR | [80] | ||
| Decreased NRF1 | GD 7.5–GD 13.5 | qPCR | [80] | ||
| Mitochondrial Genome | Decreased mtDNA copy number | GD 7.5–GD 13.5 | qPCR | [80] | |
| Ultrastructure | Mitochondrial blebbing | GD 7.5–GD 13.5 and GD 14.5 | TEM | [80,81,82] | |
| Lack of tubular cristae | GD 7.5–GD 13.5 or GD 14.5 | TEM | [81,82] | ||
| Decreased intracristal dilatation | GD 7.5–GD 13.5 | TEM | [80] | ||
| Morphology changes limited to junctional zone; little change in the labyrinth zone | GD 7.5–GD 13.5 or GD 14.5 | TEM | High-dose NAC abolished damaged morphology in junctional zone but had no effect in labyrinth zone | [81,82] | |
| Metabolism | No change in Complex III expression | GD 7.5–GD 14.5 | WB | Low-dose NAC decreased Complex I abundance | [81] |
| Decreased VDAC | GD 7.5–GD 14.5 | WB | High-dose NAC increased VDAC abundance | [80,81] | |
| Dynamics | Decreased MFN1 (Fusion) | GD 7.5–GD 13.5 | qPCR | [80] | |
| Decreased DRP1 (Fission) | GD 7.5–GD 13.5 | qPCR | [80] | ||
| ROS and Repair | Increased ROS | GD 7.5–GD 13.5 | OxiSelect In Vitro, ROS/RNS assay | [80] | |
| Increased MDA | GD 7.5–GD 13.5 | MDA ELISA | [80] | ||
| Increased cytosolic NRF2 and decreased nuclear NRF2 in basal zone | GD 7.5–GD 13.5 | WB, IHC | [80] | ||
| Reduced phosphorylated SOD1 and p-SOD1:SOD1 ratio | GD 7.5–GD 13.5 | WB | [80] | ||
| No difference in SOD2 abundance | GD 7.5–GD 13.5 | WB | [80] | ||
| No difference in GPX4 abundance | GD 7.5–GD 13.5 | WB | [82] | ||
| Less GPX4 in junctional and labyrinth zones | GD 7.5–GD 13.5 | IHC | [82] | ||
| No GPX4 in nuclei of spongiotrophoblasts, cytotrophoblasts, and synctiotrophoblasts | GD 7.5–GD 13.5 | IHC | [82] | ||
| No difference in glutathione | GD 7.5–GD 13.5 | Glutathione/glutathione + glutathione disulfide assay | [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siemers, K.M.; Klein, A.K.; Baack, M.L. Mitochondrial Dysfunction in PCOS: Insights into Reproductive Organ Pathophysiology. Int. J. Mol. Sci. 2023, 24, 13123. https://doi.org/10.3390/ijms241713123
Siemers KM, Klein AK, Baack ML. Mitochondrial Dysfunction in PCOS: Insights into Reproductive Organ Pathophysiology. International Journal of Molecular Sciences. 2023; 24(17):13123. https://doi.org/10.3390/ijms241713123
Chicago/Turabian StyleSiemers, Kyle M., Abigail K. Klein, and Michelle L. Baack. 2023. "Mitochondrial Dysfunction in PCOS: Insights into Reproductive Organ Pathophysiology" International Journal of Molecular Sciences 24, no. 17: 13123. https://doi.org/10.3390/ijms241713123