
Calcium Signaling during Cortical Apical Dendrite Initiation: A Role for Cajal-Retzius Neurons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
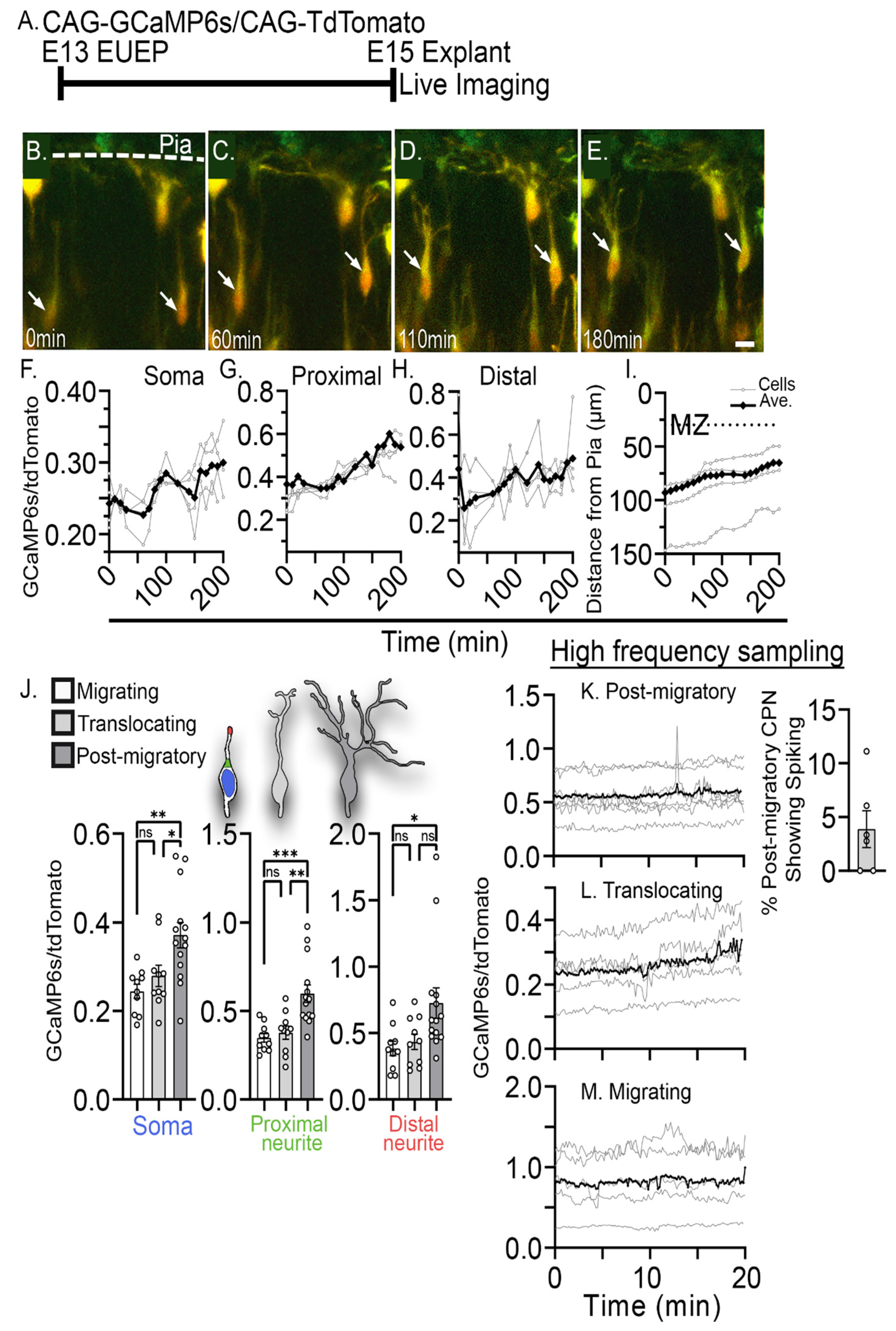
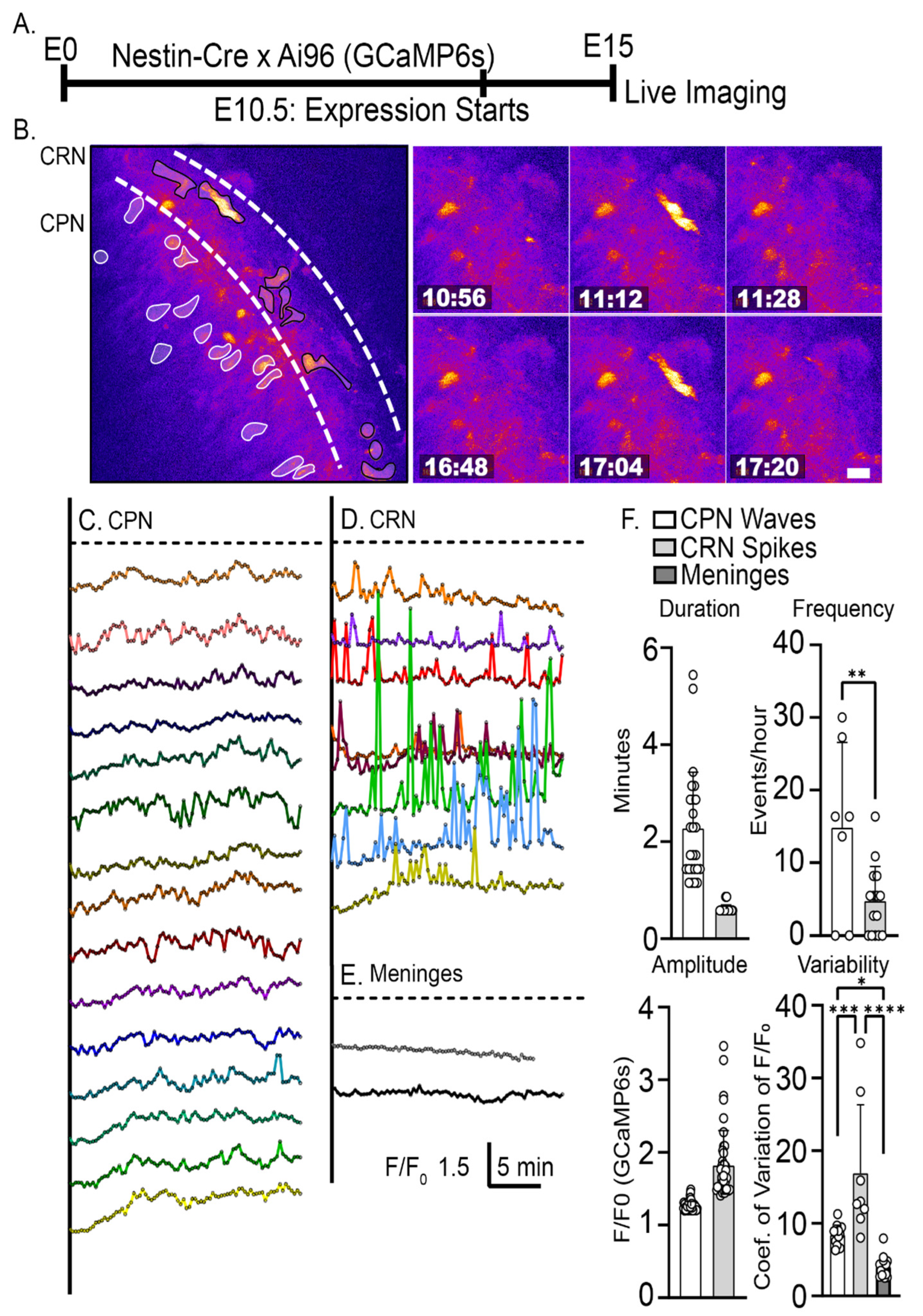
2.1. Characterization of Intracellular Calcium Signals during Dendritic Initiation and Migration Arrest
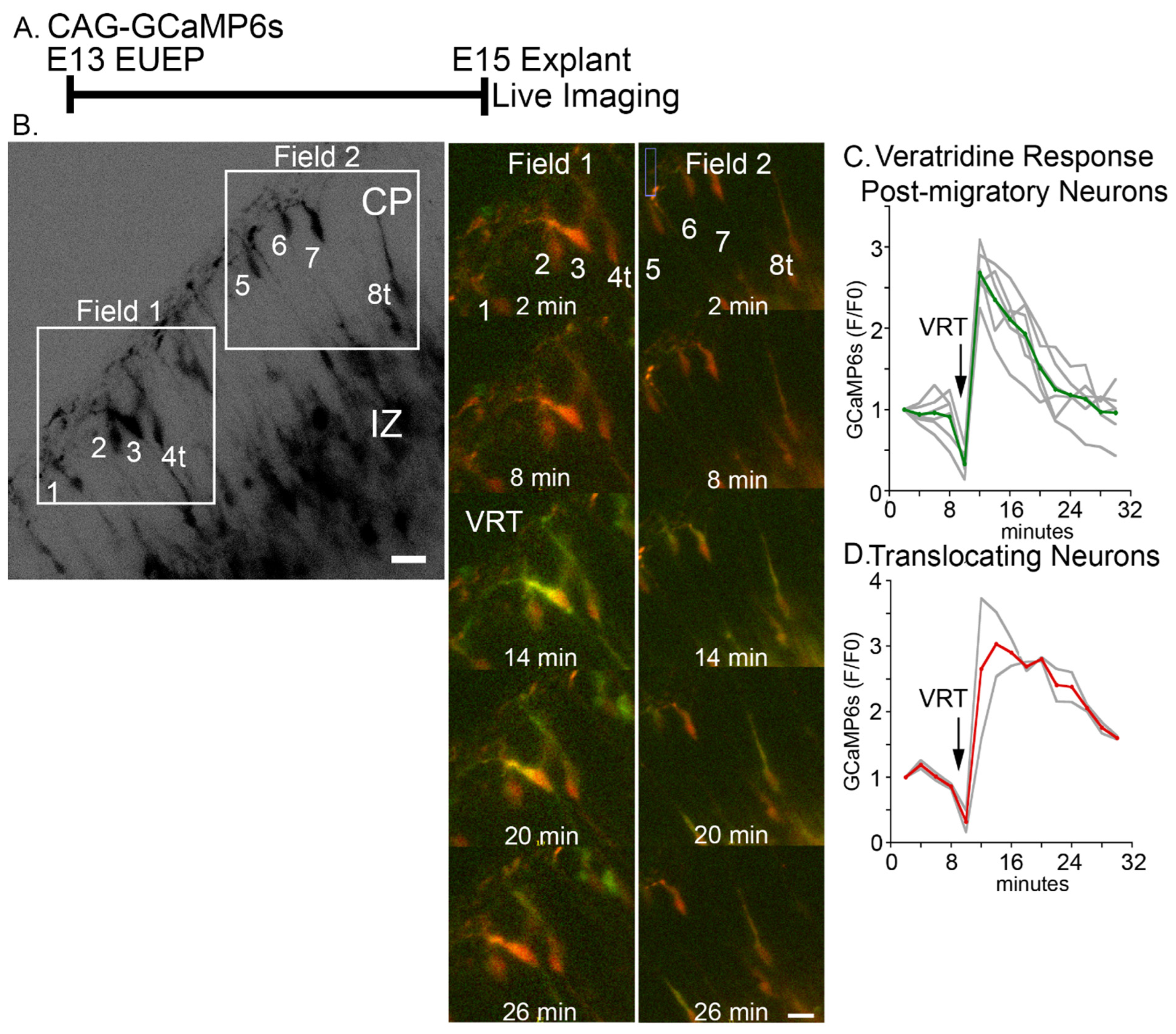
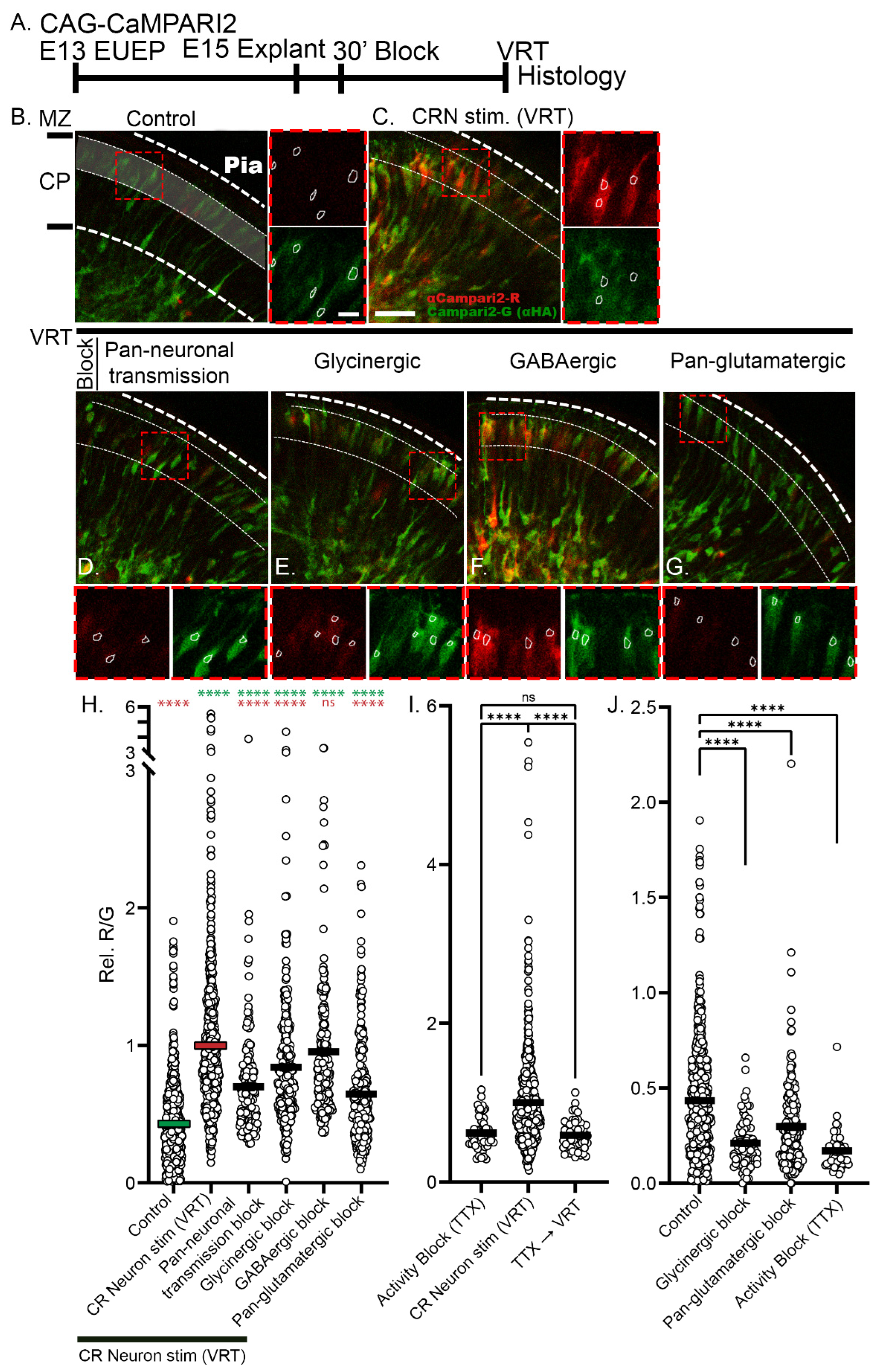
2.2. Veratridine Stimulation of CRNs Induces Rapid Calcium Transients in CPNs: Partial Block by Glutamatergic and Glycinergic Antagonists
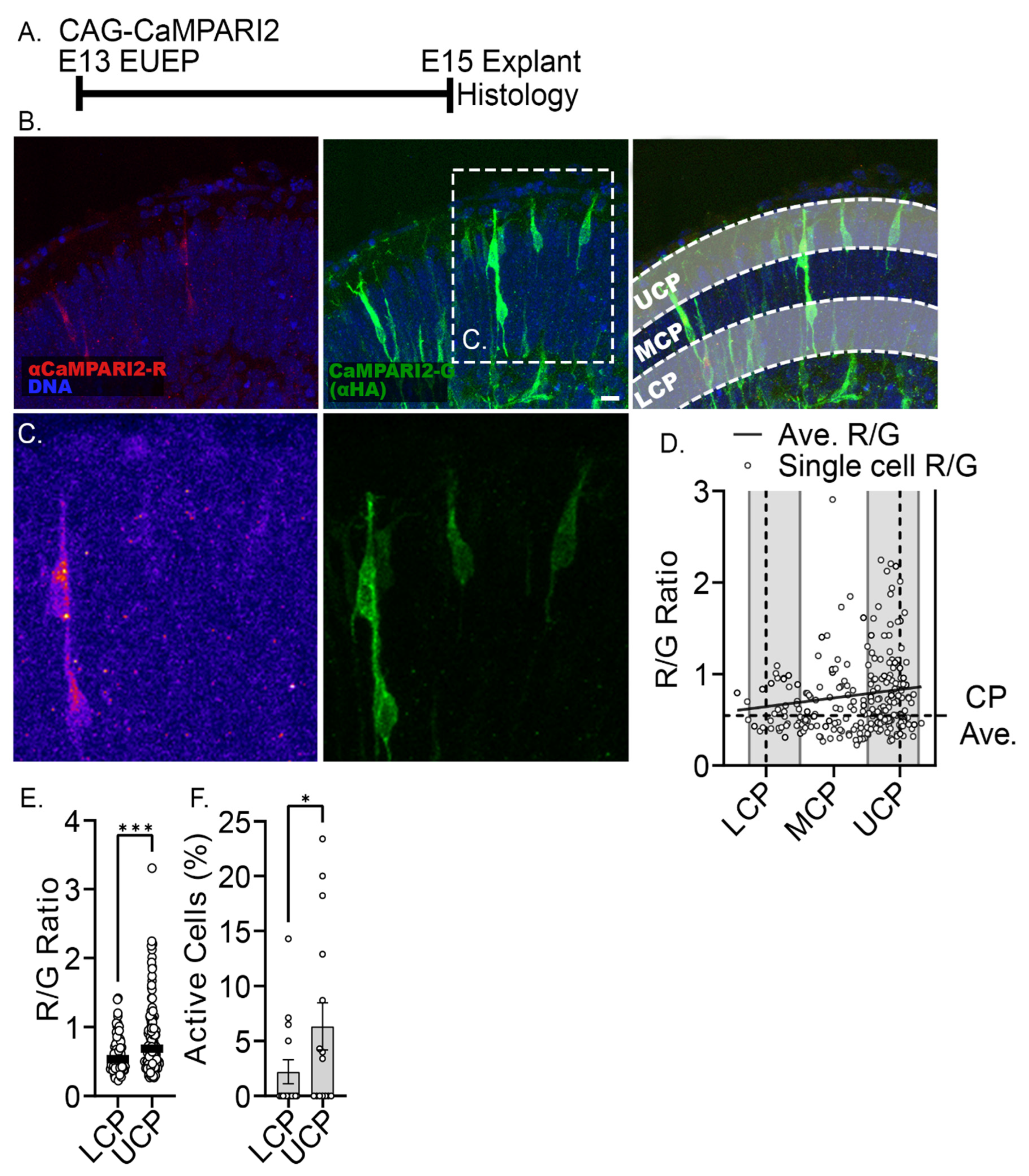
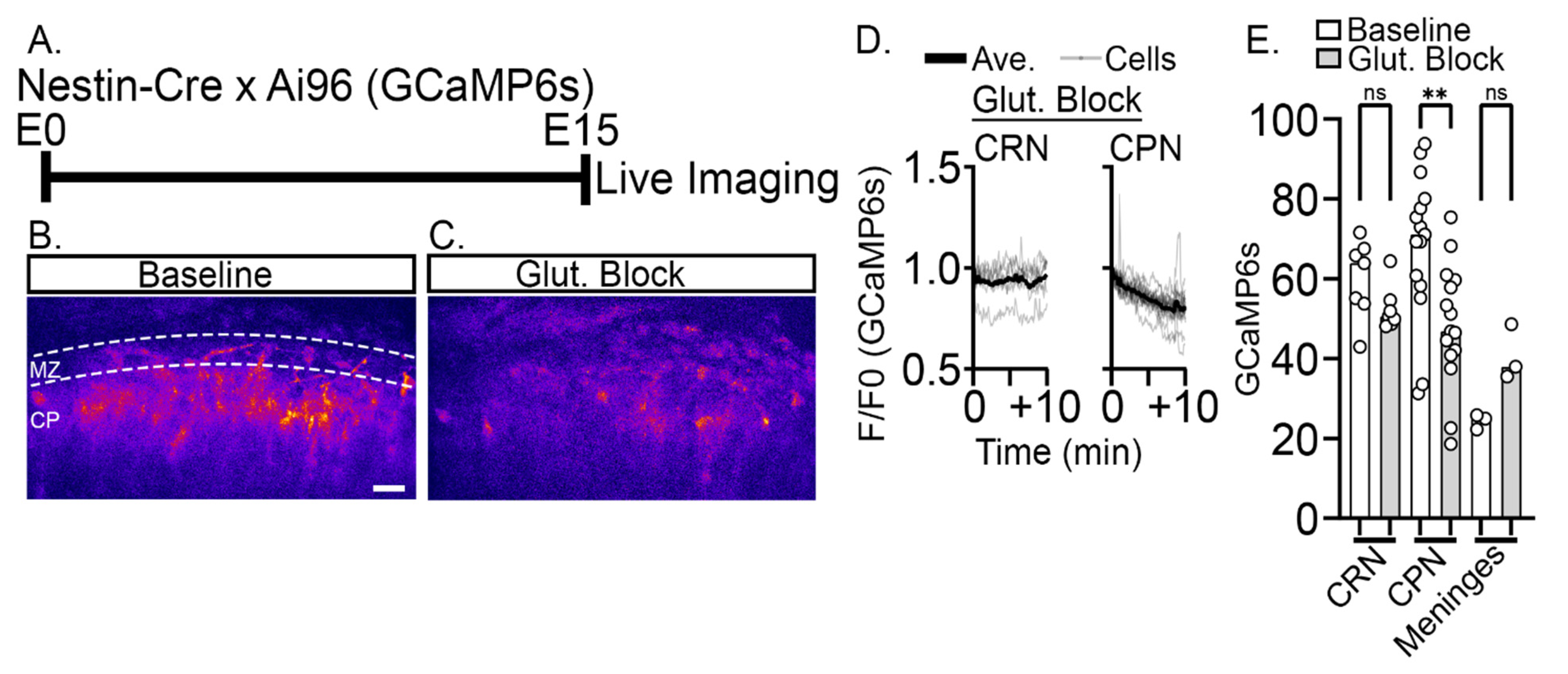
2.3. Reduction of Baseline Intracellular Calcium Signal by Glutamatergic and Activity Blockade
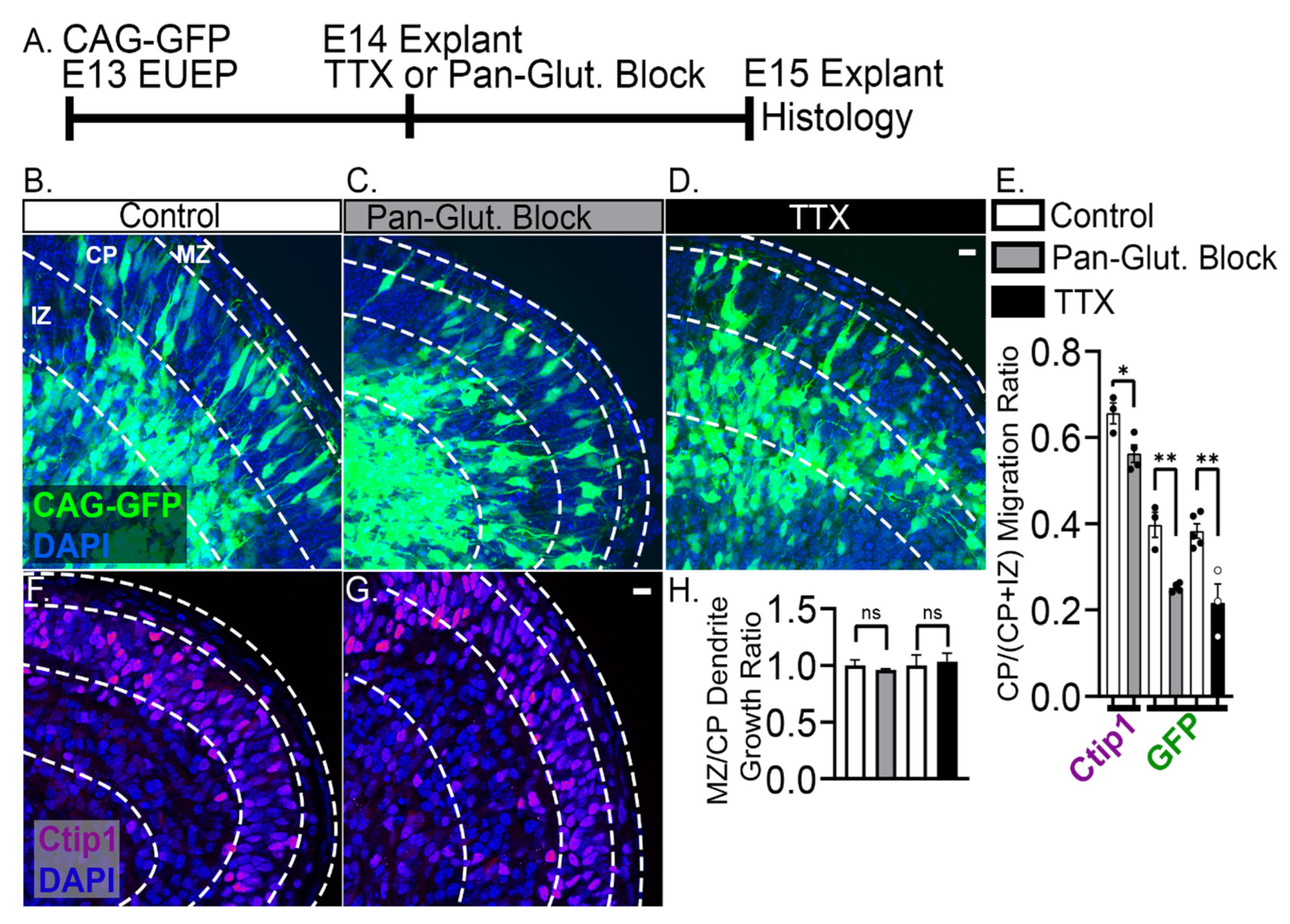
2.4. 24-h Pan-Glutamatergic and Activity Blockade Decreased Neuronal Migration into the CP
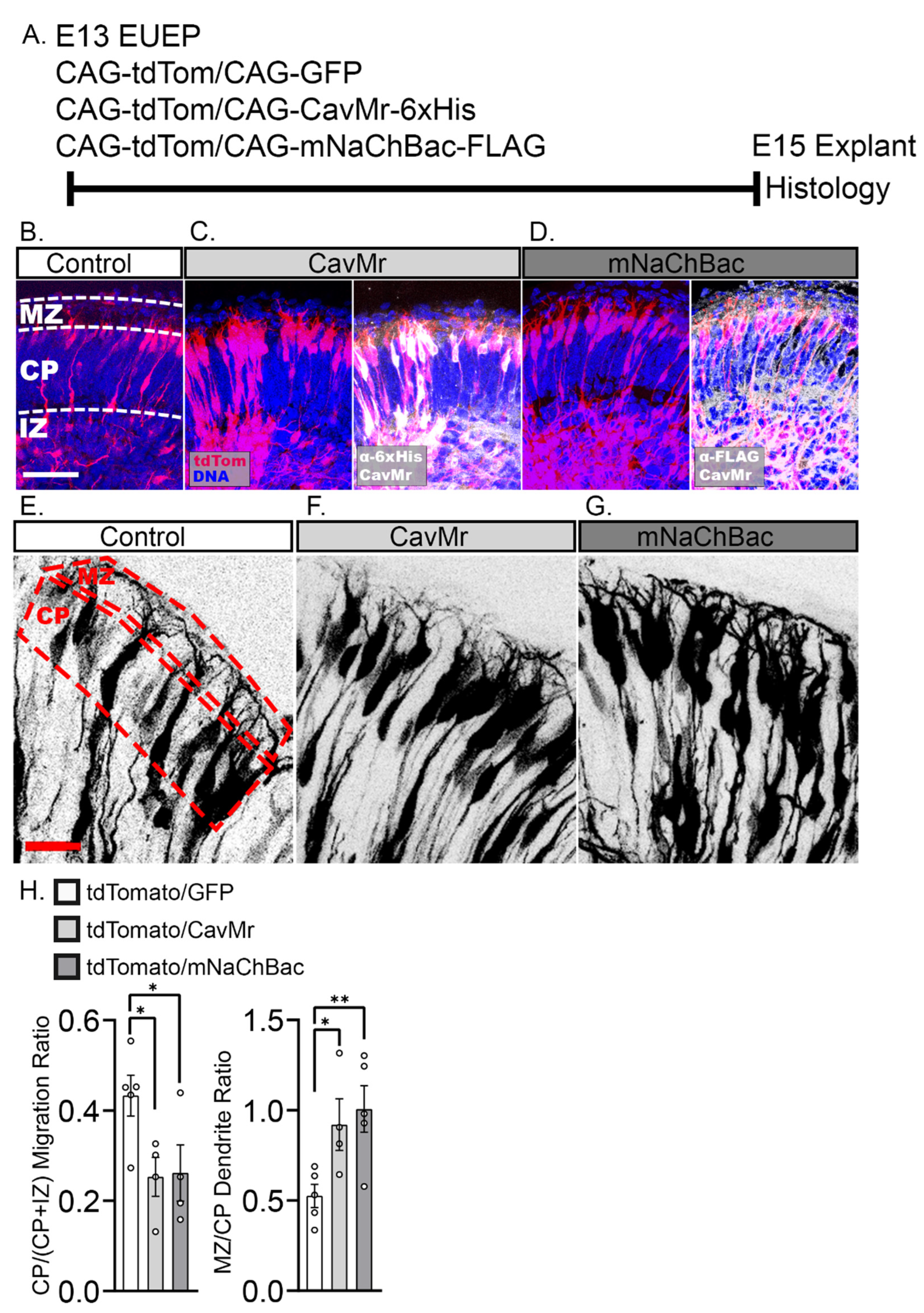
2.5. Misexpression of Voltage-Dependent Sodium and Calcium Channels Reduced Migration into the CP and Enhances Dendritic Growth in the MZ
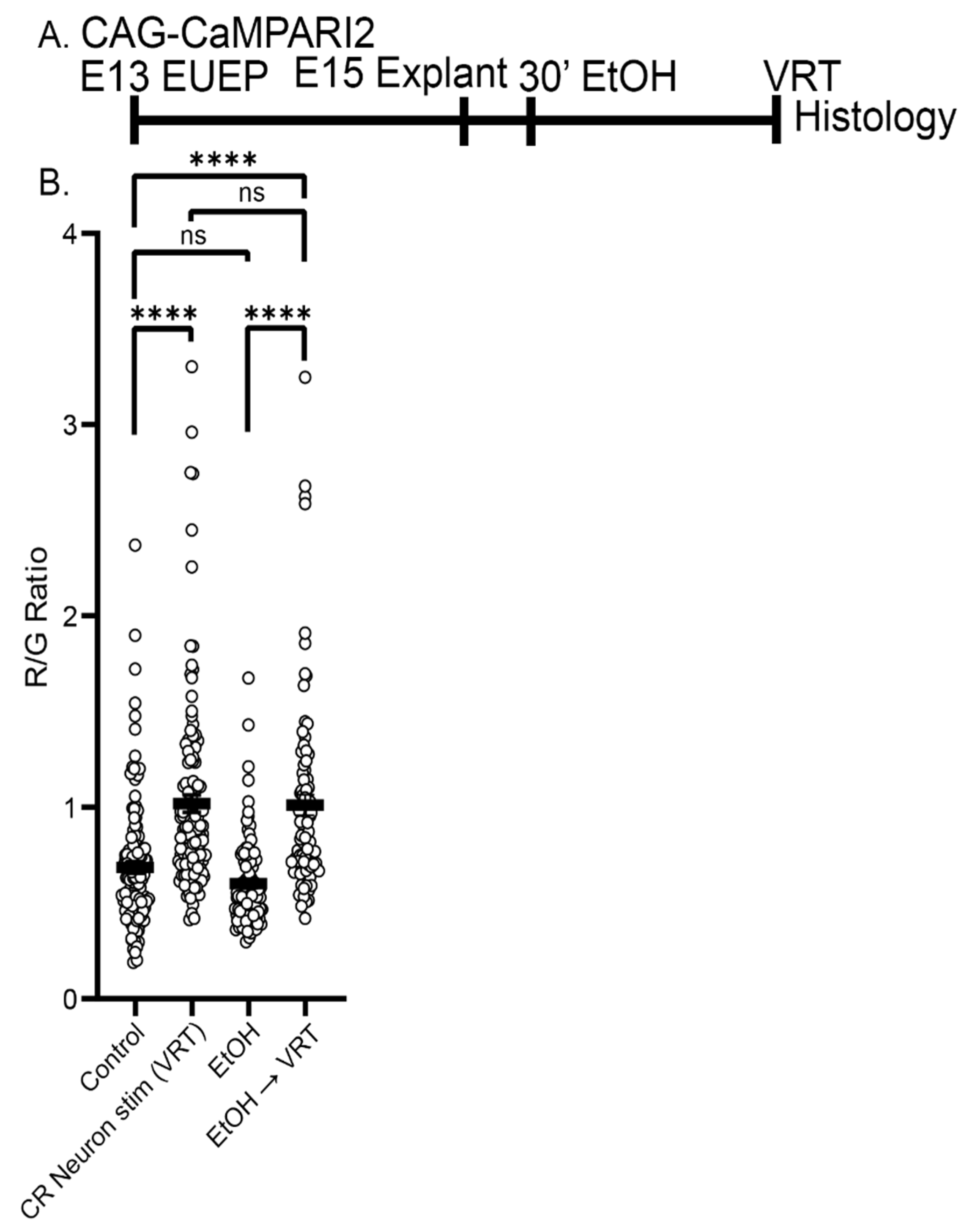
2.6. Ethanol Exposure Did Not Alter the Baseline CaMPARI2 Signal or Disrupt the Response to Veratridine
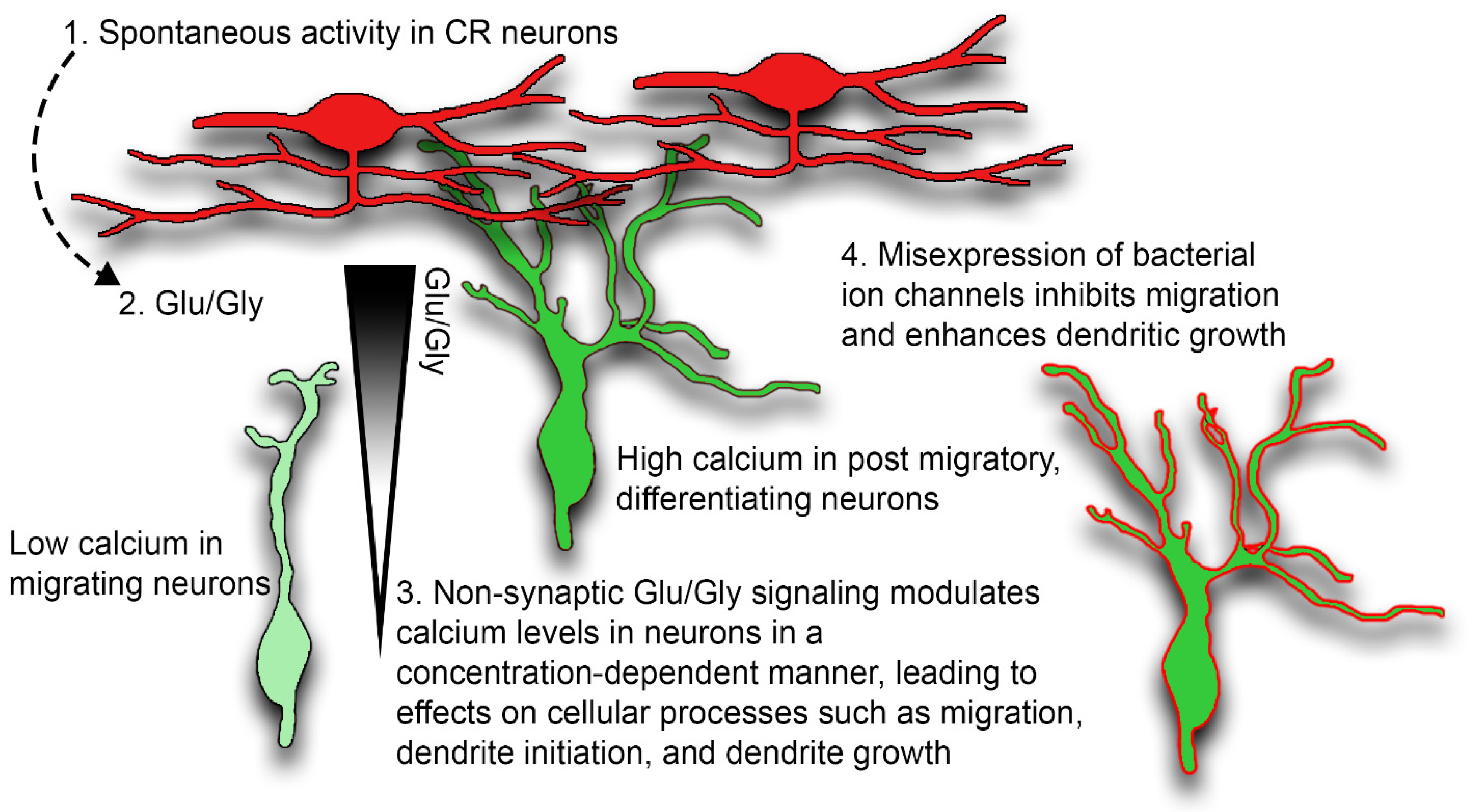
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Plasmids
4.3. Explants
4.4. Calcium Imaging and Analysis
4.5. Pharmacology
4.6. CaMPARI2 Photoconversion and Immunohistochemistry
4.7. Imaging and Data Analysis
4.8. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinto Lord, M.C.; Caviness, V.S., Jr. Determinants of cell shape and orientation: A comparative Golgi analysis of cell-axon interrelationships in the developing neocortex of normal and reeler mice. J. Comp. Neurol. 1979, 187, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.C.; Kim, S.; Walsh, C.A. Impaired neuronal positioning and dendritogenesis in the neocortex after cell-autonomous Dab1 suppression. J. Neurosci. 2006, 26, 1767–1775. [Google Scholar] [CrossRef]
- O’Dell, R.S.; Cameron, D.A.; Zipfel, W.R.; Olson, E.C. Reelin Prevents Apical Neurite Retraction during Terminal Translocation and Dendrite Initiation. J. Neurosci. 2015, 35, 10659–10674. [Google Scholar] [CrossRef]
- Marin-Padilla, M. Human cerebral cortex Cajal-Retzius neuron: Development, structure and function. A Golgi study. Front. Neuroanat. 2015, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Elorriaga, V.; Pierani, A.; Causeret, F. Cajal-retzius cells: Recent advances in identity and function. Curr. Opin. Neurobiol. 2023, 79, 102686. [Google Scholar] [CrossRef]
- Boulder-Committee. Embryonic vertebrate central nervous system: Revised terminology. The Boulder Committee. Anat. Rec. 1970, 166, 257–261. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, G.; Miao, G.G.; Chen, S.C.; Soares, H.D.; Morgan, J.I.; Curran, T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 1995, 374, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Hirotsune, S.; Takahara, T.; Sasaki, N.; Hirose, K.; Yoshiki, A.; Ohashi, T.; Kusakabe, M.; Murakami, Y.; Muramatsu, M.; Watanabe, S.; et al. The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat. Genet. 1995, 10, 77–83. [Google Scholar] [CrossRef]
- Ogawa, M.; Miyata, T.; Nakajima, K.; Yagyu, K.; Seike, M.; Ikenaka, K.; Yamamoto, H.; Mikoshiba, K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron 1995, 14, 899–912. [Google Scholar] [CrossRef]
- Niu, S.; Renfro, A.; Quattrocchi, C.C.; Sheldon, M.; D’Arcangelo, G. Reelin Promotes Hippocampal Dendrite Development through the VLDLR/ApoER2-Dab1 Pathway. Neuron 2004, 41, 71–84. [Google Scholar] [CrossRef]
- Pinto Lord, M.C.; Evrard, P.; Caviness, V.S., Jr. Obstructed neuronal migration along radial glial fibers in the neocortex of the reeler mouse: A Golgi-EM analysis. Brain Res. 1982, 256, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Aguilo, A.; Schwartz, T.H.; Kumar, V.S.; Peterlin, Z.A.; Tsiola, A.; Soriano, E.; Yuste, R. Involvement of cajal-retzius neurons in spontaneous correlated activity of embryonic and postnatal layer 1 from wild-type and reeler mice. J. Neurosci. 1999, 19, 10856–10868. [Google Scholar] [CrossRef] [PubMed]
- Sava, B.A.; David, C.S.; Teissier, A.; Pierani, A.; Staiger, J.F.; Luhmann, H.J.; Kilb, W. Electrophysiological and morphological properties of Cajal-Retzius cells with different ontogenetic origins. Neuroscience 2010, 167, 724–734. [Google Scholar] [CrossRef]
- Riva, M.; Moriceau, S.; Morabito, A.; Dossi, E.; Sanchez-Bellot, C.; Azzam, P.; Navas-Olive, A.; Gal, B.; Dori, F.; Cid, E.; et al. Aberrant survival of hippocampal Cajal-Retzius cells leads to memory deficits, gamma rhythmopathies and susceptibility to seizures in adult mice. Nat. Commun. 2023, 14, 1531. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Grayson, D.R.; Auta, J.; Sugaya, I.; Costa, E.; Guidotti, A. Reelin secretion from glutamatergic neurons in culture is independent from neurotransmitter regulation. Proc. Natl. Acad. Sci. USA 2000, 97, 3556–3561. [Google Scholar] [CrossRef]
- Nakao, Y.; Yokawa, S.; Kohno, T.; Suzuki, T.; Hattori, M. Visualization of Reelin Secretion from Primary Cultured Neurons by Bioluminescence Imaging. J. Biochem. 2022, 171, 591–598. [Google Scholar] [CrossRef]
- Engeroff, K.; Warm, D.; Bittner, S.; Blanquie, O. Different activity patterns control various stages of Reelin synthesis in the developing neocortex. Cereb. Cortex 2023, 33, 9376–9386. [Google Scholar] [CrossRef]
- Jossin, Y.; Gui, L.; Goffinet, A.M. Processing of Reelin by embryonic neurons is important for function in tissue but not in dissociated cultured neurons. J. Neurosci. 2007, 27, 4243–4252. [Google Scholar] [CrossRef]
- Derer, P.; Derer, M. Cajal-Retzius cell ontogenesis and death in mouse brain visualized with horseradish peroxidase and electron microscopy. Neuroscience 1990, 36, 839–856. [Google Scholar] [CrossRef]
- Spitzer, N.C. Electrical activity in early neuronal development. Nature 2006, 444, 707–712. [Google Scholar] [CrossRef]
- Behar, T.N.; Scott, C.A.; Greene, C.L.; Wen, X.; Smith, S.V.; Maric, D.; Liu, Q.Y.; Colton, C.A.; Barker, J.L. Glutamate acting at NMDA receptors stimulates embryonic cortical neuronal migration. J. Neurosci. 1999, 19, 4449–4461. [Google Scholar] [CrossRef]
- Behar, T.N.; Schaffner, A.E.; Scott, C.A.; O’Connell, C.; Barker, J.L. Differential response of cortical plate and ventricular zone cells to GABA as a migration stimulus. J. Neurosci. 1998, 18, 6378–6387. [Google Scholar] [CrossRef] [PubMed]
- Luhmann, H.J.; Fukuda, A.; Kilb, W. Control of cortical neuronal migration by glutamate and GABA. Front. Cell Neurosci. 2015, 9, 4. [Google Scholar] [CrossRef]
- Cornell-Bell, A.H.; Thomas, P.G.; Smith, S.J. The excitatory neurotransmitter glutamate causes filopodia formation in cultured hippocampal astrocytes. Glia 1990, 3, 322–334. [Google Scholar] [CrossRef]
- Heinen, K.; Baker, R.E.; Spijker, S.; Rosahl, T.; van Pelt, J.; Brussaard, A.B. Impaired dendritic spine maturation in GABAA receptor alpha1 subunit knock out mice. Neuroscience 2003, 122, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Portera-Cailliau, C.; Pan, D.T.; Yuste, R. Activity-regulated dynamic behavior of early dendritic protrusions: Evidence for different types of dendritic filopodia. J. Neurosci. 2003, 23, 7129–7142. [Google Scholar] [CrossRef] [PubMed]
- Bando, Y.; Irie, K.; Shimomura, T.; Umeshima, H.; Kushida, Y.; Kengaku, M.; Fujiyoshi, Y.; Hirano, T.; Tagawa, Y. Control of Spontaneous Ca2+ Transients Is Critical for Neuronal Maturation in the Developing Neocortex. Cereb. Cortex 2016, 26, 106–117. [Google Scholar] [CrossRef]
- Hurni, N.; Kolodziejczak, M.; Tomasello, U.; Badia, J.; Jacobshagen, M.; Prados, J.; Dayer, A. Transient Cell-intrinsic Activity Regulates the Migration and Laminar Positioning of Cortical Projection Neurons. Cereb. Cortex 2017, 27, 3052–3063. [Google Scholar] [CrossRef]
- Platel, J.C.; Boisseau, S.; Dupuis, A.; Brocard, J.; Poupard, A.; Savasta, M.; Villaz, M.; Albrieux, M. Na+ channel-mediated Ca2+ entry leads to glutamate secretion in mouse neocortical preplate. Proc. Natl. Acad. Sci. USA 2005, 102, 19174–19179. [Google Scholar] [CrossRef]
- Quattrocolo, G.; Maccaferri, G. Optogenetic activation of cajal-retzius cells reveals their glutamatergic output and a novel feedforward circuit in the developing mouse hippocampus. J. Neurosci. 2014, 34, 13018–13032. [Google Scholar] [CrossRef]
- Anstotz, M.; Lee, S.K.; Maccaferri, G. Glutamate released by Cajal-Retzius cells impacts specific hippocampal circuits and behaviors. Cell Rep. 2022, 39, 110822. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.W.; Wardill, T.J.; Sun, Y.; Pulver, S.R.; Renninger, S.L.; Baohan, A.; Schreiter, E.R.; Kerr, R.A.; Orger, M.B.; Jayaraman, V.; et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 2013, 499, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Shaner, N.C.; Campbell, R.E.; Steinbach, P.A.; Giepmans, B.N.; Palmer, A.E.; Tsien, R.Y. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat. Biotechnol. 2004, 22, 1567–1572. [Google Scholar] [CrossRef] [PubMed]
- Nichols, A.J.; O’Dell, R.S.; Powrozek, T.A.; Olson, E.C. Ex utero electroporation and whole hemisphere explants: A simple experimental method for studies of early cortical development. J. Vis. Exp. JoVE 2013, 3, e50271. [Google Scholar]
- Schwartz, T.H.; Rabinowitz, D.; Unni, V.; Kumar, V.S.; Smetters, D.K.; Tsiola, A.; Yuste, R. Networks of coactive neurons in developing layer 1. Neuron 1998, 20, 541–552. [Google Scholar] [CrossRef]
- Yuryev, M.; Andriichuk, L.; Leiwe, M.; Jokinen, V.; Carabalona, A.; Rivera, C. In vivo two-photon imaging of the embryonic cortex reveals spontaneous ketamine-sensitive calcium activity. Sci. Rep. 2018, 8, 16059. [Google Scholar] [CrossRef]
- Akerboom, J.; Carreras Calderon, N.; Tian, L.; Wabnig, S.; Prigge, M.; Tolo, J.; Gordus, A.; Orger, M.B.; Severi, K.E.; Macklin, J.J.; et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front. Mol. Neurosci. 2013, 6, 2. [Google Scholar] [CrossRef]
- Madisen, L.; Garner, A.R.; Shimaoka, D.; Chuong, A.S.; Klapoetke, N.C.; Li, L.; van der Bourg, A.; Niino, Y.; Egolf, L.; Monetti, C.; et al. Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 2015, 85, 942–958. [Google Scholar] [CrossRef]
- Zimmerman, L.; Parr, B.; Lendahl, U.; Cunningham, M.; McKay, R.; Gavin, B.; Mann, J.; Vassileva, G.; McMahon, A. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron 1994, 12, 11–24. [Google Scholar] [CrossRef]
- Takahashi, T.; Goto, T.; Miyama, S.; Nowakowski, R.S.; Caviness, V.S., Jr. Sequence of neuron origin and neocortical laminar fate: Relation to cell cycle of origin in the developing murine cerebral wall. J. Neurosci. 1999, 19, 10357–10371. [Google Scholar] [CrossRef]
- Gu, X.; Olson, E.C.; Spitzer, N.C. Spontaneous neuronal calcium spikes and waves during early differentiation. J. Neurosci. 1994, 14, 6325–6335. [Google Scholar] [CrossRef] [PubMed]
- Yuryev, M.; Pellegrino, C.; Jokinen, V.; Andriichuk, L.; Khirug, S.; Khiroug, L.; Rivera, C. In vivo Calcium Imaging of Evoked Calcium Waves in the Embryonic Cortex. Front. Cell Neurosci. 2015, 9, 500. [Google Scholar] [CrossRef] [PubMed]
- Moeyaert, B.; Holt, G.; Madangopal, R.; Perez-Alvarez, A.; Fearey, B.C.; Trojanowski, N.F.; Ledderose, J.; Zolnik, T.A.; Das, A.; Patel, D.; et al. Improved methods for marking active neuron populations. Nat. Commun. 2018, 9, 4440. [Google Scholar] [CrossRef]
- Fekete, A.; Franklin, L.; Ikemoto, T.; Rozsa, B.; Lendvai, B.; Sylvester Vizi, E.; Zelles, T. Mechanism of the persistent sodium current activator veratridine-evoked Ca elevation: Implication for epilepsy. J. Neurochem. 2009, 111, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Albrieux, M.; Platel, J.C.; Dupuis, A.; Villaz, M.; Moody, W.J. Early expression of sodium channel transcripts and sodium current by cajal-retzius cells in the preplate of the embryonic mouse neocortex. J. Neurosci. Off. J. Soc. Neurosci. 2004, 24, 1719–1725. [Google Scholar] [CrossRef]
- Picken Bahrey, H.L.; Moody, W.J. Early development of voltage-gated ion currents and firing properties in neurons of the mouse cerebral cortex. J. Neurophysiol. 2003, 89, 1761–1773. [Google Scholar] [CrossRef]
- Cameron, D.A.; Middleton, F.A.; Chenn, A.; Olson, E.C. Hierarchical clustering of gene expression patterns in the Eomes + lineage of excitatory neurons during early neocortical development. BMC Neurosci. 2012, 13, 90. [Google Scholar] [CrossRef]
- Narahashi, T. Tetrodotoxin: A brief history. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2008, 84, 147–154. [Google Scholar] [CrossRef]
- Ohtaka-Maruyama, C.; Okamoto, M.; Endo, K.; Oshima, M.; Kaneko, N.; Yura, K.; Okado, H.; Miyata, T.; Maeda, N. Synaptic transmission from subplate neurons controls radial migration of neocortical neurons. Science 2018, 360, 313–317. [Google Scholar] [CrossRef]
- O’Dell, R.S.; Ustine, C.J.; Cameron, D.A.; Lawless, S.M.; Williams, R.R.; Zipfel, W.R.; Olson, E.C. Layer 6 cortical neurons require Reelin-Dab1 signaling for cellular orientation, Golgi deployment, and directed neurite growth into the marginal zone. Neural Dev. 2012, 7, 25. [Google Scholar] [CrossRef]
- Shimomura, T.; Yonekawa, Y.; Nagura, H.; Tateyama, M.; Fujiyoshi, Y.; Irie, K. A native prokaryotic voltage-dependent calcium channel with a novel selectivity filter sequence. eLife 2020, 9, e52828. [Google Scholar] [CrossRef] [PubMed]
- Fabregues, I.; Ferrer, I.; Gairi, J.M.; Cahuana, A.; Giner, P. Effects of prenatal exposure to ethanol on the maturation of the pyramidal neurons in the cerebral cortex of the guinea-pig: A quantitative Golgi study. Neuropathol. Appl. Neurobiol. 1985, 11, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Kotkoskie, L.A.; Norton, S. Prenatal brain malformations following acute ethanol exposure in the rat. Alcohol. Clin. Exp. Res. 1988, 12, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Dow-Edwards, D.L. Structural and metabolic alterations in rat cerebral cortex induced by prenatal exposure to ethanol. Brain Res. 1988, 474, 316–326. [Google Scholar]
- Lovinger, D.M.; White, G.; Weight, F.F. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science 1989, 243, 1721–1724. [Google Scholar] [CrossRef]
- Olney, J.W.; Tenkova, T.; Dikranian, K.; Qin, Y.Q.; Labruyere, J.; Ikonomidou, C. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res. Dev. Brain Res. 2002, 133, 115–126. [Google Scholar] [CrossRef]
- White, G.; Lovinger, D.M.; Weight, F.F. Ethanol inhibits NMDA-activated current but does not alter GABA-activated current in an isolated adult mammalian neuron. Brain Res. 1990, 507, 332–336. [Google Scholar] [CrossRef]
- Skorput, A.G.; Yeh, H.H. Effects of ethanol exposure in utero on Cajal-Retzius cells in the developing cortex. Alcohol. Clin. Exp. Res. 2015, 39, 853–862. [Google Scholar] [CrossRef]
- Powrozek, T.A.; Olson, E.C. Ethanol-induced disruption of Golgi apparatus morphology, primary neurite number and cellular orientation in developing cortical neurons. Alcohol 2012, 46, 619–627. [Google Scholar] [CrossRef]
- Wang, D.; Enck, J.; Howell, B.W.; Olson, E.C. Ethanol Exposure Transiently Elevates but Persistently Inhibits Tyrosine Kinase Activity and Impairs the Growth of the Nascent Apical Dendrite. Mol. Neurobiol. 2019, 56, 5749–5762. [Google Scholar] [CrossRef]
- Dotti, C.G.; Sullivan, C.A.; Banker, G.A. The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 1988, 8, 1454–1468. [Google Scholar] [CrossRef] [PubMed]
- Tabata, H.; Nakajima, K. Multipolar migration: The third mode of radial neuronal migration in the developing cerebral cortex. J. Neurosci. 2003, 23, 9996–10001. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.J.; Barnes, A.P.; Hand, R.; Polleux, F.; Ehlers, M.D. TGF-beta signaling specifies axons during brain development. Cell 2010, 142, 144–157. [Google Scholar] [CrossRef]
- Xu, C.; Funahashi, Y.; Watanabe, T.; Takano, T.; Nakamuta, S.; Namba, T.; Kaibuchi, K. Radial Glial Cell-Neuron Interaction Directs Axon Formation at the Opposite Side of the Neuron from the Contact Site. J. Neurosci. 2015, 35, 14517–14532. [Google Scholar] [CrossRef] [PubMed]
- Rakic, P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J. Comp. Neurol. 1972, 145, 61–83. [Google Scholar] [CrossRef] [PubMed]
- Nadarajah, B.; Brunstrom, J.E.; Grutzendler, J.; Wong, R.O.; Pearlman, A.L. Two modes of radial migration in early development of the cerebral cortex. Nat. Neurosci. 2001, 4, 143–150. [Google Scholar] [CrossRef]
- Komuro, H.; Rakic, P. Intracellular Ca2+ fluctuations modulate the rate of neuronal migration. Neuron 1996, 17, 275–285. [Google Scholar] [CrossRef]
- Sheng, M.; McFadden, G.; Greenberg, M.E. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron 1990, 4, 571–582. [Google Scholar] [CrossRef]
- West, A.E.; Chen, W.G.; Dalva, M.B.; Dolmetsch, R.E.; Kornhauser, J.M.; Shaywitz, A.J.; Takasu, M.A.; Tao, X.; Greenberg, M.E. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 11024–11031. [Google Scholar] [CrossRef]
- Aizawa, H.; Hu, S.C.; Bobb, K.; Balakrishnan, K.; Ince, G.; Gurevich, I.; Cowan, M.; Ghosh, A. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 2004, 303, 197–202. [Google Scholar] [CrossRef]
- Willsey, A.J.; Sanders, S.J.; Li, M.; Dong, S.; Tebbenkamp, A.T.; Muhle, R.A.; Reilly, S.K.; Lin, L.; Fertuzinhos, S.; Miller, J.A.; et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 2013, 155, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.C. Analysis of preplate splitting and early cortical development illuminates the biology of neurological disease. Front. Pediatr. 2014, 2, 121. [Google Scholar] [CrossRef] [PubMed]
- Polleux, F.; Morrow, T.; Ghosh, A. Semaphorin 3A is a chemoattractant for cortical apical dendrites. Nature 2000, 404, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Whitford, K.L.; Marillat, V.; Stein, E.; Goodman, C.S.; Tessier-Lavigne, M.; Chedotal, A.; Ghosh, A. Regulation of cortical dendrite development by Slit-Robo interactions. Neuron 2002, 33, 47–61. [Google Scholar] [CrossRef]
- Kater, S.B.; Mills, L.R. Regulation of growth cone behavior by calcium. J. Neurosci. 1991, 11, 891–899. [Google Scholar] [CrossRef]
- Komuro, H.; Rakic, P. Selective role of N-type calcium channels in neuronal migration. Science 1992, 257, 806–809. [Google Scholar] [CrossRef]
- Komuro, H.; Rakic, P. Modulation of neuronal migration by NMDA receptors. Science 1993, 260, 95–97. [Google Scholar] [CrossRef]
- Roth, B.L. DREADDs for Neuroscientists. Neuron 2016, 89, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Bonhaus, D.W.; Yeh, G.C.; Skaryak, L.; McNamara, J.O. Glycine regulation of the N-methyl-D-aspartate receptor-gated ion channel in hippocampal membranes. Mol. Pharmacol. 1989, 36, 273–279. [Google Scholar]
- Hirai, K.; Yoshioka, H.; Kihara, M.; Hasegawa, K.; Sakamoto, T.; Sawada, T.; Fushiki, S. Inhibiting neuronal migration by blocking NMDA receptors in the embryonic rat cerebral cortex: A tissue culture study. Brain Res. Dev. Brain Res. 1999, 114, 63–67. [Google Scholar] [CrossRef]
- Kihara, M.; Yoshioka, H.; Hirai, K.; Hasegawa, K.; Kizaki, Z.; Sawada, T. Stimulation of N-methyl-D-aspartate (NMDA) receptors inhibits neuronal migration in embryonic cerebral cortex: A tissue culture study. Brain Res. Dev. Brain Res. 2002, 138, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Reiprich, P.; Kilb, W.; Luhmann, H.J. Neonatal NMDA receptor blockade disturbs neuronal migration in rat somatosensory cortex in vivo. Cereb. Cortex 2005, 15, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Messersmith, E.K.; Feller, M.B.; Zhang, H.; Shatz, C.J. Migration of neocortical neurons in the absence of functional NMDA receptors. Mol. Cell. Neurosci. 1997, 9, 347–357. [Google Scholar] [CrossRef]
- Iwasato, T.; Datwani, A.; Wolf, A.M.; Nishiyama, H.; Taguchi, Y.; Tonegawa, S.; Knopfel, T.; Erzurumlu, R.S.; Itohara, S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature 2000, 406, 726–731. [Google Scholar] [CrossRef]
- Maskos, U.; Brustle, O.; McKay, R.D. Long-term survival, migration, and differentiation of neural cells without functional NMDA receptors in vivo. Dev. Biol. 2001, 231, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Branchereau, P.; Cattaert, D.; Delpy, A.; Allain, A.E.; Martin, E.; Meyrand, P. Depolarizing GABA/glycine synaptic events switch from excitation to inhibition during frequency increases. Sci. Rep. 2016, 6, 21753. [Google Scholar] [CrossRef]
- Okabe, A.; Kilb, W.; Shimizu-Okabe, C.; Hanganu, I.L.; Fukuda, A.; Luhmann, H.J. Homogenous glycine receptor expression in cortical plate neurons and Cajal-Retzius cells of neonatal rat cerebral cortex. Neuroscience 2004, 123, 715–724. [Google Scholar] [CrossRef]
- Jansson, L.C.; Akerman, K.E. The role of glutamate and its receptors in the proliferation, migration, differentiation and survival of neural progenitor cells. J. Neural. Transm. 2014, 121, 819–836. [Google Scholar] [CrossRef]
- Bock, H.H.; Herz, J. Reelin activates SRC family tyrosine kinases in neurons. Curr. Biol. 2003, 13, 18–26. [Google Scholar] [CrossRef]
- Nichols, A.J.; Olson, E.C. Reelin promotes neuronal orientation and dendritogenesis during preplate splitting. Cerebral. Cortex 2010, 20, 2213–2223. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.M.; Satterstrom, F.K.; Peng, M.; Brand, H.; Collins, R.L.; Dong, S.; Wamsley, B.; Klei, L.; Wang, L.; Hao, S.P.; et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet. 2022, 54, 1320–1331. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Cepko, C.L. Electroporation and RNA interference in the rodent retina in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2004, 101, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enck, J.R.; Olson, E.C. Calcium Signaling during Cortical Apical Dendrite Initiation: A Role for Cajal-Retzius Neurons. Int. J. Mol. Sci. 2023, 24, 12965. https://doi.org/10.3390/ijms241612965
Enck JR, Olson EC. Calcium Signaling during Cortical Apical Dendrite Initiation: A Role for Cajal-Retzius Neurons. International Journal of Molecular Sciences. 2023; 24(16):12965. https://doi.org/10.3390/ijms241612965
Chicago/Turabian StyleEnck, Joshua R., and Eric C. Olson. 2023. "Calcium Signaling during Cortical Apical Dendrite Initiation: A Role for Cajal-Retzius Neurons" International Journal of Molecular Sciences 24, no. 16: 12965. https://doi.org/10.3390/ijms241612965