

Angiotensin Regulation of Vascular Homeostasis: Exploring the Role of ROS and RAS Blockers
 , , ,
, , ,
Abstract
:1. Introduction
2. Discussion
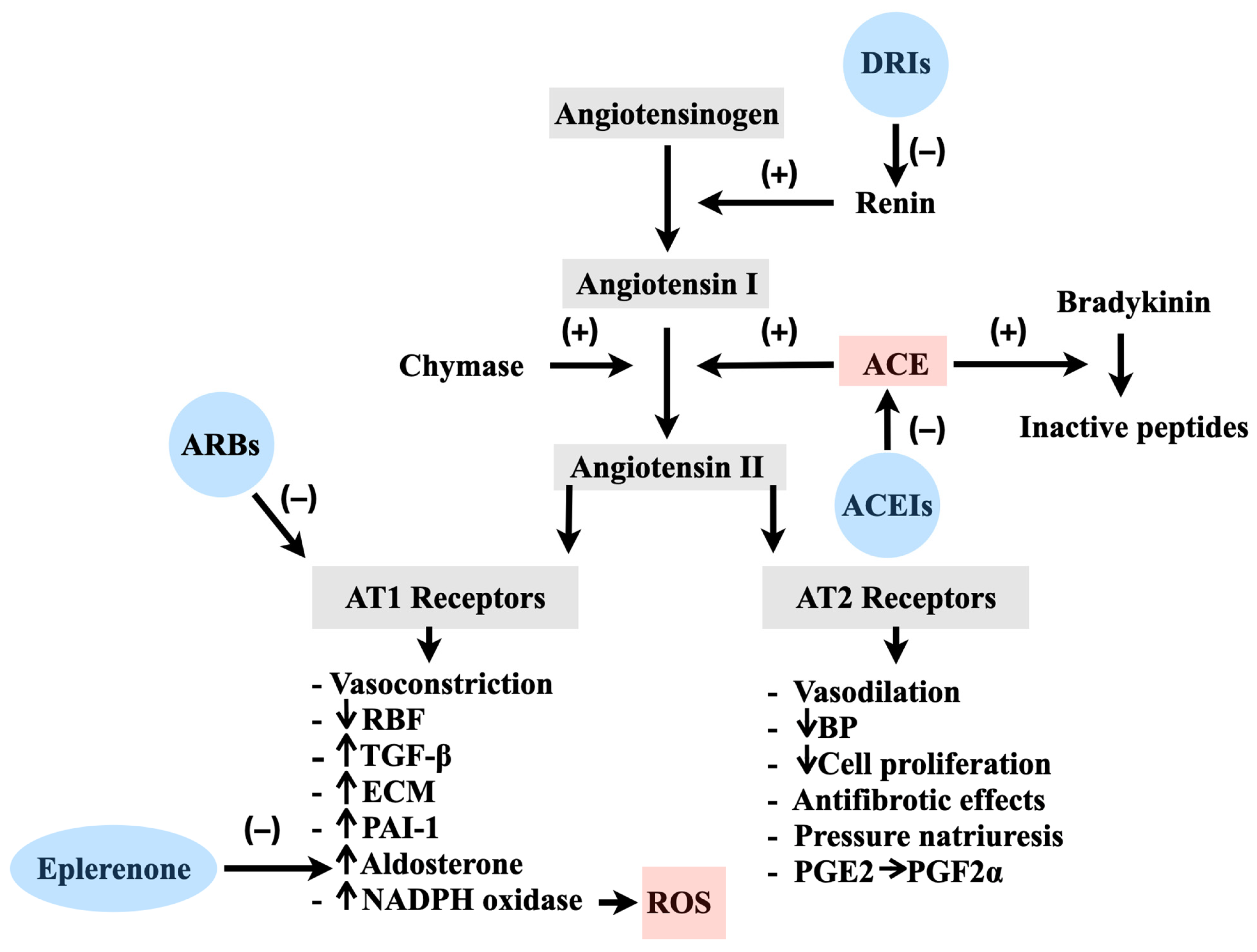
2.1. Physiology of the RAS in Vascular Regulation
2.2. Pathological Implications of the RAS and the Involvement of ROS
2.3. The Application of ACEIs and ARBs in the Treatment of Cardiovascular Disorders
2.4. Enhancing Endothelial Function and Mitigating Oxidative Stress: Exploring the Effects of ACEIs and ARBs
3. Clinical Implications
4. Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Antoniades, C.; Tousoulis, D.; Tentolouris, C.; Toutouzas, P.; Stefanadis, C. Oxidative stress, antioxidant vitamins, and atherosclerosis. From basic research to clinical practice. Herz 2003, 28, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Yamashita, S.; Okamoto, M.; Cooley, M.A.; Ozaki, K.; Everett, E.T.; Suzuki, M. Perfluorooctanoic acid-induced cell death via the dual roles of ROS-MAPK/ERK signaling in ameloblast-lineage cells. Ecotoxicol. Environ. Saf. 2023, 260, 115089. [Google Scholar] [CrossRef]
- Lee, Y.-P.; Lin, C.-R.; Chen, S.-S.; Chen, R.-J.; Wu, Y.-H.; Chen, Y.-H.; Huang, B.-M. Combination treatment of cordycepin and radiation induces MA-10 mouse Leydig tumor cell death via ROS accumulation and DNA damage. Am. J. Cancer Res. 2023, 13, 1329–1346. [Google Scholar] [PubMed]
- Wang, B.; Wang, Y.; Zhang, J.; Hu, C.; Jiang, J.; Li, Y.; Peng, Z. ROS-induced lipid peroxidation modulates cell death outcome: Mechanisms behind apoptosis, autophagy, and ferroptosis. Arch. Toxicol. 2023, 97, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.K.; Ding, H.; Jiang, M.; Yin, H.; Gollasch, M.; Huang, Y. Perivascular adipose tissue: Fine-tuner of vascular redox status and inflammation. Redox Biol. 2023, 62, 102683. [Google Scholar] [CrossRef]
- Hernández-Reséndiz, S.; Muñoz-Vega, M.; Contreras, W.E.; Crespo-Avilan, G.E.; Rodriguez-Montesinos, J.; Arias-Carrión, O.; Pérez-Méndez, O.; Boisvert, W.A.; Preissner, K.T.; Cabrera-Fuentes, H.A. Responses of Endothelial Cells Towards Ischemic Conditioning Following Acute Myocardial Infarction. Cond. Med. 2018, 1, 247–258. [Google Scholar]
- Da Silva, F.C.; de Araújo, B.J.; Cordeiro, C.S.; Arruda, V.M.; Faria, B.Q.; Guerra, J.F.D.C.; De Araújo, T.G.; Fürstenau, C.R. Endothelial dysfunction due to the inhibition of the synthesis of nitric oxide: Proposal and characterization of an in vitro cellular model. Front. Physiol. 2022, 13, 978378. [Google Scholar] [CrossRef]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; Yazbi, A.E.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci. 2022, 27, 105. [Google Scholar] [CrossRef]
- Hänze, J.; Weissmann, N.; Grimminger, F.; Seeger, W.; Rose, F. Cellular and molecular mechanisms of hypoxia-inducible factor driven vascular remodeling. Thromb. Haemost. 2007, 97, 774–787. [Google Scholar]
- Paul, M.; Poyan Mehr, A.; Kreutz, R. Physiology of local renin-angiotensin systems. Physiol. Rev. 2006, 86, 747–803. [Google Scholar] [CrossRef]
- Kasiakogias, A.; Rosei, E.A.; Camafort, M.; Ehret, G.; Faconti, L.; Ferreira, J.P.; Brguljan, J.; Januszewicz, A.; Kahan, T.; Manolis, A.; et al. Hypertension and heart failure with preserved ejection fraction: Position paper by the European Society of Hypertension. J. Hypertens. 2021, 39, 1522–1545. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, S.I.; Kumar, A.; Sowers, J.R. Mechanisms by which angiotensin-converting enzyme inhibitors prevent diabetes and cardiovascular disease. Am. J. Cardiol. 2003, 91, 30H–37H. [Google Scholar] [CrossRef] [PubMed]
- Lévy, B.I. Can angiotensin II type 2 receptors have deleterious effects in cardiovascular disease? Implications for therapeutic blockade of the renin-angiotensin system. Circulation 2004, 109, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Luginina, A.; Mishin, A.; Baidya, M.; Shukla, A.K.; Cherezov, V. Structural insights into ligand recognition and activation of angiotensin receptors. Trends Pharmacol. Sci. 2021, 42, 577–587. [Google Scholar] [CrossRef]
- Ziaja, M.; Urbanek, K.A.; Kowalska, K.; Piastowska-Ciesielska, A.W. Angiotensin II and Angiotensin Receptors 1 and 2—Multifunctional System in Cells Biology, What Do We Know? Cells 2021, 10, 381. [Google Scholar] [CrossRef]
- Duprez, D.A. Role of the renin–angiotensin–aldosterone system in vascular remodeling and inflammation: A clinical review. J. Hypertens. 2006, 24, 983–991. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Strawn, W.B. Role of the renin-angiotensin-aldosterone system and proinflammatory mediators in cardiovascular disease. Am. J. Cardiol. 2006, 98, 121–128. [Google Scholar] [CrossRef]
- Stoll, M.; Steckelings, U.M.; Paul, M.; Bottari, S.P.; Metzger, R.; Unger, T. The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J. Clin. Investig. 1995, 95, 651–657. [Google Scholar] [CrossRef]
- Steckelings, U.M.; Kaschina, E.; Unger, T. The AT2 receptor—A matter of love and hate. Peptides 2005, 26, 1401–1409. [Google Scholar] [CrossRef]
- Albiston, A.L.; McDowall, S.G.; Matsacos, D.; Sim, P.; Clune, E.; Mustafa, T.; Lee, J.; Mendelsohn, F.A.O.; Simpson, R.J.; Connolly, L.M.; et al. Evidence that the angiotensin IV (AT4) receptor is the enzyme insulin-regulated aminopeptidase. J. Biol. Chem. 2001, 276, 48623–48626. [Google Scholar] [CrossRef] [Green Version]
- Silva, I.M.S.; Assersen, K.B.; Willadsen, N.N.; Jepsen, J.; Artuc, M.; Steckelings, U.M. The role of the renin-angiotensin system in skin physiology and pathophysiology. Exp. Dermatol. 2020, 29, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.; Danser, A.H.J. Prorenin and (pro)renin receptor: A review of available data from in vitro studies and experimental models in rodents. Exp. Physiol. 2008, 93, 557–563. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.R.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, E.; Oparil, S. Role of aliskiren in cardio-renal protection and use in hypertensives with multiple risk factors. Ther. Clin. Risk Manag. 2009, 5, 459–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Ju, Q.; Sun, J.; Huang, L.; Wu, S.; Wang, S.; Li, Y.; Guan, Z.; Zhu, Q.; Xu, Y. Discovery of Novel Dual Extracellular Regulated Protein Kinases (ERK) and Phosphoinositide 3-Kinase (PI3K) Inhibitors as a Promising Strategy for Cancer Therapy. Molecules 2020, 25, 5693. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Tsukamoto, I.; Inoue, S.; Hashimoto, K.; Akagi, M. Cyclic compressive loading activates angiotensin II type 1 receptor in articular chondrocytes and stimulates hypertrophic differentiation through a G-protein-dependent pathway. FEBS Open Bio 2018, 8, 962–973. [Google Scholar] [CrossRef]
- Touyz, R.M. Reactive oxygen species and angiotensin II signaling in vascular cells: Implications in cardiovascular disease. Braz. J. Med. Biol. Res. 2004, 37, 1263–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branicky, R.; Wang, Y.; Khaki, A.; Liu, J.-L.; Kramer-Drauberg, M.; Hekimi, S. Stimulation of RAS-dependent ROS signaling extends longevity by modulating a developmental program of global gene expression. Sci. Adv. 2022, 8, eadc9851. [Google Scholar] [CrossRef]
- Mohamed, H.R.H. Acute Oral Administration of Cerium Oxide Nanoparticles Suppresses Lead Acetate–Induced Genotoxicity, Inflammation, and ROS Generation in Mice Renal and Cardiac Tissues. Biol. Trace Elem. Res. 2022, 200, 3284–3293. [Google Scholar] [CrossRef]
- Barp, C.G.; Bonaventura, D.; Assreuy, J. NO, ROS, RAS, and PVAT: More Than a Soup of Letters. Front. Physiol. 2021, 12, 640021. [Google Scholar] [CrossRef]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Laufs, U.; Bäumer, A.T.; Müller, K.; Konkol, C.; Sauer, H.; Böhm, M.; Nickenig, G. Inhibition of geranylgeranylation reduces angiotensin II-mediated free radical production in vascular smooth muscle cells: Involvement of angiotensin AT1 receptor expression and Rac1 GTPase. Mol. Pharmacol. 2001, 59, 646–654. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97. [Google Scholar] [CrossRef]
- Berk, B.C.; Corson, M.A. Angiotensin II signal transduction in vascular smooth muscle: Role of tyrosine kinases. Circ. Res. 1997, 80, 607–616. [Google Scholar] [CrossRef]
- Ren, Y.; Xie, W.; Yang, S.; Jiang, Y.; Wu, D.; Zhang, H.; Sheng, S. Angiotensin-converting enzyme 2 inhibits inflammation and apoptosis in high glucose-stimulated microvascular endothelial cell damage by regulating the JAK2/STAT3 signaling pathway. Bioengineered 2022, 13, 10802–10810. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-H.; Choi, J.-W.; Lee, M.-K.; Choi, Y.-H.; Nam, T.-J. Effect of Cyclophilin from Pyropia Yezoensis on the Proliferation of Intestinal Epithelial Cells by Epidermal Growth Factor Receptor/Ras Signaling Pathway. Mar. Drugs 2019, 17, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-H.; Chung, J.-K. Akt: Versatile mediator of cell survival and beyond. Korean Soc. Biochem. Mol. Biol.-BMB Rep. 2002, 35, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Azouz, A.A.; Abdel-Rahman, D.M.; Messiha, B.A.S. Balancing renal Ang-II/Ang-(1-7) by xanthenone; an ACE2 activator; contributes to the attenuation of Ang-II/p38 MAPK/NF-κB p65 and Bax/caspase-3 pathways in amphotericin B-induced nephrotoxicity in rats. Toxicol. Mech. Methods 2023, 33, 452–462. [Google Scholar] [CrossRef]
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006, 20, 1546–1548. [Google Scholar] [CrossRef] [Green Version]
- Struthers, A.D.; MacDonald, T.M. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc. Res. 2004, 61, 663–670. [Google Scholar] [CrossRef] [Green Version]
- Dehe, L.; Mousa, S.A.; Aboryag, N.; Shaqura, M.; Beyer, A.; Schäfer, M.; Treskatsch, S. Identification of Mineralocorticoid Receptors, Aldosterone, and Its Processing Enzyme CYP11B2 on Parasympathetic and Sympathetic Neurons in Rat Intracardiac Ganglia. Front. Neuroanat. 2021, 15, 802359. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Han, W.; Shi, S.; Hu, Y. The effects of ACEI/ARB, aldosterone receptor antagonists and statins on preventing recurrence of atrial fibrillation: A protocol for systematic review and network meta-analysis. Medicine 2021, 100, e24280. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, R.T.; Kevak, R.M.; Kang, P.M.; Frishman, W.H. Angiotensin II receptor blockade: An innovative approach to cardiovascular pharmacotherapy. J. Clin. Pharmacol. 1993, 33, 1023–1038. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.P.; Votteas, V. Role of perindopril in the prevention of stroke. Recent Pat. Cardiovasc. Drug Discov. 2006, 1, 283–289. [Google Scholar] [CrossRef]
- Mogensen, U.M.; Gong, J.; Jhund, P.S.; Shen, L.; Køber, L.; Desai, A.S.; Lefkowitz, M.P.; Packer, M.; Rouleau, J.L.; Solomon, S.D.; et al. Effect of sacubitril/valsartan on recurrent events in the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM-HF). Eur. J. Heart Fail. 2018, 20, 760–768. [Google Scholar] [CrossRef]
- Hornig, B.; Landmesser, U.; Kohler, C.; Ahlersmann, D.; Spiekermann, S.; Christoph, A.; Tatge, H.; Drexler, H. Comparative effect of ace inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease: Role of superoxide dismutase. Circulation 2001, 103, 799–805. [Google Scholar] [CrossRef] [Green Version]
- Yusuf, S.; Sleight, P.; Pogue, J.; Bosch, J.; Davies, R.; Dagenais, G.; Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N. Engl. J. Med. 2000, 342, 145–153. [Google Scholar] [CrossRef]
- Schmieder, R.E.; Martus, P.; Klingbeil, A. Reversal of left ventricular hypertrophy in essential hypertension. A meta-analysis of randomized double-blind studies. JAMA 1996, 275, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.M. The EURopean Trial on Reduction of Cardiac Events with Perindopril in Stable Coronary Artery Disease Investigators. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: Randomised, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet 2003, 362, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E.; Domanski, M.J.; Fowler, S.E.; Geller, N.L.; Gersh, B.J.; Hsia, J.; Pfeffer, M.A.; Rice, M.M.; Rosenberg, Y.D.; Rouleau, J.L.; et al. Angiotensin-converting-enzyme inhibition in stable coronary artery disease. N. Engl. J. Med. 2004, 351, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef]
- Warnholtz, A.; Nickenig, G.; Schulz, E.; Macharzina, R.; Bräsen, J.H.; Skatchkov, M.; Heitzer, T.; Stasch, J.P.; Griendling, K.K.; Harrison, D.G.; et al. Increased NADH-oxidase-mediated superoxide production in the early stages of atherosclerosis: Evidence for involvement of the renin-angiotensin system. Circulation 1999, 99, 2027–2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cernecka, H.; Doka, G.; Srankova, J.; Pivackova, L.; Malikova, E.; Galkova, K.; Kyselovic, J.; Krenek, P.; Klimas, J. Ramipril restores PPARβ/δ and PPARγ expressions and reduces cardiac NADPH oxidase but fails to restore cardiac function and accompanied myosin heavy chain ratio shift in severe anthracycline-induced cardiomyopathy in rat. Eur. J. Pharmacol. 2016, 791, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Petersen, S.V.; Poulsen, N.B.; Linneberg Matthiesen, C.; Vilhardt, F. Novel and Converging Ways of NOX2 and SOD3 in Trafficking and Redox Signaling in Macrophages. Antioxidants 2021, 10, 172. [Google Scholar] [CrossRef] [PubMed]
- Walczak-Gałęzewska, M.; Szulińska, M.; Miller-Kasprzak, E.; Pupek-Musialik, D.; Bogdański, P. The effect of nebivolol and ramipril on selected biochemical parameters, arterial stiffness, and circadian profile of blood pressure in young men with primary hypertension: A 12-week prospective randomized, open-label study trial. Medicine 2018, 97, e11717. [Google Scholar] [CrossRef]
- Ridker, P.M.; Danielson, E.; Rifai, N.; Glynn, R.J.; Val-MARC Investigators. Valsartan, blood pressure reduction, and C-reactive protein: Primary report of the Val-MARC trial. Hypertension 2006, 48, 73–79. [Google Scholar] [CrossRef] [Green Version]
- Ghiadoni, L.; Versari, D.; Magagna, A.; Kardasz, I.; Plantinga, Y.; Giannarelli, C.; Taddei, S.; Salvetti, A. Ramipril dose-dependently increases nitric oxide availability in the radial artery of essential hypertension patients. J. Hypertens. 2007, 25, 361–366. [Google Scholar] [CrossRef]
- Ghiadoni, L.; Virdis, A.; Magagna, A.; Taddei, S.; Salvetti, A. Effect of the angiotensin II type 1 receptor blocker candesartan on endothelial function in patients with essential hypertension. Hypertension 2000, 35 Pt 2, 501–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffrin, E.L.; Deng, L.Y. Comparison of effects of angiotensin I– converting enzyme inhibition and β-blockade for 2 years on function of small arteries from hypertensive patients. Hypertension 1995, 25 Pt 2, 699–703. [Google Scholar] [CrossRef]
- Schmieder, R.E.; Delles, C.; Mimran, A.; Fauvel, J.P.; Ruilope, L.M. Impact of telmisartan versus ramipril on renal endothelial function in patients with hypertension and type 2 diabetes. Diabetes Care 2007, 30, 1351–1356. [Google Scholar] [CrossRef] [Green Version]
- Goh, K.L.; Bhaskaran, K.; Minassian, C.; Evans, S.J.; Smeeth, L.; Douglas, I.J. Angiotensin receptor blockers and risk of dementia: Cohort study in UK Clinical Practice Research Datalink. Br. J. Clin. Pharmacol. 2015, 79, 337–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, Y.; Ye, P.; Zhu, L.; Kong, X.; Qu, X.; Zhang, J.; Luo, J.; Yang, H.; Chen, S. Low shear stress induces endothelial reactive oxygen species via the AT1R/eNOS/NO pathway. J. Cell. Physiol. 2018, 233, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Suzuki, J.; Yamawaki, H.; Sharma, V.K.; Sheu, S.-S.; Berk, B.C. Losartan metabolite EXP3179 activates Akt and endothelial nitric oxide synthase via vascular endothelial growth factor receptor-2 in endothelial cells: Angiotensin II type 1 receptor-independent effects of EXP3179. Circulation 2005, 112, 1798–1805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, M.; Fujimoto, S.; Arakawa, S.; Yada, T.; Namikoshi, T.; Haruna, Y.; Horike, H.; Sasaki, T.; Kashihara, N. Angiotensin II type 1 receptor blocker ameliorates uncoupled endothelial nitric oxide synthase in rats with experimental diabetic nephropathy. Nephrol. Dial. Transplant. 2008, 23, 3806–3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imanishi, T.; Tsujioka, H.; Ikejima, H.; Kuroi, A.; Takarada, S.; Kitabata, H.; Tanimoto, T.; Muragaki, Y.; Mochizuki, S.; Goto, M.; et al. Renin inhibitor aliskiren improves impaired nitric oxide bioavailability and protects against atherosclerotic changes. Hypertension 2008, 52, 563–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussberger, J.; Aubert, J.-F.; Bouzourene, K.; Pellegrin, M.; Hayoz, D.; Mazzolai, L. Renin inhibition by aliskiren prevents atherosclerosis progression: Comparison with irbesartan, atenolol, and amlodipine. Hypertension 2008, 51, 1306–1311. [Google Scholar] [CrossRef] [Green Version]
- Honjo, T.; Yamaoka-Tojo, M.; Inoue, N. Pleiotropic effects of ARB in vascular metabolism--focusing on atherosclerosis-based cardiovascular disease. Curr. Vasc. Pharmacol. 2011, 9, 145–152. [Google Scholar] [CrossRef]
- Fleming, I. Signaling by the angiotensin-converting enzyme. Circ. Res. 2006, 98, 887–896. [Google Scholar] [CrossRef]
- Horne, M.C.; Hell, J.W. Angiotensin II signalling kicks out p27Kip1: Casein kinase 2 augmentation of Cav1.2 L-type Ca2+ channel activity in immature ventricular cardiomyocytes. J. Physiol. 2017, 595, 4131–4132. [Google Scholar] [CrossRef]
- Kashihara, T.; Nakada, T.; Kojima, K.; Takeshita, T.; Yamada, M. Angiotensin II activates CaV1.2 Ca2+ channels through β-arrestin2 and casein kinase 2 in mouse immature cardiomyocytes. J. Physiol. 2017, 595, 4207–4225. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I.; Kohlstedt, K.; Busse, R. New fACEs to the renin-angiotensin system. Physiology 2005, 20, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hu, H.; Wang, Y.; Xue, M.; Li, X.; Cheng, W.; Xuan, Y.; Yin, J.; Yang, N.; Yan, S. Valsartan Upregulates Kir2.1 in Rats Suffering from Myocardial Infarction via Casein Kinase 2. Cardiovasc. Drugs Ther. 2015, 29, 209–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daub, S.; Lutgens, E.; Münzel, T.; Daiber, A. CD40/CD40L and Related Signaling Pathways in Cardiovascular Health and Disease—The Pros and Cons for Cardioprotection. Int. J. Mol. Sci. 2020, 21, 8533. [Google Scholar] [CrossRef] [PubMed]
- Senchenkova, E.Y.; Russell, J.; Vital, S.A.; Yildirim, A.; Orr, A.W.; Granger, D.N.; Gavins, F.N.E. A critical role for both CD40 and VLA5 in angiotensin II–mediated thrombosis and inflammation. FASEB J. 2018, 32, 3448–3456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tousoulis, D.; Antoniades, C.; Nikolopoulou, A.; Koniari, K.; Vasiliadou, C.; Marinou, K.; Koumallos, N.; Papageorgiou, N.; Stefanadi, E.; Siasos, G.; et al. Interaction between cytokines and sCD40L in patients with stable and unstable coronary syndromes. Eur. J. Clin. Investig. 2007, 37, 623–628. [Google Scholar] [CrossRef]
- Antoniades, C.; Antonopoulos, A.S.; Tousoulis, D.; Stefanadis, C. Adiponectin: From obesity to cardiovascular disease. Obes. Rev. 2009, 10, 269–279. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Liu, L.-Y.; Wu, J.-X.; Chen, S.-X.; Sun, Y.-X. Comparison of captopril and enalapril to study the role of the sulfhydryl-group in improvement of endothelial dysfunction with ACE inhibitors in high dieted methionine mice. J. Cardiovasc. Pharmacol. 2006, 47, 82–88. [Google Scholar] [CrossRef]
- Buikema, H.; Monnink, S.H.J.; Tio, R.A.; Crijns, H.J.G.M.; de Zeeuw, D.; van Gilst, W.H. Comparison of zofenopril and lisinopril to study the role of the sulfhydryl-group in improvement of endothelial dysfunction with ACE-inhibitors in experimental heart failure. Br. J. Pharmacol. 2000, 130, 1999–2007. [Google Scholar] [CrossRef] [Green Version]
- Julius, S.; Kjeldsen, S.E.; Weber, M.; Brunner, H.R.; Ekman, S.; Hansson, L.; Hua, T.; Laragh, J.; McInnes, G.T.; Mitchell, L.; et al. Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: The VALUE randomised trial. Lancet 2004, 363, 2022–2031. [Google Scholar] [CrossRef]
- Strauss, M.H.; Hall, A.S. Angiotensin receptor blockers may increase risk of myocardial infarction: Unraveling the ARB-MI paradox. Circulation 2006, 114, 838–854. [Google Scholar] [CrossRef] [Green Version]
- Senbonmatsu, T.; Ichihara, S.; Price, E., Jr.; Gaffney, F.A.; Inagami, T. Evidence for angiotensin II type 2 receptor-mediated cardiac myocyte enlargement during in vivo pressure overload. J. Clin. Investig. 2000, 106, R25–R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amore, A.; Black, M.J.; Thomas, W.G. The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor-mediated hypertrophy. Hypertension 2005, 46, 1347–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trocha, M.; Szuba, A.; Merwid-Lad, A.; Sozański, T. Effect of selected drugs on plasma asymmetric dimethylarginine (ADMA) levels. Pharmazie 2010, 65, 562–571. [Google Scholar] [PubMed]
- Kalinowski, L.; Malinski, T. Endothelial NADH/NADPH-dependent enzymatic sources of superoxide production: Relationship to endothelial dysfunction. Acta Biochim. Pol. 2004, 51, 459–469. [Google Scholar] [CrossRef]
- Wang, C.; Luo, Z.; Carter, G.; Wellstein, A.; Jose, P.A.; Tomlinson, J.; Leiper, J.; Welch, W.J.; Wilcox, C.S.; Wang, D. NRF2 prevents hypertension, increased ADMA, microvascular oxidative stress, and dysfunction in mice with two weeks of ANG II infusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R399–R406. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, H.; Liu, H.; Zhang, X.; Ran, G.; He, H.; Liu, X. Modeling Disease Progression: Angiotensin II Indirectly Inhibits Nitric Oxide Production via ADMA Accumulation in Spontaneously Hypertensive Rats. Front. Physiol. 2016, 7, 555. [Google Scholar] [CrossRef] [Green Version]
- Shahin, N.N.; Abdelkader, N.F.; Safar, M.M. A Novel Role of Irbesartan in Gastroprotection against Indomethacin-Induced Gastric Injury in Rats: Targeting DDAH/ADMA and EGFR/ERK Signaling. Sci. Rep. 2018, 8, 4280. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.-F.; Xiong, Y.; Guo, Z. A reduction of endogenous asymmetric dimethylarginine contributes to the effect of captopril on endothelial dysfunction induced by homocysteine in rats. Eur. J. Pharmacol. 2005, 508, 167–175. [Google Scholar] [CrossRef]
- Ito, A.; Egashira, K.; Narishige, T.; Muramatsu, K.; Takeshita, A. Renin-angiotensin system is involved in the mechanism of increased serum asymmetric dimethylarginine in essential hypertension. Jpn. Circ. J. 2001, 65, 775–778. [Google Scholar] [CrossRef] [Green Version]
- Napoli, C.; Sica, V.; de Nigris, F.; Pignalosa, O.; Condorelli, M.; Ignarro, L.J.; Liguori, A. Sulfhydryl angiotensin-converting enzyme inhibition induces sustained reduction of systemic oxidative stress and improves the nitric oxide pathway in patients with essential hypertension. Am. Heart J. 2004, 148, 172. [Google Scholar] [CrossRef]
- Ito, A.; Egashira, K.; Narishige, T.; Muramatsu, K.; Takeshita, A. Angiotensin-converting enzyme activity is involved in the mechanism of increased endogenous nitric oxide synthase inhibitor in patients with type 2 diabetes mellitus. Circ. J. 2002, 66, 811–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibal, L.; Agarwal, S.C.; Home, P.D.; Boger, R.H. The Role of Asymmetric Dimethylarginine (ADMA) in Endothelial Dysfunction and Cardiovascular Disease. Curr. Cardiol. Rev. 2010, 6, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-W.; Hsu, N.-W.; Wu, T.-C.; Lin, S.-J.; Chang, M.-S. Long-term angiotensin-converting enzyme inhibition reduces plasma asymmetric dimethylarginine and improves endothelial nitric oxide bioavailability and coronary microvascular function in patients with syndrome X. Am. J. Cardiol. 2002, 90, 974–982. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Author | Agents | Population | Period | Outcome |
|---|---|---|---|---|
| Walczak-Galezewska et al. [54] | Ramipril vs. Nebivolol | Hypertension | 12 weeks | Ramipril demonstrated a decrease in CRP levels in comparison to nebivolol. |
| Ridker et al. [55] | HCTZ/Valsartan vs. Valsartan | Hypertension | 6 weeks | CRP levels were reduced solely through treatment with valsartan. |
| Ghiadoni et al. [56] | Lisopril | Ventricular hypertrophy | 3 years | Endothelial function showed improvement with lisinopril treatment when compared to the initial baseline. |
| Ghiadoni et al. [57] | Candesartan | Hypertension | 1 year | Endothelial function demonstrated improvement with candesartan treatment when compared to the initial baseline. |
| Schiffrin et al. [58] | Losartan | Hypertension | 1 year | Endothelial function showed improvement with losartan treatment in comparison to the initial baseline. |
| Schmieder et al. [59] | Telmisaran | Hypertension and DM | 9 weeks | Endothelial function demonstrated improvement with telmisartan treatment in comparison to the initial baseline. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koumallos, N.; Sigala, E.; Milas, T.; Baikoussis, N.G.; Aragiannis, D.; Sideris, S.; Tsioufis, K. Angiotensin Regulation of Vascular Homeostasis: Exploring the Role of ROS and RAS Blockers. Int. J. Mol. Sci. 2023, 24, 12111. https://doi.org/10.3390/ijms241512111
Koumallos N, Sigala E, Milas T, Baikoussis NG, Aragiannis D, Sideris S, Tsioufis K. Angiotensin Regulation of Vascular Homeostasis: Exploring the Role of ROS and RAS Blockers. International Journal of Molecular Sciences. 2023; 24(15):12111. https://doi.org/10.3390/ijms241512111
Chicago/Turabian StyleKoumallos, Nikolaos, Evangelia Sigala, Theodoros Milas, Nikolaos G. Baikoussis, Dimitrios Aragiannis, Skevos Sideris, and Konstantinos Tsioufis. 2023. "Angiotensin Regulation of Vascular Homeostasis: Exploring the Role of ROS and RAS Blockers" International Journal of Molecular Sciences 24, no. 15: 12111. https://doi.org/10.3390/ijms241512111