
ATAD3A: A Key Regulator of Mitochondria-Associated Diseases

Abstract
:1. Introduction
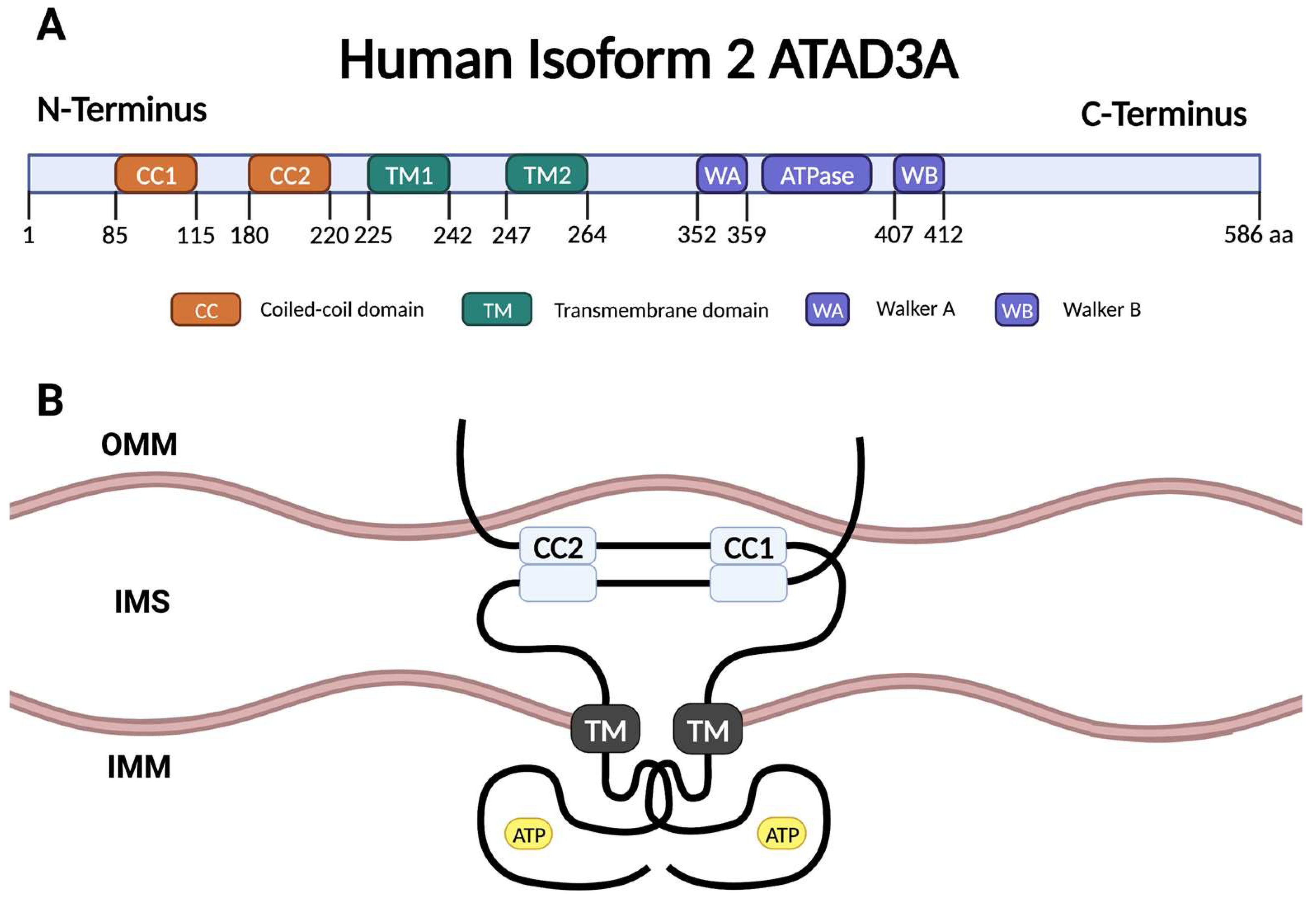
2. ATAD3A Structure
3. ATAD3A and Mitochondrial Structure
3.1. ATAD3A and mtDNA
3.2. ATAD3A and Mitochondrial Cristae Morphology
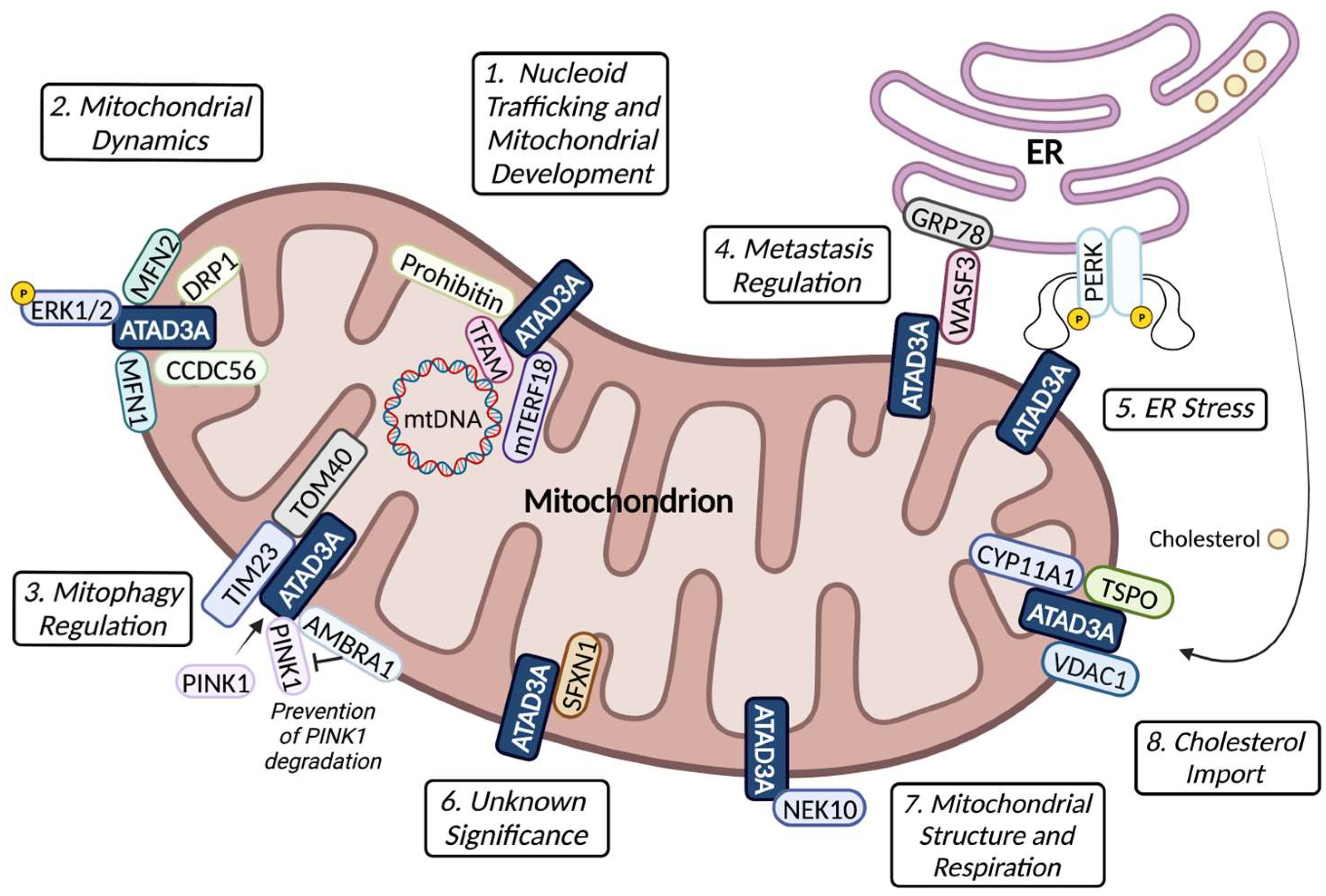
4. ATAD3A–Protein Interactions and Function
5. ATAD3A and Cholesterol Homeostasis
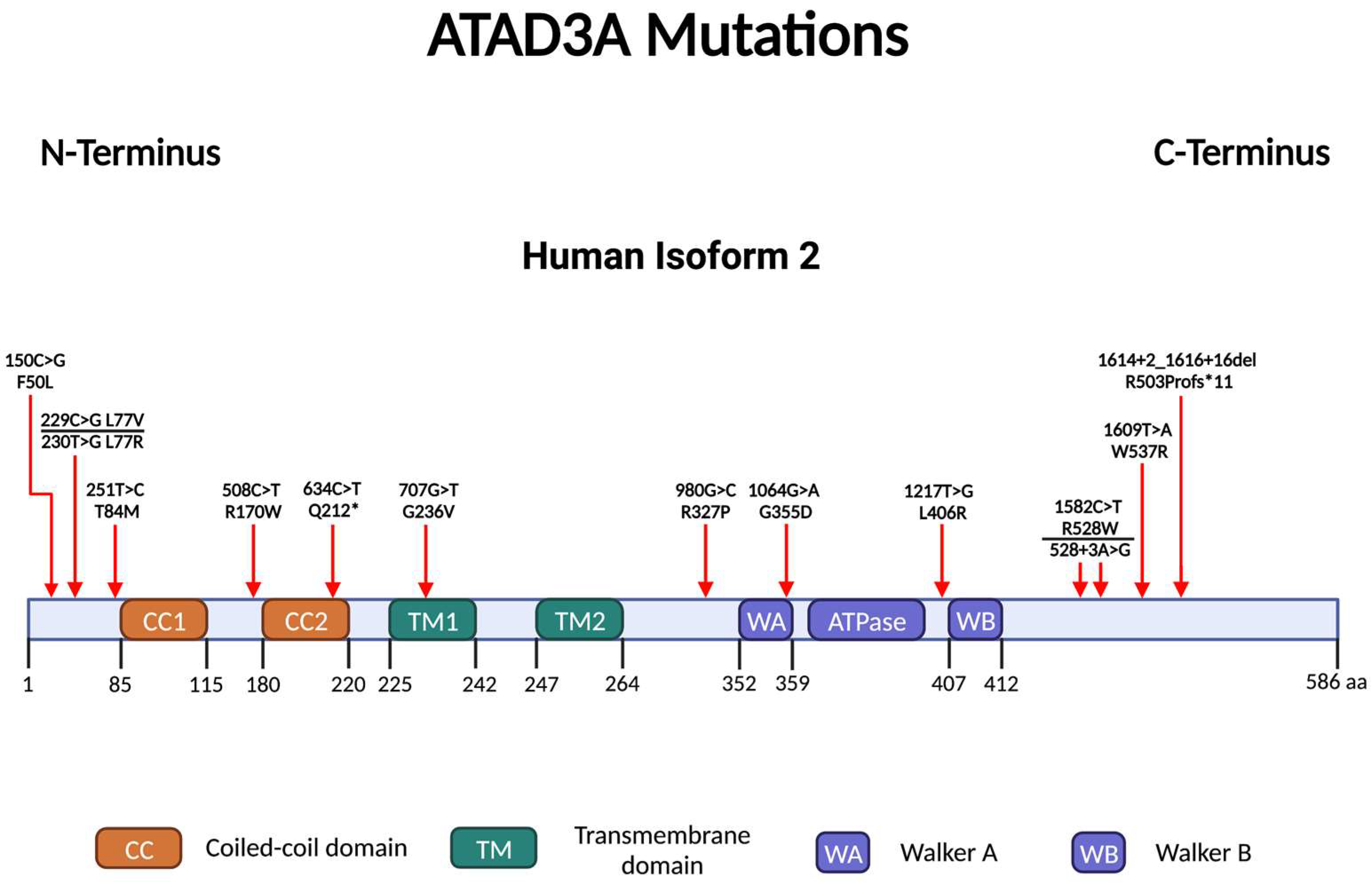
6. ATAD3A Mutations, Mitochondrial Abnormalities and Neurological Diseases
6.1. ATAD3A Point Mutations
6.1.1. Walker A/B Motif
6.1.2. C-Terminus
6.1.3. N-Terminus
6.2. Copy Number Variants
7. ATAD3A and Mitochondria in Cancer
7.1. ATAD3A Expression and Cancer
7.2. ATAD3A, Mitochondria and Drug Resistance
8. Conclusions
9. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATAD3A | ATPase family AAA-domain-containing protein 3A |
| ATP | Adenosine triphosphate |
| AMBRA1 | Beclin-1-regulated autophagy |
| CCDC56 | Coiled-coil domain-containing protein 56 |
| cGAS | Cyclic GMP-AMP synthase |
| coIP-MS | Co-immunoprecipitation followed by shotgun mass spectrometry |
| CYP11A1 | Cytochrome P450 family 11 subfamily A member 1 |
| D-loop | Displacement loop |
| DRP1 | Dynamin-related protein 1 |
| GBM | Glioblastoma multiforme |
| GRP78 | Glucose-regulated protein |
| HCC | Hepatocellular carcinoma |
| HNSCC | Head and neck squamous cell carcinomas |
| IMM | Inner mitochondrial membrane |
| ISG | Interferon-stimulated gene |
| LDL | Low-density lipoprotein |
| LDs | Lipid droplets |
| MAMs | Mitochondrial-associated membranes |
| MFN1 | Fusion proteins mitofusin-1 |
| mtDNA | Mitochondrial DNA |
| mTERF | Mitochondrial transcription termination factor |
| mTOR | Mammalian target of rapamycin |
| NEK10 | NIMA-related kinase 10 |
| OMM | Outer mitochondrial membrane |
| PERK | Protein-kinase-R-like endoplasmic reticulum kinase |
| SFXN1 | Sideroflexin 1 |
| SREBP-1c | Sterol regulatory element binding protein 1c |
| STING | Stimulator of interferon genes |
| TFAM | Mitochondrial transcription factor A |
| TIM23 | Translocase of inner mitochondrial membrane 23 |
| TOM40 | Outer mitochondrial membrane 40 |
| TSPO | Translocator protein |
| VDAC | Voltage-dependent anion channel |
| WASF3 | Wiskott-Aldrich syndrome protein family member 3 |
References
- White, S.R.; Lauring, B. AAA+ ATPases: Achieving diversity of function with conserved machinery. Traffic 2007, 8, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Ogura, T.; Wilkinson, A.J. AAA+ superfamily ATPases: Common structure–diverse function. Genes Cells 2001, 6, 575–597. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Mao, C.C.; Reyes, A.; Sembongi, H.; Di Re, M.; Granycome, C.; Clippingdale, A.B.; Fearnley, I.M.; Harbour, M.; Robinson, A.J.; et al. The AAA+ protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J. Cell Biol. 2007, 176, 141–146. [Google Scholar] [CrossRef]
- Issop, L.; Fan, J.; Lee, S.; Rone, M.B.; Basu, K.; Mui, J.; Papadopoulos, V. Mitochondria-associated membrane formation in hormone-stimulated Leydig cell steroidogenesis: Role of ATAD3. Endocrinology 2015, 156, 334–345. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Ren, X.; Li, H.; Shull, A.; Kim, J.; Cowell, J.K. Mitochondrial ATAD3A combines with GRP78 to regulate the WASF3 metastasis-promoting protein. Oncogene 2016, 35, 333–343. [Google Scholar] [CrossRef] [Green Version]
- Peralta, S.; Goffart, S.; Williams, S.L.; Diaz, F.; Garcia, S.; Nissanka, N.; Area-Gomez, E.; Pohjoismäki, J.; Moraes, C.T. ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J. Cell Sci. 2018, 131, jcs217075. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Yuan, X.; Liu, L.; Zhang, M.; Qu, B.; Zhen, Z.; Gao, X. Mitochondrial ATAD3A regulates milk biosynthesis and proliferation of mammary epithelial cells from dairy cow via the mTOR pathway. Cell Biol. Int. 2018, 42, 533–542. [Google Scholar] [CrossRef]
- Li, S.; Lamarche, F.; Charton, R.; Delphin, C.; Gires, O.; Hubstenberger, A.; Schlattner, U.; Rousseau, D. Expression analysis of ATAD3 isoforms in rodent and human cell lines and tissues. Gene 2014, 535, 60–69. [Google Scholar] [CrossRef]
- Lang, L.; Loveless, R.; Teng, Y. Emerging Links between Control of Mitochondrial Protein ATAD3A and Cancer. Int. J. Mol. Sci. 2020, 21, 7917. [Google Scholar] [CrossRef]
- Gilquin, B.; Taillebourg, E.; Cherradi, N.; Hubstenberger, A.; Gay, O.; Merle, N.; Assard, N.; Fauvarque, M.O.; Tomohiro, S.; Kuge, O.; et al. The AAA+ ATPase ATAD3A controls mitochondrial dynamics at the interface of the inner and outer membranes. Mol. Cell. Biol. 2010, 30, 1984–1996. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Lang, L.; Shay, C. ATAD3A on the Path to Cancer. Adv. Exp. Med. Biol. 2019, 1134, 259–269. [Google Scholar]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid. Med. Cell. Longev. 2017, 2017, 8060949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, S.; Kadowaki, T.; Kumaki, N.; Tang, X.; Tokuda, Y.; Yoshimura, S.; Takekoshi, S.; Osamura, R.Y. Analysis of gene alterations of mitochondrial DNA D-loop regions to determine breast cancer clonality. Br. J. Cancer 2012, 107, 2016–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.B.; Yang, Y.H.; Lee, W.C.; Liou, C.W.; Lin, T.K.; Chung, Y.H.; Chuang, L.Y.; Yang, C.H.; Chang, H.W. Sequence-based polymorphisms in the mitochondrial D-loop and potential SNP predictors for chronic dialysis. PLoS ONE 2012, 7, e41125. [Google Scholar] [CrossRef] [Green Version]
- Iborra, F.J.; Kimura, H.; Cook, P.R. The functional organization of mitochondrial genomes in human cells. BMC Biol. 2004, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, T.; Ban-Ishihara, R.; Ota, A.; Ishihara, N. Mitochondrial nucleoid trafficking regulated by the inner-membrane AAA-ATPase ATAD3A modulates respiratory complex formation. Proc. Natl. Acad. Sci. USA 2022, 119, e2210730119. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Cooper, H.M.; Reyes, A.; Di Re, M.; Sembongi, H.; Litwin, T.R.; Gao, J.; Neuman, K.C.; Fearnley, I.M.; Spinazzola, A.; et al. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 2012, 40, 6109–6121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Schulz, V.; Brings, L.; Schoeller, T.; Kühn, K.; Vierling, E. mTERF18 and ATAD3 are required for mitochondrial nucleoid structure and their disruption confers heat tolerance in Arabidopsis thaliana. New Phytol. 2021, 232, 2026–2042. [Google Scholar] [CrossRef]
- Bruser, C.; Keller-Findeisen, J.; Jakobs, S. The TFAM-to-mtDNA ratio defines inner-cellular nucleoid populations with distinct activity levels. Cell Rep. 2021, 37, 110000. [Google Scholar] [CrossRef]
- Kasashima, K.; Sumitani, M.; Satoh, M.; Endo, H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp. Cell Res. 2008, 314, 988–996. [Google Scholar] [CrossRef]
- Frazier, A.E.; Compton, A.G.; Kishita, Y.; Hock, D.H.; Welch, A.E.; Amarasekera, S.S.; Rius, R.; Formosa, L.E.; Imai-Okazaki, A.; Francis, D.; et al. Fatal perinatal mitochondrial cardiac failure caused by recurrent de novo duplications in the ATAD3 locus. Med 2021, 2, 49–73. [Google Scholar] [CrossRef]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; Di Rago, J.P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Shu, L.; Huang, X.; Yu, J.; Li, L.; Gong, L.; Yang, M.; Wu, Z.; Gao, Z.; Zhao, Y.; et al. OPA1 and MICOS Regulate mitochondrial crista dynamics and formation. Cell Death Dis. 2020, 11, 940. [Google Scholar] [CrossRef]
- Chen, L.; Li, Y.; Sottas, C.; Lazaris, A.; Petrillo, S.K.; Metrakos, P.; Li, L.; Ishida, Y.; Saito, T.; Garza, S.; et al. Loss of mitochondrial ATPase ATAD3A contributes to nonalcoholic fatty liver disease through accumulation of lipids and damaged mitochondria. J. Biol. Chem. 2022, 298, 102008. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, X.; Hu, D.; Prosdocimo, D.A.; Hoppel, C.; Jain, M.K.; Ramachandran, R.; Qi, X. ATAD3A oligomerization causes neurodegeneration by coupling mitochondrial fragmentation and bioenergetics defects. Nat. Commun. 2019, 10, 1371. [Google Scholar] [CrossRef] [Green Version]
- Goller, T.; Seibold, U.K.; Kremmer, E.; Voos, W.; Kolanus, W. Atad3 function is essential for early post-implantation development in the mouse. PLoS ONE 2013, 8, e54799. [Google Scholar] [CrossRef] [Green Version]
- Ban-Ishihara, R.; Tomohiro-Takamiya, S.; Tani, M.; Baudier, J.; Ishihara, N.; Kuge, O. COX assembly factor ccdc56 regulates mitochondrial morphology by affecting mitochondrial recruitment of Drp1. FEBS Lett. 2015, 589 Pt B, 3126–3132. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.Y.; Novak, K.; Haqqi, T.M. ERK1/2-mediated activation of DRP1 regulates mitochondrial dynamics and apoptosis in chondrocytes. Osteoarthr. Cartil. 2022, 30, 315–328. [Google Scholar] [CrossRef]
- Lang, L.; Loveless, R.; Dou, J.; Lam, T.; Chen, A.; Wang, F.; Sun, L.; Juarez, J.; Qin, Z.S.; Saba, N.F.; et al. ATAD3A mediates activation of RAS-independent mitochondrial ERK1/2 signaling, favoring head and neck cancer development. J. Exp. Clin. Cancer Res. 2022, 41, 43. [Google Scholar] [CrossRef]
- Jin, G.; Xu, C.; Zhang, X.; Long, J.; Rezaeian, A.H.; Liu, C.; Furth, M.E.; Kridel, S.; Pasche, B.; Bian, X.W.; et al. Atad3a suppresses Pink1-dependent mitophagy to maintain homeostasis of hematopoietic progenitor cells. Nat. Immunol. 2018, 19, 29–40. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Romagnoli, A.; Ciccosanti, F.; Refolo, G.; Consalvi, V.; Arena, G.; Valente, E.M.; Piacentini, M.; Fimia, G.M. AMBRA1 regulates mitophagy by interacting with ATAD3A and promoting PINK1 stability. Autophagy 2022, 18, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Loveless, R.; Teng, Y. Targeting WASF3 Signaling in Metastatic Cancer. Int. J. Mol. Sci. 2021, 22, 836. [Google Scholar] [CrossRef]
- Hughes, D.T.; Brar, K.K.; Morris, J.; Subramanian, K.; Krishna, S.; Gao, F.; Rieder, L.S.; Freeman, J.L.; Smith, H.L.; Jukes-Jones, R.; et al. PERK-ATAD3A interaction protects mitochondrial proteins synthesis during ER stress. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tifoun, N.; Bekhouche, M.; De las Heras, J.M.; Guillaume, A.; Bouleau, S.; Guénal, I.; Mignotte, B.; Le Floch, N. A High-Throughput Search for SFXN1 Physical Partners Led to the Identification of ATAD3, HSD10 and TIM50. Biology 2022, 11, 1298. [Google Scholar] [CrossRef] [PubMed]
- Peres de Oliveira, A.; Basei, F.L.; Slepicka, P.F.; de Castro Ferezin, C.; Melo-Hanchuk, T.D.; de Souza, E.E.; Lima, T.I.; Dos Santos, V.T.; Mendes, D.; Silveira, L.R.; et al. NEK10 interactome and depletion reveal new roles in mitochondria. Proteome Sci. 2020, 18, 4. [Google Scholar] [CrossRef] [PubMed]
- Rone, M.B.; Midzak, A.S.; Issop, L.; Rammouz, G.; Jagannathan, S.; Fan, J.; Ye, X.; Blonder, J.; Veenstra, T.; Papadopoulos, V. Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol. Endocrinol. 2012, 26, 1868–1882. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Hu, D.; Wang, R.; Sun, X.; Ropelewski, P.; Hubler, Z.; Lundberg, K.; Wang, Q.; Adams, D.J.; Xu, R.; et al. ATAD3A oligomerization promotes neuropathology and cognitive deficits in Alzheimer’s disease models. Nat. Commun. 2022, 13, 1121. [Google Scholar] [CrossRef]
- Vance, J.E. Cellular itinerary of LDL cholesterol. Proc. Natl. Acad. Sci. USA 2022, 119, e2122584119. [Google Scholar] [CrossRef]
- Cooper, H.M.; Yang, Y.; Ylikallio, E.; Khairullin, R.; Woldegebriel, R.; Lin, K.L.; Euro, L.; Palin, E.; Wolf, A.; Trokovic, R.; et al. ATPase-deficient mitochondrial inner membrane protein ATAD3A disturbs mitochondrial dynamics in dominant hereditary spastic paraplegia. Hum. Mol. Genet. 2017, 26, 1432–1443. [Google Scholar] [CrossRef]
- Yap, Z.Y.; Park, Y.H.; Wortmann, S.B.; Gunning, A.C.; Ezer, S.; Lee, S.; Duraine, L.; Wilichowski, E.; Wilson, K.; Mayr, J.A.; et al. Functional interpretation of ATAD3A variants in neuro-mitochondrial phenotypes. Genome Med. 2021, 13, 55. [Google Scholar] [CrossRef]
- Harel, T.; Yoon, W.H.; Garone, C.; Gu, S.; Coban-Akdemir, Z.; Eldomery, M.K.; Posey, J.E.; Jhangiani, S.N.; Rosenfeld, J.A.; Cho, M.T.; et al. Recurrent De Novo and Biallelic Variation of ATAD3A, Encoding a Mitochondrial Membrane Protein, Results in Distinct Neurological Syndromes. Am. J. Hum. Genet. 2016, 99, 831–845. [Google Scholar] [CrossRef] [Green Version]
- Hanes, I.; McMillan, H.J.; Ito, Y.; Kernohan, K.D.; Lazier, J.; Lines, M.A.; Dyment, D.A. A splice variant in ATAD3A expands the clinical and genetic spectrum of Harel-Yoon syndrome. Neurol. Genet. 2020, 6, e452. [Google Scholar] [CrossRef]
- Al Madhoun, A.; Alnaser, F.; Melhem, M.; Nizam, R.; Al-Dabbous, T.; Al-Mulla, F. Ketogenic diet attenuates cerebellar atrophy progression in a subject with a biallelic variant at the ATAD3A locus. Appl. Clin. Genet. 2019, 12, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Tawfik, C.A.; Zaitoun, R.; Farag, A.A. Harel Yoon syndrome: A novel mutation in ATAD3A gene and expansion of the clinical spectrum. Ophthalmic Genet. 2023, 44, 226–233. [Google Scholar] [CrossRef]
- Peralta, S.; González-Quintana, A.; Ybarra, M.; Delmiro, A.; Pérez-Pérez, R.; Docampo, J.; Arenas, J.; Blázquez, A.; Ugalde, C.; Martín, M.A. Novel ATAD3A recessive mutation associated to fatal cerebellar hypoplasia with multiorgan involvement and mitochondrial structural abnormalities. Mol. Genet. Metab. 2019, 128, 452–462. [Google Scholar] [CrossRef]
- Dorison, N.; Gaignard, P.; Bayot, A.; Gelot, A.; Becker, P.H.; Fourati, S.; Lebigot, E.; Charles, P.; Wai, T.; Therond, P.; et al. Mitochondrial dysfunction caused by novel ATAD3A mutations. Mol. Genet. Metab. 2020, 131, 107–113. [Google Scholar] [CrossRef]
- Skopkova, M.; Stufkova, H.; Rambani, V.; Stranecky, V.; Brennerova, K.; Kolnikova, M.; Pietrzykova, M.; Karhanek, M.; Noskova, L.; Tesarova, M.; et al. ATAD3A-related pontocerebellar hypoplasia: New patients and insights into phenotypic variability. Orphanet J. Rare Dis. 2023, 18, 92. [Google Scholar] [CrossRef]
- Peeters-Scholte, C.M.; Adama van Scheltema, P.N.; Klumper, F.J.; Everwijn, S.M.; Koopmans, M.; Hoffer, M.J.; Koopmann, T.T.; Ruivenkamp, C.A.; Steggerda, S.J.; Van Der Knaap, M.S.; et al. Genotype-phenotype correlation in ATAD3A deletions: Not just of scientific relevance. Brain 2017, 140, e66. [Google Scholar] [CrossRef]
- Desai, R.; Frazier, A.E.; Durigon, R.; Patel, H.; Jones, A.W.; Dalla Rosa, I.; Lake, N.J.; Compton, A.G.; Mountford, H.S.; Tucker, E.J.; et al. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 2017, 140, 1595–1610. [Google Scholar] [CrossRef] [Green Version]
- Gunning, A.C.; Strucinska, K.; Oreja, M.M.; Parrish, A.; Caswell, R.; Stals, K.L.; Durigon, R.; Durlacher-Betzer, K.; Cunningham, M.H.; Grochowski, C.M.; et al. Recurrent De Novo NAHR Reciprocal Duplications in the ATAD3 Gene Cluster Cause a Neurogenetic Trait with Perturbed Cholesterol and Mitochondrial Metabolism. Am. J. Hum. Genet. 2020, 106, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Azova, S.; Rajabi, F.; Modi, B.P.; Mansfield, L.; Jonas, M.M.; Drobysheva, A.; Boyd, T.K.; Wassner, A.J.; Smith, J.R. Graves’ disease in a five-month-old boy with an unusual treatment course. J. Pediatr. Endocrinol. Metab. JPEM 2021, 34, 401–406. [Google Scholar] [CrossRef]
- Ebihara, T.; Nagatomo, T.; Sugiyama, Y.; Tsuruoka, T.; Osone, Y.; Shimura, M.; Tajika, M.; Ichimoto, K.; Naruke, Y.; Akiyama, N.; et al. Severe spinal cord hypoplasia due to a novel ATAD3A compound heterozygous deletion. Mol. Genet. Metab. Rep. 2022, 33, 100912. [Google Scholar] [CrossRef]
- Lepelley, A.; Della Mina, E.; Van Nieuwenhove, E.; Waumans, L.; Fraitag, S.; Rice, G.I.; Dhir, A.; Frémond, M.L.; Rodero, M.P.; Seabra, L.; et al. Enhanced cGAS-STING-dependent interferon signaling associated with mutations in ATAD3A. J. Exp. Med. 2021, 218, e20201560. [Google Scholar] [CrossRef]
- Chen, Y.; Rong, S.; Luo, H.; Huang, B.; Hu, F.; Chen, M.; Li, C. Ketogenic Diet Attenuates Refractory Epilepsy of Harel-Yoon Syndrome with ATAD3A Variants: A Case Report and Review of Literature. Pediatr. Neurol. 2023, 143, 79–83. [Google Scholar] [CrossRef]
- Davies, T.F.; Andersen, S.; Latif, R.; Nagayama, Y.; Barbesino, G.; Brito, M.; Eckstein, A.K.; Stagnaro-Green, A.; Kahaly, G.J. Graves’ disease. Nat. Rev. Dis. Primers 2020, 6, 52. [Google Scholar] [CrossRef]
- Zhao, W.; Zeng, H.; Zhang, X.; Liu, F.; Pan, J.; Zhao, J.; Zhao, J.; Li, L.; Bao, Y.; Liu, F.; et al. A high thyroid stimulating hormone level is associated with diabetic peripheral neuropathy in type 2 diabetes patients. Diabetes Res. Clin. Pract. 2016, 115, 122–129. [Google Scholar] [CrossRef]
- Joy Mathew, C.; Jose, M.T.; Elshaikh, A.O.; Shah, L.; Lee, R.; Cancarevic, I. Is Hyperthyroidism a Possible Etiology of Early Onset Dementia? Cureus 2020, 12, e10603. [Google Scholar] [CrossRef]
- Zhang, T.; Nie, Y.; Gu, J.; Cai, K.; Chen, X.; Li, H.; Wang, J. Identification of Mitochondrial-Related Prognostic Biomarkers Associated with Primary Bile Acid Biosynthesis and Tumor Microenvironment of Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 587479. [Google Scholar] [CrossRef]
- Liu, X.; Li, G.; Ai, L.; Ye, Q.; Yu, T.; Yang, B. Prognostic value of ATAD3 gene cluster expression in hepatocellular carcinoma. Oncol. Lett. 2019, 18, 1304–1310. [Google Scholar] [CrossRef]
- Xie, Q.; Wang, D.; Luo, X.; Li, Z.; Hu, A.; Yang, H.; Tang, J.; Gao, P.; Sun, T.; Kong, L. Proteome profiling of formalin-fixed, paraffin-embedded lung adenocarcinoma tissues using a tandem mass tag-based quantitative proteomics approach. Oncol. Lett. 2021, 22, 706. [Google Scholar] [CrossRef]
- Li, Q.; Chu, Y.; Li, S.; Yu, L.; Deng, H.; Liao, C.; Liao, X.; Yang, C.; Qi, M.; Cheng, J.; et al. The oncoprotein MUC1 facilitates breast cancer progression by promoting Pink1-dependent mitophagy via ATAD3A destabilization. Cell Death Dis. 2022, 13, 899. [Google Scholar] [CrossRef]
- Huang, K.H.; Chow, K.C.; Chang, H.W.; Lin, T.Y.; Lee, M.C. ATPase family AAA domain containing 3A is an anti-apoptotic factor and a secretion regulator of PSA in prostate cancer. Int. J. Mol. Med. 2011, 28, 9–15. [Google Scholar] [CrossRef]
- Fang, H.Y.; Chang, C.L.; Hsu, S.H.; Huang, C.Y.; Chiang, S.F.; Chiou, S.H.; Huang, C.H.; Hsiao, Y.T.; Lin, T.Y.; Chiang, I.P.; et al. ATPase family AAA domain-containing 3A is a novel anti-apoptotic factor in lung adenocarcinoma cells. J. Cell Sci. 2010, 123 Pt 7, 1171–1180. [Google Scholar] [CrossRef] [Green Version]
- You, W.C.; Chiou, S.H.; Huang, C.Y.; Chiang, S.F.; Yang, C.L.; Sudhakar, J.N.; Lin, T.Y.; Chiang, I.P.; Shen, C.C.; Cheng, W.Y.; et al. Mitochondrial protein ATPase family, AAA domain containing 3A correlates with radioresistance in glioblastoma. Neuro-Oncology 2013, 15, 1342–1352. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.C.; Hung, Y.C.; Lin, T.Y.; Chang, H.W.; Chiang, I.; Chen, Y.Y.; Chow, K.C. Human papillomavirus infection and expression of ATPase family AAA domain containing 3A, a novel anti-autophagy factor, in uterine cervical cancer. Int. J. Mol. Med. 2011, 28, 689–696. [Google Scholar]
- Xie, X.Q.; Yang, Y.; Wang, Q.; Liu, H.F.; Fang, X.Y.; Li, C.L.; Jiang, Y.Z.; Wang, S.; Zhao, H.Y.; Miao, J.Y.; et al. Targeting ATAD3A-PINK1-mitophagy axis overcomes chemoimmunotherapy resistance by redirecting PD-L1 to mitochondria. Cell Res. 2023, 33, 215–228. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Type | Protein Domain | Name | Effects | Diseases/Symptoms | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| Nature | ATAD3A Protein | Mitochondria | ||||||
| Cell Type | Effects | |||||||
| Point mutations | Walker A motif | c.1064G>A (p.Gly355Asp) | Dominant-negative | Reduced ATPase activity | Neuron differentiated from fibroblast | Fragmentation and accumulation of mitochondria, increase of autophagy | Hereditary spastic paraplegia (HSP) and axonal neuropathy and dyskinetic cerebral palsy | [39] |
| Adjacent to Walker A motif | c.980G>C (p.Arg327Pro) Drosophila p.Arg333Pro | Loss-of-function | Reduced expression in head | Embryo | Increase in mitochondrial content and size | Congenital cataract, hypertrophic cardiomyopathy, and elevated 3-methylglutaconate in urine | [40] | |
| Adjacent to Walker B motif | c.1217T>G (p.Leu406Arg) | N/A * | Reduced expression in fibroblasts | Fibroblast | Reduction in mitochondrial cristae content and mitochondrial size | Fatal neonatal cerebellar hypoplasia, seizures, axial hypotonia, hypertrophic cardiomyopathy, hepatomegaly, congenital cataract, and dysmorphic facies | [45] | |
| C-terminus | c.1582C>T (p.Arg528Trp) | Dominant-negative | N/A | Fibroblast | Reduction of mitochondrial content (not significant) and elevation of mitophagy | Global developmental delay, hypotonia, optic atrophy, axonal neuropathy, and hypertrophic cardiomyopathy (Harel-Yoon syndrome) | [41] | |
| 528+3A>G | Loss-of-function | Premature termination and reduced expression in fibroblasts | N/A | N/A | Harel-Yoon syndrome | [42] | ||
| c.1609T>A (p.Trp537Arg) and c.1614+2_1616+16del (p.Arg503Profs*11) † | Not dominant-negative for p.Arg503Profs*11 | Premature termination for p.Arg503Profs*11 and reduced expression in fibroblasts | Skeletal muscle | Abnormal mitochondrial morphology, increased number and size of mitochondria, disrupted cristae network, and decreased activity of complex I | Axonal sensory–motor neuropathy and neonatal cataract | [46] | ||
| N-terminus | c.230T>G (p.Leu77Arg) and c.634C>T (p.Gln212*) † | N/A | Premature termination for c.634C>T (p.Gln212*) | Cardiomyocyte | Increased amount of mitochondria | Abnormalities in basal ganglia and white matter, corneal clouding, cataract, GFAP-positive gliosis in retina, and depletion of ganglion cells in retina | [48] | |
| c.251T>C (p.Thr84Me) | Potential loss-of-function | N/A | N/A | N/A | Harel-Yoon syndrome without cardiomyopathy or optic atrophy | [43] | ||
| c.229C>G (p.Leu77Val) Drosophila p.Leu83Val | Partial loss-of-function | Unaffected expression in head | Larvae | Unaffected mitochondrial content | Global developmental delay, congenital cataract, and hypertrophic cardiomyopathy | [40] | ||
| Adult muscle | Smaller mitochondria, loosened and fragmented cristae, and potentially increased autophagy and mitophagy | |||||||
| c.150C>G (p.Phe50Leu) Drosophila p.Phe56Leu | Loss-of-function | Unaffected expression in head | Embryo | Abnormal increase in mitochondrial content and size | Hypotonia, cloudy corneas, hypertrophic cardiomyopathy, cerebellar atrophy/hypoplasia, and elevated 3-methylglutaconate in urine | |||
| c.508C>T (p.Arg170Trp) Drosophila p.Arg176Trp | Partial loss-of-function | Unaffected expression in head | Larvae | Unaffected mitochondrial content | Moderate–severe learning difficulties, ataxia, muscle wasting, cerebellar atrophy/hypoplasia, and hearing loss | |||
| Adult muscle | Smaller mitochondria, loosened and fragmented cristae, and potentially increased autophagy and mitophagy | |||||||
| c.707G>T (p.Gly236Val) Drosophila p.Gly242Val | Loss-of-function | Complete loss of protein in head | Embryo | Abnormal increase in mitochondrial content and size | Central hypotonia, increased peripheral tone, hypertrophic cardiomyopathy, and cerebellar atrophy/hypoplasia | |||
| c.229C>G (p.Leu77Val) and exon 3–4 deletion † | N/A | Reduced expression in fibroblasts | Fibroblast | Reduction in the activity of complex IV, levels of subunits COX2 and complex V, and mitochondrial proteosynthesis rate | Bilateral congenital cataracts, strabismus, residual nystagmus, ophthalmoplegia, hypotonia, hyporeflexia, axonal sensory–motor neuropathy, growth delay, cerebellar syndrome, etc. | [47] | ||
| Muscle | Reduction in protein expression of complexes I, IV, and V | |||||||
| Copy number variants | N/A | Biallelic deletions in the region encoding the ATAD3C, ATAD3B and ATAD3A genes | N/A | ATAD3A-ATAD3B chimeric protein and reduced ATAD3A expression in fibroblasts | Fibroblast | Aggregation and enlargement of mtDNA, slow mtDNA synthesis, and increase of free cholesterol | Fatal congenital pontocerebellar hypoplasia | [49] |
| Intergenic duplications of ATAD3A gene cluster | N/A | ATAD3A-ATAD3C chimeric protein (maybe nonfunctional) and ATAD3B duplication | Fibroblast | Aggregation of mitochondria, accumulation of mtDNA, and increase of free cholesterol | Lethal metabolic disorder characterized by cardiomyopathy, corneal opacities, encephalopathy, hypotonia, and seizures | [50] | ||
| Duplication of the ATAD3 gene cluster | N/A | N/A | N/A | N/A | Graves’ disease | [51] | ||
| 68 kbp duplications in ATAD3 locus | Dominant-negative | ATAD3A-ATAD3C chimeric protein | Liver and heart | Disturbance of the oligomerization of ATAD3A and reduction of the activity of complexes I and IV Heart only: decreased expression of complexes I, III, IV, and V | Lethal perinatal cardiomyopathy, persistent hyperlactacidemia, frequent corneal clouding or cataracts, and encephalopathy | [21] | ||
| 38 kbp ATAD3B/3A deletion and 19 bp deletion in ATAD3A exon 6 † | Potential loss-of-function | Premature termination for 19 bp deletion in ATAD3A exon 6 | Myocardium | Complex I deficiency | Severe spinal cord hypoplasia and pontocerebellar hypoplasia | [52] | ||
| Liver | Normal respiratory chain enzyme activity | |||||||
| c.624_644del (p.Glu209_Glu215del) | Loss-of-function | N/A | N/A | Suggested mitochondrial dysfunction | Harel-Yoon syndrome, fatigable ptosis, facial weakness, progressive bulbar palsy, and obsessive-compulsive disorder | [44] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Li, Y.; Zambidis, A.; Papadopoulos, V. ATAD3A: A Key Regulator of Mitochondria-Associated Diseases. Int. J. Mol. Sci. 2023, 24, 12511. https://doi.org/10.3390/ijms241512511
Chen L, Li Y, Zambidis A, Papadopoulos V. ATAD3A: A Key Regulator of Mitochondria-Associated Diseases. International Journal of Molecular Sciences. 2023; 24(15):12511. https://doi.org/10.3390/ijms241512511
Chicago/Turabian StyleChen, Liting, Yuchang Li, Alexander Zambidis, and Vassilios Papadopoulos. 2023. "ATAD3A: A Key Regulator of Mitochondria-Associated Diseases" International Journal of Molecular Sciences 24, no. 15: 12511. https://doi.org/10.3390/ijms241512511