
Pediatric Type 1 Diabetes: Mechanisms and Impact of Technologies on Comorbidities and Life Expectancy

Abstract
:1. Introduction
2. T1D Pathophysiology
3. Continuous Glucose Monitoring Systems (CGM)
4. Insulin Pumps
5. Hybrid Systems and Artificial Pancreas (AP)
6. Impact of Technologies on Diabetes-Related Comorbidities
6.1. Cardiovascular Disease and Type 1 Diabetes
6.2. Diabetic Nephropathy
6.3. Diabetic Retinopathy
6.4. Diabetic Neuropathy
6.5. Impact of T1D on Bone Health
6.5.1. DKK-1 and Sclerostin
6.5.2. Irisin
7. Estimated Life Expectancy
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nathan, D.M. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: Overview. Diabetes Care 2014, 37, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.R.; Chen, Y.T.; Sheu, W.H. Glycemic variability and diabetes retinopathy: A missing link. J. Diabetes Its Complicat. 2015, 29, 302–306. [Google Scholar] [CrossRef]
- Nevo-Shenker, M.; Shalitin, S. The Impact of Hypo- and Hyperglycemia on Cognition and Brain Development in Young Children with Type 1 Diabetes. Horm. Res. Paediatr. 2021, 94, 115–123. [Google Scholar] [CrossRef]
- De Bock, M.; Codner, E.; Craig, M.E.; Huynh, T.; Maahs, D.M.; Mahmud, F.H.; Marcovecchio, L.; DiMeglio, L.A. ISPAD Clinical Practice Consensus Guidelines 2022: Glycemic targets and glucose monitoring for children, adolescents, and young people with diabetes. Pediatr. Diabetes 2022, 23, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Bergenstal, R.M.; Tamborlane, W.V.; Ahmann, A.; Buse, J.B.; Dailey, G.; Davis, S.N.; Joyce, C.; Peoples, T.; Perkins, B.A.; Welsh, J.B.; et al. Effectiveness of sensor-augmented insulin-pump therapy in type 1 diabetes. N. Engl. J. Med. 2010, 363, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gale, E.A.M. The Discovery of Type 1 Diabetes. Diabetes 2001, 50, 217–226. [Google Scholar] [CrossRef] [Green Version]
- Herold, K.C.; Gitelman, S.E.; Ehlers, M.R.; Gottlieb, P.A.; Greenbaum, C.J.; Hagopian, W.; Boyle, K.D.; Keyes-Elstein, L.; Aggarwal, S.; Phippard, D.; et al. Teplizumab (anti-CD3 mAb) treatment preserves C-peptide responses in patients with new-onset type 1 diabetes in a randomized controlled trial: Metabolic and immunologic features at baseline identify a subgroup of responders. Diabetes 2013, 62, 3766–3774. [Google Scholar] [CrossRef] [Green Version]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Quattrin, T.; Haller, M.J.; Steck, A.K.; Felner, E.I.; Li, Y.; Xia, Y.; Leu, J.H.; Zoka, R.; Hedrick, J.A.; Rigby, M.R.; et al. Golimumab and Beta-Cell Function in Youth with New-Onset Type 1 Diabetes. N. Engl. J. Med. 2020, 383, 2007–2017. [Google Scholar] [CrossRef]
- Pociot, F. Type 1 diabetes genome-wide association studies: Not to be lost in translation. Clin. Transl. Immunol. 2017, 6, e162. [Google Scholar] [CrossRef] [Green Version]
- Wilcox, N.S.; Rui, J.; Hebrok, M.; Herold, K.C. Life and death of β cells in Type 1 diabetes: A comprehensive review. J. Autoimmun. 2016, 71, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.; Colon-Negron, K.; Papa, F.R. Endoplasmic reticulum stress, degeneration of pancreatic islet β-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol. Metab. 2019, 27, S60–S68. [Google Scholar] [CrossRef] [PubMed]
- Szabat, M.; Page, M.M.; Panzhinskiy, E.; Skovsø, S.; Mojibian, M.; Fernandez-Tajes, J.; Bruin, J.E.; Bround, M.J.; Lee, J.T.; Xu, E.E.; et al. Reduced Insulin Production Relieves Endoplasmic Reticulum Stress and Induces β Cell Proliferation. Cell Metab. 2016, 23, 179–193. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Brandizzi, F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol. 2013, 23, 547–555. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; van Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.J.; Shah, A.; Ntranos, V.; Van Gool, F.; Atkinson, M.; Bhushan, A. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab. 2019, 29, 1045–1060.e10. [Google Scholar] [CrossRef]
- Horwitz, E.; Krogvold, L.; Zhitomirsky, S.; Swisa, A.; Fischman, M.; Lax, T.; Dahan, T.; Hurvitz, N.; Weinberg-Corem, N.; Klochendler, A.; et al. β-Cell DNA Damage Response Promotes Islet Inflammation in Type 1 Diabetes. Diabetes 2018, 67, 2305–2318. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Ramsey, M.R.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N.E. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef]
- Helman, A.; Klochendler, A.; Azazmeh, N.; Gabai, Y.; Horwitz, E.; Anzi, S.; Swisa, A.; Condiotti, R.; Granit, R.Z.; Nevo, Y.; et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med. 2016, 22, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguayo-Mazzucato, C.; Andle, J.; Lee, T.B., Jr.; Midha, A.; Talemal, L.; Chipashvili, V.; Hollister-Lock, J.; van Deursen, J.; Weir, G.; Bonner-Weir, S. Acceleration of β Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab. 2019, 30, 129–142.e4. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Sims, E.K.; Syed, F.; Nyalwidhe, J.; Bahnson, H.T.; Haataja, L.; Speake, C.; Morris, M.A.; Balamurugan, A.N.; Mirmira, R.G.; Nadler, J.; et al. Abnormalities in proinsulin processing in islets from individuals with longstanding T1D. Transl. Res. J. Lab. Clin. Med. 2019, 213, 90–99. [Google Scholar] [CrossRef]
- Wasserfall, C.; Nick, H.S.; Campbell-Thompson, M.; Beachy, D.; Haataja, L.; Kusmartseva, I.; Posgai, A.; Beery, M.; Rhodes, C.; Bonifacio, E.; et al. Persistence of Pancreatic Insulin mRNA Expression and Proinsulin Protein in Type 1 Diabetes Pancreata. Cell Metab. 2017, 26, 568–575.e3. [Google Scholar] [CrossRef] [Green Version]
- Lam, C.J.; Chatterjee, A.; Shen, E.; Cox, A.R.; Kushner, J.A. Low-Level Insulin Content Within Abundant Non-β Islet Endocrine Cells in Long-standing Type 1 Diabetes. Diabetes 2019, 68, 598–608. [Google Scholar] [CrossRef] [Green Version]
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting Adult Pancreatic Islet α Cells into β Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.C.; Taylor, A.J.; Verchere, C.B. Islet prohormone processing in health and disease. Diabetes Obes. Metab. 2018, 20 (Suppl. 2), 64–76. [Google Scholar] [CrossRef] [Green Version]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugliani, M.; Mossuto, S.; Grano, F.; Suleiman, M.; Marselli, L.; Boggi, U.; De Simone, P.; Eizirik, D.L.; Cnop, M.; Marchetti, P.; et al. Modulation of Autophagy Influences the Function and Survival of Human Pancreatic Beta Cells Under Endoplasmic Reticulum Stress Conditions and in Type 2 Diabetes. Front. Endocrinol. 2019, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muralidharan, C.; Conteh, A.M.; Marasco, M.R.; Crowder, J.J.; Kuipers, J.; de Boer, P.; Linnemann, A.K. Pancreatic beta cell autophagy is impaired in type 1 diabetes. Diabetologia 2021, 64, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Stimpson, S.E.; Fernandez-Bueno, G.A.; Mathews, C.E. Mitochondrial Reactive Oxygen Species and Type 1 Diabetes. Antioxid. Redox Signal. 2018, 29, 1361–1372. [Google Scholar] [CrossRef]
- Pickup, J.C.; Keen, H.; Parsons, J.A.; Alberti, K.G. Continuous subcutaneous insulin infusion: An approach to achieving normoglycaemia. Br. Med. J. 1978, 1, 204–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhardsson, P.; Schwandt, A.; Witsch, M.; Kordonouri, O.; Svensson, J.; Forsander, G.; Battelino, T.; Veeze, H.; Danne, T. The SWEET Project 10-Year Benchmarking in 19 Countries Worldwide Is Associated with Improved HbA1c and Increased Use of Diabetes Technology in Youth with Type 1 Diabetes. Diabetes Technol. Ther. 2021, 23, 491–499. [Google Scholar] [CrossRef]
- Basu, A.; Dube, S.; Veettil, S.; Slama, M.; Kudva, Y.C.; Peyser, T.; Carter, R.E.; Cobelli, C.; Basu, R. Time lag of glucose from intravascular to interstitial compartment in type 1 diabetes. J. Diabetes Sci. Technol. 2015, 9, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keenan, D.B.; Mastrototaro, J.J.; Voskanyan, G.; Steil, G.M. Delays in minimally invasive continuous glucose monitoring devices: A review of current technology. J. Diabetes Sci. Technol. 2009, 3, 1207–1214. [Google Scholar] [CrossRef] [Green Version]
- Helton, K.L.; Ratner, B.D.; Wisniewski, N.A. Biomechanics of the sensor-tissue interface-effects of motion, pressure, and design on sensor performance and foreign body response-part II: Examples and application. J. Diabetes Sci. Technol. 2011, 5, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Heinemann, L.; Schoemaker, M.; Schmelzeisen-Redecker, G.; Hinzmann, R.; Kassab, A.; Freckmann, G.; Reiterer, F.; Del Re, L. Benefits and Limitations of MARD as a Performance Parameter for Continuous Glucose Monitoring in the Interstitial Space. J. Diabetes Sci. Technol. 2020, 14, 135–150. [Google Scholar] [CrossRef]
- Alva, S.; Bailey, T.; Brazg, R.; Budiman, E.S.; Castorino, K.; Christiansen, M.P.; Forlenza, G.; Kipnes, M.; Liljenquist, D.R.; Liu, H. Accuracy of a 14-Day Factory-Calibrated Continuous Glucose Monitoring System With Advanced Algorithm in Pediatric and Adult Population With Diabetes. J. Diabetes Sci. Technol. 2022, 16, 70–77. [Google Scholar] [CrossRef]
- Christiansen, M.P.; Klaff, L.J.; Bailey, T.S.; Brazg, R.; Carlson, G.; Tweden, K.S. A Prospective Multicenter Evaluation of the Accuracy and Safety of an Implanted Continuous Glucose Sensor: The PRECISION Study. Diabetes Technol. Ther. 2019, 21, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, V.N.; Laffel, L.M.; Wadwa, R.P.; Garg, S.K. Performance of a Factory-Calibrated Real-Time Continuous Glucose Monitoring System Utilizing an Automated Sensor Applicator. Diabetes Technol. Ther. 2018, 20, 428–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafri, R.Z.; Balliro, C.A.; El-Khatib, F.; Maheno, M.M.; Hillard, M.A.; O’Donovan, A.; Selagamsetty, R.; Zheng, H.; Damiano, E.R.; Russell, S.J. A Three-Way Accuracy Comparison of the Dexcom G5, Abbott Freestyle Libre Pro, and Senseonics Eversense Continuous Glucose Monitoring Devices in a Home-Use Study of Subjects with Type 1 Diabetes. Diabetes Technol. Ther. 2020, 22, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.S.; Alva, S. Landscape of Continuous Glucose Monitoring (CGM) and Integrated CGM: Accuracy Considerations. Diabetes Technol. Ther. 2021, 23, S5–S11. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.K.; Liljenquist, D.; Bode, B.; Christiansen, M.P.; Bailey, T.S.; Brazg, R.L.; Denham, D.S.; Chang, A.R.; Akturk, H.K.; Dehennis, A.; et al. Evaluation of Accuracy and Safety of the Next-Generation Up to 180-Day Long-Term Implantable Eversense Continuous Glucose Monitoring System: The PROMISE Study. Diabetes Technol. Ther. 2022, 24, 84–92. [Google Scholar] [CrossRef]
- Bailey, T.; Bode, B.W.; Christiansen, M.P.; Klaff, L.J.; Alva, S. The Performance and Usability of a Factory-Calibrated Flash Glucose Monitoring System. Diabetes Technol. Ther. 2015, 17, 787–794. [Google Scholar] [CrossRef]
- Battelino, T.; Danne, T.; Bergenstal, R.M.; Amiel, S.A.; Beck, R.; Biester, T.; Bosi, E.; Buckingham, B.A.; Cefalu, W.T.; Close, K.L.; et al. Clinical Targets for Continuous Glucose Monitoring Data Interpretation: Recommendations From the International Consensus on Time in Range. Diabetes Care 2019, 42, 1593–1603. [Google Scholar] [CrossRef] [Green Version]
- Pickup, J.C.; Keen, H.; Parsons, J.A.; Alberti, K.G.; Rowe, A.S. Continuous subcutaneous insulin infusion: Improved blood-glucose and intermediary-metabolite control in diabetics. Lancet 1979, 1, 1255–1257. [Google Scholar] [CrossRef]
- Tauschmann, M.; Forlenza, G.; Hood, K.; Cardona-Hernandez, R.; Giani, E.; Hendrieckx, C.; DeSalvo, D.J.; Laffel, L.M.; Saboo, B.; Wheeler, B.J.; et al. ISPAD Clinical Practice Consensus Guidelines 2022: Diabetes technologies: Glucose monitoring. Pediatr. Diabetes 2022, 23, 1390–1405. [Google Scholar] [CrossRef]
- Cherubini, V.; Bonfanti, R.; Casertano, A.; De Nitto, E.; Iannilli, A.; Lombardo, F.; Maltoni, G.; Marigliano, M.; Bassi, M.; Minuto, N.; et al. Time In Range in Children with Type 1 Diabetes Using Treatment Strategies Based on Nonautomated Insulin Delivery Systems in the Real World. Diabetes Technol. Ther. 2020, 22, 509–515. [Google Scholar] [CrossRef]
- Prahalad, P.; Schwandt, A.; Besançon, S.; Mohan, M.; Obermannova, B.; Kershaw, M.; Bonfanti, R.; Lyckå, A.P.; Hanas, R.; Casteels, K. Hemoglobin A1c trajectories in the first 18 months after diabetes diagnosis in the SWEET diabetes registry. Pediatr. Diabetes 2022, 23, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Trevitt, S.; Simpson, S.; Wood, A. Artificial Pancreas Device Systems for the Closed-Loop Control of Type 1 Diabetes: What Systems Are in Development? J. Diabetes Sci. Technol. 2016, 10, 714–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Rashid, M.; Hobbs, N.; Brandt, R.; Askari, M.R.; Cinar, A. Incorporating Prior Information in Adaptive Model Predictive Control for Multivariable Artificial Pancreas Systems. J. Diabetes Sci. Technol. 2022, 16, 19–28. [Google Scholar] [CrossRef]
- Nimri, R.; Phillip, M. Artificial pancreas: Fuzzy logic and control of glycemia. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 251–256. [Google Scholar] [CrossRef] [PubMed]
- El-Khatib, F.H.; Balliro, C.; Hillard, M.A.; Magyar, K.L.; Ekhlaspour, L.; Sinha, M.; Mondesir, D.; Esmaeili, A.; Hartigan, C.; Thompson, M.J.; et al. Home use of a bihormonal bionic pancreas versus insulin pump therapy in adults with type 1 diabetes: A multicentre randomised crossover trial. Lancet 2017, 389, 369–380. [Google Scholar] [CrossRef] [Green Version]
- Hanaire, H.; Lassmann-Vague, V.; Jeandidier, N.; Renard, E.; Tubiana-Rufi, N.; Vambergue, A.; Raccah, D.; Pinget, M.; Guerci, B. Treatment of diabetes mellitus using an external insulin pump: The state of the art. Diabetes Metab. 2008, 34, 401–423. [Google Scholar] [CrossRef]
- Weisman, A.; Bai, J.W.; Cardinez, M.; Kramer, C.K.; Perkins, B.A. Effect of artificial pancreas systems on glycaemic control in patients with type 1 diabetes: A systematic review and meta-analysis of outpatient randomised controlled trials. Lancet. Diabetes Endocrinol. 2017, 5, 501–512. [Google Scholar] [CrossRef]
- Bergenstal, R.M.; Garg, S.; Weinzimer, S.A.; Buckingham, B.A.; Bode, B.W.; Tamborlane, W.V.; Kaufman, F.R. Safety of a Hybrid Closed-Loop Insulin Delivery System in Patients With Type 1 Diabetes. JAMA 2016, 316, 1407–1408. [Google Scholar] [CrossRef]
- Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar]
- Diabetes Control and Complications Trial Research Group. Effect of intensive diabetes treatment on the development and progression of long-term complications in adolescents with insulin-dependent diabetes mellitus: Diabetes Control and Complications Trial. J. Pediatr. 1994, 125, 177–188. [Google Scholar] [CrossRef]
- Rawshani, A.; Sattar, N.; Franzén, S.; Rawshani, A.; Hattersley, A.T.; Svensson, A.M.; Eliasson, B.; Gudbjörnsdottir, S. Excess mortality and cardiovascular disease in young adults with type 1 diabetes in relation to age at onset: A nationwide, register-based cohort study. Lancet 2018, 392, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das Evcimen, N.; King, G.L. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol. Res. 2007, 55, 498–510. [Google Scholar] [CrossRef]
- Koike, N.; Takamura, T.; Kaneko, S. Induction of reactive oxygen species from isolated rat glomeruli by protein kinase C activation and TNF-alpha stimulation, and effects of a phosphodiesterase inhibitor. Life Sci. 2007, 80, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Sharma, H.; Lencioni, M.; Narendran, P. Cardiovascular disease in type 1 diabetes. Cardiovasc. Endocrinol. Metab. 2019, 8, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Negre-Salvayre, A.; Salvayre, R.; Augé, N.; Pamplona, R.; Portero-Otín, M. Hyperglycemia and glycation in diabetic complications. Antioxid. Redox Signal. 2009, 11, 3071–3109. [Google Scholar] [CrossRef]
- Xu, B.; Ji, Y.; Yao, K.; Cao, Y.X.; Ferro, A. Inhibition of human endothelial cell nitric oxide synthesis by advanced glycation end-products but not glucose: Relevance to diabetes. Clin. Sci. 2005, 109, 439–446. [Google Scholar] [CrossRef]
- Vlassara, H.; Fuh, H.; Makita, Z.; Krungkrai, S.; Cerami, A.; Bucala, R. Exogenous advanced glycosylation end products induce complex vascular dysfunction in normal animals: A model for diabetic and aging complications. Proc. Natl. Acad. Sci. USA 1992, 89, 12043–12047. [Google Scholar] [CrossRef]
- Quehenberger, P.; Bierhaus, A.; Fasching, P.; Muellner, C.; Klevesath, M.; Hong, M.; Stier, G.; Sattler, M.; Schleicher, E.; Speiser, W.; et al. Endothelin 1 transcription is controlled by nuclear factor-kappaB in AGE-stimulated cultured endothelial cells. Diabetes 2000, 49, 1561–1570. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Wang, K.; Penn, M.S.; Marso, S.P.; Lauer, M.A.; Forudi, F.; Zhou, X.; Qu, W.; Lu, Y.; Stern, D.M.; et al. Receptor for AGE (RAGE) mediates neointimal formation in response to arterial injury. Circulation 2003, 107, 2238–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, C.L.; Knight, S.C. Advanced glycation: A novel outlook on atherosclerosis. Curr. Pharm. Des. 2007, 13, 3681–3687. [Google Scholar] [CrossRef] [PubMed]
- Van Eupen, M.G.; Schram, M.T.; Colhoun, H.M.; Scheijen, J.L.; Stehouwer, C.D.; Schalkwijk, C.G. Plasma levels of advanced glycation endproducts are associated with type 1 diabetes and coronary artery calcification. Cardiovasc. Diabetol. 2013, 12, 149. [Google Scholar] [CrossRef] [Green Version]
- Hanssen, N.M.J.; Scheijen, J.; Jorsal, A.; Parving, H.H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D.A.; Schalkwijk, C.G. Higher Plasma Methylglyoxal Levels Are Associated With Incident Cardiovascular Disease in Individuals With Type 1 Diabetes: A 12-Year Follow-up Study. Diabetes 2017, 66, 2278–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanssen, N.M.; Wouters, K.; Huijberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a rupture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, H.; Motoyama, T.; Hirashima, O.; Hirai, N.; Miyao, Y.; Sakamoto, T.; Kugiyama, K.; Ogawa, H.; Yasue, H. Hyperglycemia rapidly suppresses flow-mediated endothelium-dependent vasodilation of brachial artery. J. Am. Coll. Cardiol. 1999, 34, 146–154. [Google Scholar] [CrossRef] [Green Version]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [PubMed]
- Skyler, J.S.; Bergenstal, R.; Bonow, R.O.; Buse, J.; Deedwania, P.; Gale, E.A.; Howard, B.V.; Kirkman, M.S.; Kosiborod, M.; Reaven, P.; et al. Intensive glycemic control and the prevention of cardiovascular events: Implications of the ACCORD, ADVANCE, and VA diabetes trials: A position statement of the American Diabetes Association and a scientific statement of the American College of Cardiology Foundation and the American Heart Association. Circulation 2009, 119, 351–357. [Google Scholar]
- Meza, C.A.; La Favor, J.D.; Kim, D.H.; Hickner, R.C. Endothelial Dysfunction: Is There a Hyperglycemia-Induced Imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [Green Version]
- Alatawi, Z.; Mirghani, H. The Association Between Glycemic Variability and Myocardial Infarction: A Review and Meta-Analysis of Prospective Studies and Randomized Trials. Cureus 2020, 12, e11556. [Google Scholar] [CrossRef]
- Sun, J.; Xu, Y.; Dai, Z.; Sun, Y. Intermittent high glucose enhances proliferation of vascular smooth muscle cells by upregulating osteopontin. Mol. Cell. Endocrinol. 2009, 313, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Novials, A.; Ortega, E.; La Sala, L.; Pujadas, G.; Testa, R.; Bonfigli, A.R.; Esposito, K.; Giugliano, D. Evidence that hyperglycemia after recovery from hypoglycemia worsens endothelial function and increases oxidative stress and inflammation in healthy control subjects and subjects with type 1 diabetes. Diabetes 2012, 61, 2993–2997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.W.; Park, K.H.; Zhou, Y.J. The Impact of Hypoglycemia on the Cardiovascular System: Physiology and Pathophysiology. Angiology 2016, 67, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Schofield, J.; Ho, J.; Soran, H. Cardiovascular Risk in Type 1 Diabetes Mellitus. Diabetes Ther. Res. Treat. Educ. Diabetes Relat. Disord. 2019, 10, 773–789. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, A.; Januszewski, A.; O’Neal, D. The early detection of atherosclerosis in type 1 diabetes: Why, how and what to do about it. Cardiovasc. Endocrinol. Metab. 2019, 8, 14–27. [Google Scholar] [CrossRef]
- Pauley, M.E.; Tommerdahl, K.L.; Snell-Bergeon, J.K.; Forlenza, G.P. Continuous Glucose Monitor, Insulin Pump, and Automated Insulin Delivery Therapies for Type 1 Diabetes: An Update on Potential for Cardiovascular Benefits. Curr. Cardiol. Rep. 2022, 24, 2043–2056. [Google Scholar] [CrossRef]
- Steineck, I.; Cederholm, J.; Eliasson, B.; Rawshani, A.; Eeg-Olofsson, K.; Svensson, A.M.; Zethelius, B.; Avdic, T.; Landin-Olsson, M.; Jendle, J.; et al. Insulin pump therapy, multiple daily injections, and cardiovascular mortality in 18,168 people with type 1 diabetes: Observational study. BMJ (Clin. Res. Ed.) 2015, 350, h3234. [Google Scholar]
- Tubili, C.; Folco, U.D.; Nardone, M.R.; Clementi, A. A Single-Center Long-Term Continuous Subcutaneous Insulin Infusion (CSII) Experience: Higher Fractional Use Is Associated With Less Diabetes Complications. J. Diabetes Sci. Technol. 2017, 11, 1057–1058. [Google Scholar] [CrossRef]
- Faienza, M.F.; Brunetti, G.; Delvecchio, M.; Zito, A.; De Palma, F.; Cortese, F.; Nitti, A.; Massari, E.; Gesualdo, M.; Ricci, G.; et al. Vascular Function and Myocardial Performance Indices in Children Born Small for Gestational Age. Circ. J. Off. J. Jpn. Circ. Soc. 2016, 80, 958–963. [Google Scholar] [CrossRef] [Green Version]
- Giordano, P.; Muggeo, P.; Delvecchio, M.; Carbonara, S.; Romano, A.; Altomare, M.; Ricci, G.; Valente, F.; Zito, A.; Scicchitano, P.; et al. Endothelial dysfunction and cardiovascular risk factors in childhood acute lymphoblastic leukemia survivors. Int. J. Cardiol. 2017, 228, 621–627. [Google Scholar] [CrossRef]
- Faienza, M.F.; Scicchitano, P.; Lamparelli, R.; Zaza, P.; Cecere, A.; Brunetti, G.; Cortese, F.; Valente, F.; Delvecchio, M.; Giordano, P.; et al. Vascular and Myocardial Function in Young People with Type 1 Diabetes Mellitus: Insulin Pump Therapy Versus Multiple Daily Injections Insulin Regimen. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2022, 130, 415–422. [Google Scholar]
- KDOQI. KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2007, 49 (Suppl. 2), S12–S154. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Jick, S.; Breitenstein, S.; Michel, A. Prevalence of Diabetes and Diabetic Nephropathy in a Large U.S. Commercially Insured Pediatric Population, 2002–2013. Diabetes Care 2016, 39, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Vehik, K.; Hamman, R.F.; Lezotte, D.; Norris, J.M.; Klingensmith, G.; Bloch, C.; Rewers, M.; Dabelea, D. Increasing incidence of type 1 diabetes in 0- to 17-year-old Colorado youth. Diabetes Care 2007, 30, 503–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, L.N.; Wang, W.; Loomba, L.; Afkarian, M.; Butani, L. Diabetic kidney disease in children and adolescents: An update. Pediatr. Nephrol. 2022, 37, 2583–2597. [Google Scholar] [CrossRef] [PubMed]
- Bogdanović, R. Diabetic nephropathy in children and adolescents. Pediatr. Nephrol. 2008, 23, 507–525. [Google Scholar] [CrossRef]
- Alleyn, C.R.; Volkening, L.K.; Wolfson, J.; Rodriguez-Ventura, A.; Wood, J.R.; Laffel, L.M. Occurrence of microalbuminuria in young people with Type 1 diabetes: Importance of age and diabetes duration. Diabet. Med. A J. Br. Diabet. Assoc. 2010, 27, 532–537. [Google Scholar] [CrossRef] [Green Version]
- Noh, H.; King, G.L. The role of protein kinase C activation in diabetic nephropathy. Kidney Int. Suppl. 2007, 72, S49–S53. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.S.; Ho, E.C.; Lam, K.S.; Chung, S.K. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. JASN 2003, 14 (Suppl. 3), S233–S236. [Google Scholar] [CrossRef] [Green Version]
- Sanajou, D.; Ghorbani Haghjo, A.; Argani, H.; Aslani, S. AGE-RAGE axis blockade in diabetic nephropathy: Current status and future directions. Eur. J. Pharmacol. 2018, 833, 158–164. [Google Scholar] [CrossRef]
- Kang, J.S.; Lee, S.J.; Lee, J.H.; Kim, J.H.; Son, S.S.; Cha, S.K.; Lee, E.S.; Chung, C.H.; Lee, E.Y. Angiotensin II-mediated MYH9 downregulation causes structural and functional podocyte injury in diabetic kidney disease. Sci. Rep. 2019, 9, 7679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Li, Y.; Jiang, T.; Yuan, Y.; Li, R.; Xu, Z.; Zhong, X.; Jia, G.; Liu, Y.; Xie, L.; et al. Reduction of cellular stress is essential for Fibroblast growth factor 1 treatment for diabetic nephropathy. J. Cell. Mol. Med. 2018, 22, 6294–6303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiMeglio, L.A.; Acerini, C.L.; Codner, E.; Craig, M.E.; Hofer, S.E.; Pillay, K.; Maahs, D.M. ISPAD Clinical Practice Consensus Guidelines 2018: Glycemic control targets and glucose monitoring for children, adolescents, and young adults with diabetes. Pediatr. Diabetes 2018, 19 (Suppl. 27), 105–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, R.W.; Bergenstal, R.M.; Riddlesworth, T.D.; Kollman, C.; Li, Z.; Brown, A.S.; Close, K.L. Validation of Time in Range as an Outcome Measure for Diabetes Clinical Trials. Diabetes Care 2019, 42, 400–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludvigsson, J.; Samuelsson, U. Continuous insulin infusion (CSII) or modern type of multiple daily injections (MDI) in diabetic children and adolescents a critical review on a controversial issue. Pediatr. Endocrinol. Rev. PER 2007, 5, 666–678. [Google Scholar] [PubMed]
- Blair, J.; McKay, A.; Ridyard, C.; Thornborough, K.; Bedson, E.; Peak, M.; Didi, M.; Annan, F.; Gregory, J.W.; Hughes, D.; et al. Continuous subcutaneous insulin infusion versus multiple daily injections in children and young people at diagnosis of type 1 diabetes: The SCIPI RCT. Health Technol. Assess. 2018, 22, 1–112. [Google Scholar] [CrossRef]
- Schiel, R.; Burgard, D.; Perenthaler, T.; Stein, G.; Kramer, G.; Steveling, A. Use and Effectiveness of Continuous Subcutaneous Insulin Infusion (CSII) and Multiple Daily Insulin Injection Therapy (MIT) in Children, Adolescents and Young Adults with Type 1 Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2016, 124, 99–104. [Google Scholar] [CrossRef]
- Karageorgiou, V.; Papaioannou, T.G.; Bellos, I.; Alexandraki, K.; Tentolouris, N.; Stefanadis, C.; Chrousos, G.P.; Tousoulis, D. Effectiveness of artificial pancreas in the non-adult population: A systematic review and network meta-analysis. Metab. Clin. Exp. 2019, 90, 20–30. [Google Scholar] [CrossRef]
- Forrester, J.V.; Kuffova, L.; Delibegovic, M. The Role of Inflammation in Diabetic Retinopathy. Front. Immunol. 2020, 11, 583687. [Google Scholar] [CrossRef]
- Yerramothu, P.; Vijay, A.K.; Willcox, M.D.P. Inflammasomes, the eye and anti-inflammasome therapy. Eye 2018, 32, 491–505. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Kern, T.S. Inflammation in diabetic retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidyula, V.R.; Boden, G.; Rao, A.K. Platelet and monocyte activation by hyperglycemia and hyperinsulinemia in healthy subjects. Platelets 2006, 17, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Portillo, J.A.; Greene, J.A.; Okenka, G.; Miao, Y.; Sheibani, N.; Kern, T.S.; Subauste, C.S. CD40 promotes the development of early diabetic retinopathy in mice. Diabetologia 2014, 57, 2222–2231. [Google Scholar] [PubMed]
- Samuels, I.S.; Portillo, J.C.; Miao, Y.; Kern, T.S.; Subauste, C.S. Loss of CD40 attenuates experimental diabetes-induced retinal inflammation but does not protect mice from electroretinogram defects. Vis. Neurosci. 2017, 34, E009. [Google Scholar] [CrossRef]
- Serra, A.M.; Waddell, J.; Manivannan, A.; Xu, H.; Cotter, M.; Forrester, J.V. CD11b+ bone marrow-derived monocytes are the major leukocyte subset responsible for retinal capillary leukostasis in experimental diabetes in mouse and express high levels of CCR5 in the circulation. Am. J. Pathol. 2012, 181, 719–727. [Google Scholar] [CrossRef]
- Giulietti, A.; van Etten, E.; Overbergh, L.; Stoffels, K.; Bouillon, R.; Mathieu, C. Monocytes from type 2 diabetic patients have a pro-inflammatory profile. 1,25-Dihydroxyvitamin D(3) works as anti-inflammatory. Diabetes Res. Clin. Pract. 2007, 77, 47–57. [Google Scholar] [CrossRef]
- Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. Jama 2002, 287, 2563–2569. [CrossRef]
- Lin, T.; Gubitosi-Klug, R.A.; Channa, R.; Wolf, R.M. Pediatric Diabetic Retinopathy: Updates in Prevalence, Risk Factors, Screening, and Management. Curr. Diab. Rep. 2021, 21, 56. [Google Scholar] [CrossRef]
- Keel, S.; Itsiopoulos, C.; Koklanis, K.; Vukicevic, M.; Cameron, F.; Brazionis, L. Prevalence and risk factors for diabetic retinopathy in a hospital-based population of Australian children and adolescents with type 1 diabetes. J. Pediatr. Endocrinol. Metab. JPEM 2016, 29, 1135–1142. [Google Scholar] [CrossRef]
- Cahill, M.; Wallace, D.; Travers, S.; Lipinski, H.; Aldington, S.; Costigan, C.; Mooney, D. Detection and prevalence of early diabetic retinopathy in juvenile diabetics with diabetes for 10 years or more. Eye 2000, 14, 847–850. [Google Scholar] [CrossRef]
- Holl, R.W.; Lang, G.E.; Grabert, M.; Heinze, E.; Lang, G.K.; Debatin, K.M. Diabetic retinopathy in pediatric patients with type-1 diabetes: Effect of diabetes duration, prepubertal and pubertal onset of diabetes, and metabolic control. J. Pediatr. 1998, 132, 790–794. [Google Scholar] [CrossRef]
- Donaghue, K.C.; Fung, A.T.; Hing, S.; Fairchild, J.; King, J.; Chan, A.; Howard, N.J.; Silink, M. The effect of prepubertal diabetes duration on diabetes. Microvascular complications in early and late adolescence. Diabetes Care 1997, 20, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, D.P.; Bebu, I.; Aiello, L.P.; Sivitz, W.; Gubitosi-Klug, R.; Malone, J.; White, N.H.; Danis, R.; Wallia, A.; Gao, X.; et al. Risk Factors for Retinopathy in Type 1 Diabetes: The DCCT/EDIC Study. Diabetes Care 2019, 42, 875–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gołębiewska, J.; Olechowski, A.; Wysocka-Mincewicz, M.; Odrobina, D.; Baszyńska-Wilk, M.; Groszek, A.; Szalecki, M.; Hautz, W. Optical coherence tomography angiography vessel density in children with type 1 diabetes. PLoS ONE 2017, 12, e0186479. [Google Scholar] [CrossRef]
- Wang, S.Y.; Andrews, C.A.; Herman, W.H.; Gardner, T.W.; Stein, J.D. Incidence and Risk Factors for Developing Diabetic Retinopathy among Youths with Type 1 or Type 2 Diabetes throughout the United States. Ophthalmology 2017, 124, 424–430. [Google Scholar] [PubMed] [Green Version]
- Aptel, F.; Denis, P.; Rouberol, F.; Thivolet, C. Screening of diabetic retinopathy: Effect of field number and mydriasis on sensitivity and specificity of digital fundus photography. Diabetes Metab. 2008, 34, 290–293. [Google Scholar] [CrossRef]
- Kirkizlar, E.; Serban, N.; Sisson, J.A.; Swann, J.L.; Barnes, C.S.; Williams, M.D. Evaluation of telemedicine for screening of diabetic retinopathy in the Veterans Health Administration. Ophthalmology 2013, 120, 2604–2610. [Google Scholar] [PubMed]
- Abràmoff, M.D.; Lavin, P.T.; Birch, M.; Shah, N.; Folk, J.C. Pivotal trial of an autonomous AI-based diagnostic system for detection of diabetic retinopathy in primary care offices. NPJ Digit. Med. 2018, 1, 39. [Google Scholar] [PubMed] [Green Version]
- Wolf, R.M.; Liu, T.Y.A.; Thomas, C.; Prichett, L.; Zimmer-Galler, I.; Smith, K.; Abramoff, M.D.; Channa, R. The SEE Study: Safety, Efficacy, and Equity of Implementing Autonomous Artificial Intelligence for Diagnosing Diabetic Retinopathy in Youth. Diabetes Care 2021, 44, 781–787. [Google Scholar] [CrossRef]
- Wysocka-Mincewicz, M.; Baszyńska-Wilk, M.; Gołębiewska, J.; Olechowski, A.; Byczyńska, A.; Hautz, W.; Szalecki, M. Influence of Metabolic Parameters and Treatment Method on OCT Angiography Results in Children with Type 1 Diabetes. J. Diabetes Res. 2020, 2020, 4742952. [Google Scholar] [CrossRef]
- Zabeen, B.; Craig, M.E.; Virk, S.A.; Pryke, A.; Chan, A.K.; Cho, Y.H.; Benitez-Aguirre, P.Z.; Hing, S.; Donaghue, K.C. Insulin Pump Therapy Is Associated with Lower Rates of Retinopathy and Peripheral Nerve Abnormality. PLoS ONE 2016, 11, e0153033. [Google Scholar] [CrossRef] [Green Version]
- Laffel, L.M.; Kanapka, L.G.; Beck, R.W.; Bergamo, K.; Clements, M.A.; Criego, A.; DeSalvo, D.J.; Goland, R.; Hood, K.; Liljenquist, D.; et al. Effect of Continuous Glucose Monitoring on Glycemic Control in Adolescents and Young Adults With Type 1 Diabetes: A Randomized Clinical Trial. JAMA 2020, 323, 2388–2396. [Google Scholar] [CrossRef]
- Selvarajah, D.; Kar, D.; Khunti, K.; Davies, M.J.; Scott, A.R.; Walker, J.; Tesfaye, S. Diabetic peripheral neuropathy: Advances in diagnosis and strategies for screening and early intervention. Lancet. Diabetes Endocrinol. 2019, 7, 938–948. [Google Scholar] [CrossRef] [Green Version]
- Braffett, B.H.; Gubitosi-Klug, R.A.; Albers, J.W.; Feldman, E.L.; Martin, C.L.; White, N.H.; Orchard, T.J.; Lopes-Virella, M.; Lachin, J.M.; Pop-Busui, R. Risk Factors for Diabetic Peripheral Neuropathy and Cardiovascular Autonomic Neuropathy in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study. Diabetes 2020, 69, 1000–1010. [Google Scholar] [CrossRef]
- Tesfaye, S.; Stevens, L.K.; Stephenson, J.M.; Fuller, J.H.; Plater, M.; Ionescu-Tirgoviste, C.; Nuber, A.; Pozza, G.; Ward, J.D. Prevalence of diabetic peripheral neuropathy and its relation to glycaemic control and potential risk factors: The EURODIAB IDDM Complications Study. Diabetologia 1996, 39, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Boulton, A.J.; Vinik, A.I.; Arezzo, J.C.; Bril, V.; Feldman, E.L.; Freeman, R.; Malik, R.A.; Maser, R.E.; Sosenko, J.M.; Ziegler, D. Diabetic neuropathies: A statement by the American Diabetes Association. Diabetes Care 2005, 28, 956–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pop-Busui, R.; Boulton, A.J.; Feldman, E.L.; Bril, V.; Freeman, R.; Malik, R.A.; Sosenko, J.M.; Ziegler, D. Diabetic Neuropathy: A Position Statement by the American Diabetes Association. Diabetes Care 2017, 40, 136–154. [Google Scholar] [CrossRef] [Green Version]
- Said, G.; Slama, G.; Selva, J. Progressive centripetal degeneration of axons in small fibre diabetic polyneuropathy. Brain A J. Neurol. 1983, 106, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Perez, E.; Schönberger, T.; Sumalla, M.; Stierstorfer, B.; Solà, R.; Doods, H.; Serra, J.; Gorodetskaya, N. Behavioural, morphological and electrophysiological assessment of the effects of type 2 diabetes mellitus on large and small nerve fibres in Zucker diabetic fatty, Zucker lean and Wistar rats. Eur. J. Pain 2018, 22, 1457–1472. [Google Scholar] [CrossRef]
- Malik, R.A.; Tesfaye, S.; Newrick, P.G.; Walker, D.; Rajbhandari, S.M.; Siddique, I.; Sharma, A.K.; Boulton, A.J.; King, R.H.; Thomas, P.K.; et al. Sural nerve pathology in diabetic patients with minimal but progressive neuropathy. Diabetologia 2005, 48, 578–585. [Google Scholar] [CrossRef]
- Smith, S.; Normahani, P.; Lane, T.; Hohenschurz-Schmidt, D.; Oliver, N.; Davies, A.H. Pathogenesis of Distal Symmetrical Polyneuropathy in Diabetes. Life 2022, 12, 1074. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Sato, R.; Kanda, T. Blood-Nerve Barrier (BNB) Pathology in Diabetic Peripheral Neuropathy and In Vitro Human BNB Model. Int. J. Mol. Sci. 2020, 22, 62. [Google Scholar] [CrossRef]
- Kanda, T. Biology of the blood-nerve barrier and its alteration in immune mediated neuropathies. J. Neurol. Neurosurg. Psychiatry 2013, 84, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Ubogu, E.E. The molecular and biophysical characterization of the human blood-nerve barrier: Current concepts. J. Vasc. Res. 2013, 50, 289–303. [Google Scholar] [CrossRef] [Green Version]
- Rechthand, E.; Smith, Q.R.; Latker, C.H.; Rapoport, S.I. Altered blood-nerve barrier permeability to small molecules in experimental diabetes mellitus. J. Neuropathol. Exp. Neurol. 1987, 46, 302–314. [Google Scholar] [CrossRef]
- Poduslo, J.F.; Curran, G.L.; Dyck, P.J. Increase in albumin, IgG, and IgM blood-nerve barrier indices in human diabetic neuropathy. Proc. Natl. Acad. Sci. USA 1988, 85, 4879–4883. [Google Scholar] [CrossRef]
- Mizisin, A.P.; Weerasuriya, A. Homeostatic regulation of the endoneurial microenvironment during development, aging and in response to trauma, disease and toxic insult. Acta Neuropathol. 2011, 121, 291–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellogg, A.P.; Wiggin, T.D.; Larkin, D.D.; Hayes, J.M.; Stevens, M.J.; Pop-Busui, R. Protective effects of cyclooxygenase-2 gene inactivation against peripheral nerve dysfunction and intraepidermal nerve fiber loss in experimental diabetes. Diabetes 2007, 56, 2997–3005. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.M.; Perrone, L.; Sullivan, K.A.; Backus, C.; Sastry, A.M.; Lastoskie, C.; Feldman, E.L. Receptor for advanced glycation end products activation injures primary sensory neurons via oxidative stress. Endocrinology 2007, 148, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Callaghan, B.C.; Little, A.A.; Feldman, E.L.; Hughes, R.A. Enhanced glucose control for preventing and treating diabetic neuropathy. Cochrane Database Syst. Rev. 2012, 6, CD007543. [Google Scholar] [CrossRef]
- Gouveri, E.; Papanas, N. The Emerging Role of Continuous Glucose Monitoring in the Management of Diabetic Peripheral Neuropathy: A Narrative Review. Diabetes Ther. Res. Treat. Educ. Diabetes Relat. Disord. 2022, 13, 931–952. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, P. Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—A meta-analysis. Osteoporos. Int. A J. Establ. Result Coop. Between Eur. Found. Osteoporos. Natl. Osteoporos. Found. USA 2007, 18, 427–444. [Google Scholar] [CrossRef]
- Hothersall, E.J.; Livingstone, S.J.; Looker, H.C.; Ahmed, S.F.; Cleland, S.; Leese, G.P.; Lindsay, R.S.; McKnight, J.; Pearson, D.; Philip, S.; et al. Contemporary risk of hip fracture in type 1 and type 2 diabetes: A national registry study from Scotland. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2014, 29, 1054–1060. [Google Scholar] [CrossRef] [Green Version]
- Weber, D.R.; Haynes, K.; Leonard, M.B.; Willi, S.M.; Denburg, M.R. Type 1 diabetes is associated with an increased risk of fracture across the life span: A population-based cohort study using The Health Improvement Network (THIN). Diabetes Care 2015, 38, 1913–1920. [Google Scholar] [CrossRef] [Green Version]
- Vavanikunnel, J.; Charlier, S.; Becker, C.; Schneider, C.; Jick, S.S.; Meier, C.R.; Meier, C. Association Between Glycemic Control and Risk of Fracture in Diabetic Patients: A Nested Case-Control Study. J. Clin. Endocrinol. Metab. 2019, 104, 1645–1654. [Google Scholar] [CrossRef] [Green Version]
- Maahs, D.M.; West, N.A.; Lawrence, J.M.; Mayer-Davis, E.J. Epidemiology of type 1 diabetes. Endocrinol. Metab. Clin. N. Am. 2010, 39, 481–497. [Google Scholar] [CrossRef] [Green Version]
- Baxter-Jones, A.D.; Faulkner, R.A.; Forwood, M.R.; Mirwald, R.L.; Bailey, D.A. Bone mineral accrual from 8 to 30 years of age: An estimation of peak bone mass. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2011, 26, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Baum, T.; Yap, S.P.; Karampinos, D.C.; Nardo, L.; Kuo, D.; Burghardt, A.J.; Masharani, U.B.; Schwartz, A.V.; Li, X.; Link, T.M. Does vertebral bone marrow fat content correlate with abdominal adipose tissue, lumbar spine bone mineral density, and blood biomarkers in women with type 2 diabetes mellitus? J. Magn. Reson. Imaging JMRI 2012, 35, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsch, J.M.; Li, X.; Baum, T.; Yap, S.P.; Karampinos, D.C.; Schwartz, A.V.; Link, T.M. Bone marrow fat composition as a novel imaging biomarker in postmenopausal women with prevalent fragility fractures. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2013, 28, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Sheu, Y.; Amati, F.; Schwartz, A.V.; Danielson, M.E.; Li, X.; Boudreau, R.; Cauley, J.A. Vertebral bone marrow fat, bone mineral density and diabetes: The Osteoporotic Fractures in Men (MrOS) study. Bone 2017, 97, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.W.; Greenblatt, L.; Eajazi, A.; Torriani, M.; Bredella, M.A. Marrow adipose tissue composition in adults with morbid obesity. Bone 2017, 97, 38–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalrahaman, N.; McComb, C.; Foster, J.E.; McLean, J.; Lindsay, R.S.; McClure, J.; McMillan, M.; Drummond, R.; Gordon, D.; McKay, G.A.; et al. Deficits in Trabecular Bone Microarchitecture in Young Women With Type 1 Diabetes Mellitus. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2015, 30, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Shepherd, S.; McMillan, M.; McNeilly, J.; Foster, J.; Wong, S.C.; Robertson, K.J.; Ahmed, S.F. Skeletal Fragility and Its Clinical Determinants in Children With Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 3585–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faienza, M.F.; Ventura, A.; Delvecchio, M.; Fusillo, A.; Piacente, L.; Aceto, G.; Colaianni, G.; Colucci, S.; Cavallo, L.; Grano, M.; et al. High Sclerostin and Dickkopf-1 (DKK-1) Serum Levels in Children and Adolescents With Type 1 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2017, 102, 1174–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faienza, M.F.; Brunetti, G.; Sanesi, L.; Colaianni, G.; Celi, M.; Piacente, L.; D’Amato, G.; Schipani, E.; Colucci, S.; Grano, M. High irisin levels are associated with better glycemic control and bone health in children with Type 1 diabetes. Diabetes Res. Clin. Pract. 2018, 141, 10–17. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, N.; Fu, Z.; Zhang, Q. Progress of Wnt Signaling Pathway in Osteoporosis. Biomolecules 2023, 13, 483. [Google Scholar] [CrossRef]
- Mao, B.; Wu, W.; Li, Y.; Hoppe, D.; Stannek, P.; Glinka, A.; Niehrs, C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature 2001, 411, 321–325. [Google Scholar] [CrossRef]
- Bafico, A.; Liu, G.; Yaniv, A.; Gazit, A.; Aaronson, S.A. Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/Arrow. Nat. Cell Biol. 2001, 3, 683–686. [Google Scholar] [CrossRef]
- Semënov, M.V.; Zhang, X.; He, X. DKK1 antagonizes Wnt signaling without promotion of LRP6 internalization and degradation. J. Biol. Chem. 2008, 283, 21427–21432. [Google Scholar] [CrossRef] [Green Version]
- Mao, B.; Wu, W.; Davidson, G.; Marhold, J.; Li, M.; Mechler, B.M.; Delius, H.; Hoppe, D.; Stannek, P.; Walter, C.; et al. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature 2002, 417, 664–667. [Google Scholar] [CrossRef]
- Yamamoto, H.; Sakane, H.; Yamamoto, H.; Michiue, T.; Kikuchi, A. Wnt3a and Dkk1 regulate distinct internalization pathways of LRP6 to tune the activation of beta-catenin signaling. Dev. Cell 2008, 15, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Niehrs, C. Kremen2 modulates Dickkopf2 activity during Wnt/LRP6 signaling. Gene 2003, 302, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sarosi, I.; Cattley, R.C.; Pretorius, J.; Asuncion, F.; Grisanti, M.; Morony, S.; Adamu, S.; Geng, Z.; Qiu, W.; et al. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone 2006, 39, 754–766. [Google Scholar] [CrossRef]
- Li, X.; Grisanti, M.; Fan, W.; Asuncion, F.J.; Tan, H.L.; Dwyer, D.; Han, C.Y.; Yu, L.; Lee, J.; Lee, E.; et al. Dickkopf-1 regulates bone formation in young growing rodents and upon traumatic injury. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2011, 26, 2610–2621. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Joiner, D.M.; Oyserman, S.M.; Sharma, P.; Goldstein, S.A.; He, X.; Hauschka, P.V. Bone mass is inversely proportional to Dkk1 levels in mice. Bone 2007, 41, 331–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balemans, W.; Devogelaer, J.P.; Cleiren, E.; Piters, E.; Caussin, E.; Van Hul, W. Novel LRP5 missense mutation in a patient with a high bone mass phenotype results in decreased DKK1-mediated inhibition of Wnt signaling. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2007, 22, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Albiol, L.; Büttner, A.; Pflanz, D.; Mikolajewicz, N.; Birkhold, A.I.; Kramer, I.; Kneissel, M.; Duda, G.N.; Checa, S.; Willie, B.M. Effects of Long-Term Sclerostin Deficiency on Trabecular Bone Mass and Adaption to Limb Loading Differ in Male and Female Mice. Calcif. Tissue Int. 2020, 106, 415–430. [Google Scholar] [CrossRef]
- Pflanz, D.; Birkhold, A.I.; Albiol, L.; Thiele, T.; Julien, C.; Seliger, A.; Thomson, E.; Kramer, I.; Kneissel, M.; Duda, G.N.; et al. Sost deficiency led to a greater cortical bone formation response to mechanical loading and altered gene expression. Sci. Rep. 2017, 7, 9435. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Ebeling, M.; Patel, N.; Van Hul, E.; Olson, P.; Dioszegi, M.; Lacza, C.; Wuyts, W.; Van Den Ende, J.; Willems, P.; et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST). Hum. Mol. Genet. 2001, 10, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Balemans, W.; Patel, N.; Ebeling, M.; Van Hul, E.; Wuyts, W.; Lacza, C.; Dioszegi, M.; Dikkers, F.G.; Hildering, P.; Willems, P.J.; et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J. Med. Genet. 2002, 39, 91–97. [Google Scholar] [CrossRef]
- Li, X.; Ominsky, M.S.; Niu, Q.T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef]
- Van Lierop, A.H.; Appelman-Dijkstra, N.M.; Papapoulos, S.E. Sclerostin deficiency in humans. Bone 2017, 96, 51–62. [Google Scholar] [CrossRef]
- Loots, G.G.; Kneissel, M.; Keller, H.; Baptist, M.; Chang, J.; Collette, N.M.; Ovcharenko, D.; Plajzer-Frick, I.; Rubin, E.M. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005, 15, 928–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, X.; Rhee, Y.; Condon, K.W.; Bivi, N.; Allen, M.R.; Dwyer, D.; Stolina, M.; Turner, C.H.; Robling, A.G.; Plotkin, L.I.; et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 2012, 50, 209–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, D.G.; Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Hayes, T.; Skonier, J.E.; Shpektor, D.; Jonas, M.; Kovacevich, B.R.; Staehling-Hampton, K.; et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003, 22, 6267–6276. [Google Scholar] [CrossRef] [Green Version]
- Robling, A.G.; Niziolek, P.J.; Baldridge, L.A.; Condon, K.W.; Allen, M.R.; Alam, I.; Mantila, S.M.; Gluhak-Heinrich, J.; Bellido, T.M.; Harris, S.E.; et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J. Biol. Chem. 2008, 283, 5866–5875. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Jiang, X.; Dai, Z.; Guo, X.; Weng, T.; Wang, J.; Li, Y.; Feng, G.; Gao, X.; He, L. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/beta-catenin signaling. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2009, 24, 1651–1661. [Google Scholar] [CrossRef]
- Spatz, J.M.; Wein, M.N.; Gooi, J.H.; Qu, Y.; Garr, J.L.; Liu, S.; Barry, K.J.; Uda, Y.; Lai, F.; Dedic, C.; et al. The Wnt Inhibitor Sclerostin Is Up-regulated by Mechanical Unloading in Osteocytes in Vitro. J. Biol. Chem. 2015, 290, 16744–16758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poole, K.E.; van Bezooijen, R.L.; Loveridge, N.; Hamersma, H.; Papapoulos, S.E.; Löwik, C.W.; Reeve, J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1842–1844. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, G.; Todisco, S.; Grano, M. Antibody Treatment and Osteoporosis: Clinical Perspective. In Innovative Bioceramics in Translational Medicine II: Surgical Applications; Springer: Berlin/Heidelberg, Germany, 2022; pp. 111–126. [Google Scholar]
- Kurban, S.; Selver Eklioglu, B.; Selver, M.B. Investigation of the relationship between serum sclerostin and dickkopf-1 protein levels with bone turnover in children and adolescents with type-1 diabetes mellitus. J. Pediatr. Endocrinol. Metab. JPEM 2022, 35, 673–679. [Google Scholar] [CrossRef]
- Tsentidis, C.; Gourgiotis, D.; Kossiva, L.; Marmarinos, A.; Doulgeraki, A.; Karavanaki, K. Increased levels of Dickkopf-1 are indicative of Wnt/β-catenin downregulation and lower osteoblast signaling in children and adolescents with type 1 diabetes mellitus, contributing to lower bone mineral density. Osteoporos. Int. A J. Establ. Result Coop. Between Eur. Found. Osteoporos. Natl. Osteoporos. Found. USA 2017, 28, 945–953. [Google Scholar] [CrossRef]
- Tsentidis, C.; Gourgiotis, D.; Kossiva, L.; Marmarinos, A.; Doulgeraki, A.; Karavanaki, K. Sclerostin distribution in children and adolescents with type 1 diabetes mellitus and correlation with bone metabolism and bone mineral density. Pediatr. Diabetes 2016, 17, 289–299. [Google Scholar] [CrossRef]
- Catalano, A.; Pintaudi, B.; Morabito, N.; Di Vieste, G.; Giunta, L.; Bruno, M.L.; Cucinotta, D.; Lasco, A.; Di Benedetto, A. Gender differences in sclerostin and clinical characteristics in type 1 diabetes mellitus. Eur. J. Endocrinol. 2014, 171, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, T.; Hofbauer, L.C.; Rauner, M.; Lodes, S.; Kästner, B.; Franke, S.; Kiehntopf, M.; Lehmann, T.; Müller, U.A.; Wolf, G.; et al. Clinical and endocrine correlates of circulating sclerostin levels in patients with type 1 diabetes mellitus. Clin. Endocrinol. 2014, 80, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Gennari, L.; Merlotti, D.; Valenti, R.; Ceccarelli, E.; Ruvio, M.; Pietrini, M.G.; Capodarca, C.; Franci, M.B.; Campagna, M.S.; Calabrò, A.; et al. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J. Clin. Endocrinol. Metab. 2012, 97, 1737–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, Y.; Dai, B.; Guo, X.; Liu, D. Cleavage of FNDC5 and insights into its maturation process. Mol. Cell. Endocrinol. 2020, 510, 110840. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, R.; Meng, Y.; Li, S.; Donelan, W.; Zhao, Y.; Qi, L.; Zhang, M.; Wang, X.; Cui, T.; et al. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes 2014, 63, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Kou, W.; Xu, X.; Zhou, S.; Luan, P.; Xu, X.; Li, H.; Zhuang, J.; Wang, J.; Zhao, Y.; et al. FNDC5/Irisin inhibits pathological cardiac hypertrophy. Clin. Sci. 2019, 133, 611–627. [Google Scholar] [CrossRef]
- Boström, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Boström, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Colaianni, G.; Cuscito, C.; Mongelli, T.; Pignataro, P.; Buccoliero, C.; Liu, P.; Lu, P.; Sartini, L.; Di Comite, M.; Mori, G.; et al. The myokine irisin increases cortical bone mass. Proc. Natl. Acad. Sci. USA 2015, 112, 12157–12162. [Google Scholar] [CrossRef]
- Oranger, A.; Zerlotin, R.; Buccoliero, C.; Sanesi, L.; Storlino, G.; Schipani, E.; Kozloff, K.M.; Mori, G.; Colaianni, G.; Colucci, S.; et al. Irisin Modulates Inflammatory, Angiogenic, and Osteogenic Factors during Fracture Healing. Int. J. Mol. Sci. 2023, 24, 1809. [Google Scholar] [CrossRef] [PubMed]
- Gregory, G.A.; Robinson, T.I.G.; Linklater, S.E.; Wang, F.; Colagiuri, S.; de Beaufort, C.; Donaghue, K.C.; Magliano, D.J.; Maniam, J.; Orchard, T.J.; et al. Global incidence, prevalence, and mortality of type 1 diabetes in 2021 with projection to 2040: A modelling study. Lancet. Diabetes Endocrinol. 2022, 10, 741–760. [Google Scholar] [CrossRef] [PubMed]
- Ward, Z.J.; Yeh, J.M.; Reddy, C.L.; Gomber, A.; Ross, C.; Rittiphairoj, T.; Manne-Goehler, J.; Abdalla, A.T.; Abdullah, M.A.; Ahmed, A.; et al. Estimating the total incidence of type 1 diabetes in children and adolescents aged 0–19 years from 1990 to 2050: A global simulation-based analysis. Lancet. Diabetes Endocrinol. 2022, 10, 848–858. [Google Scholar] [CrossRef] [PubMed]



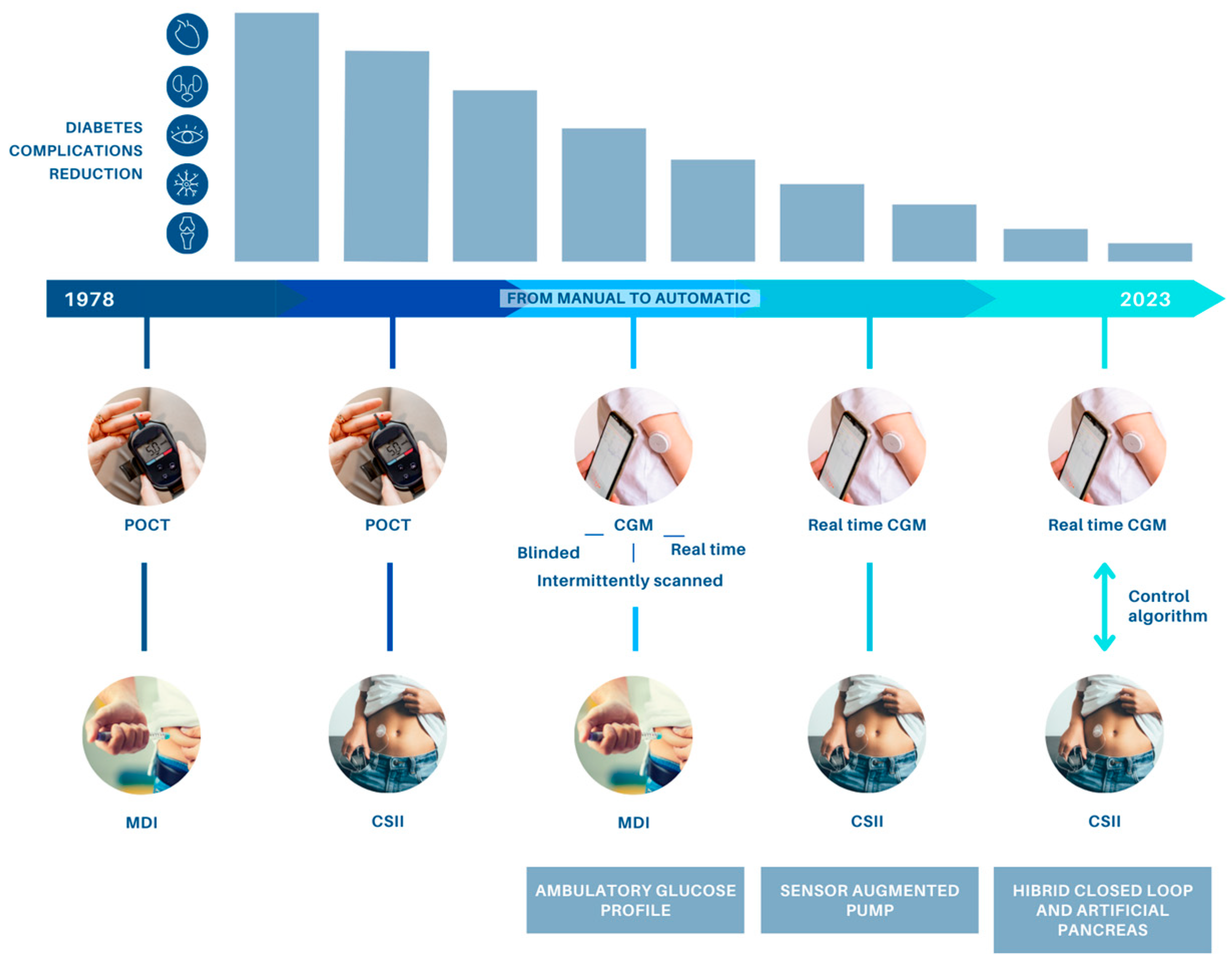{kind=link}
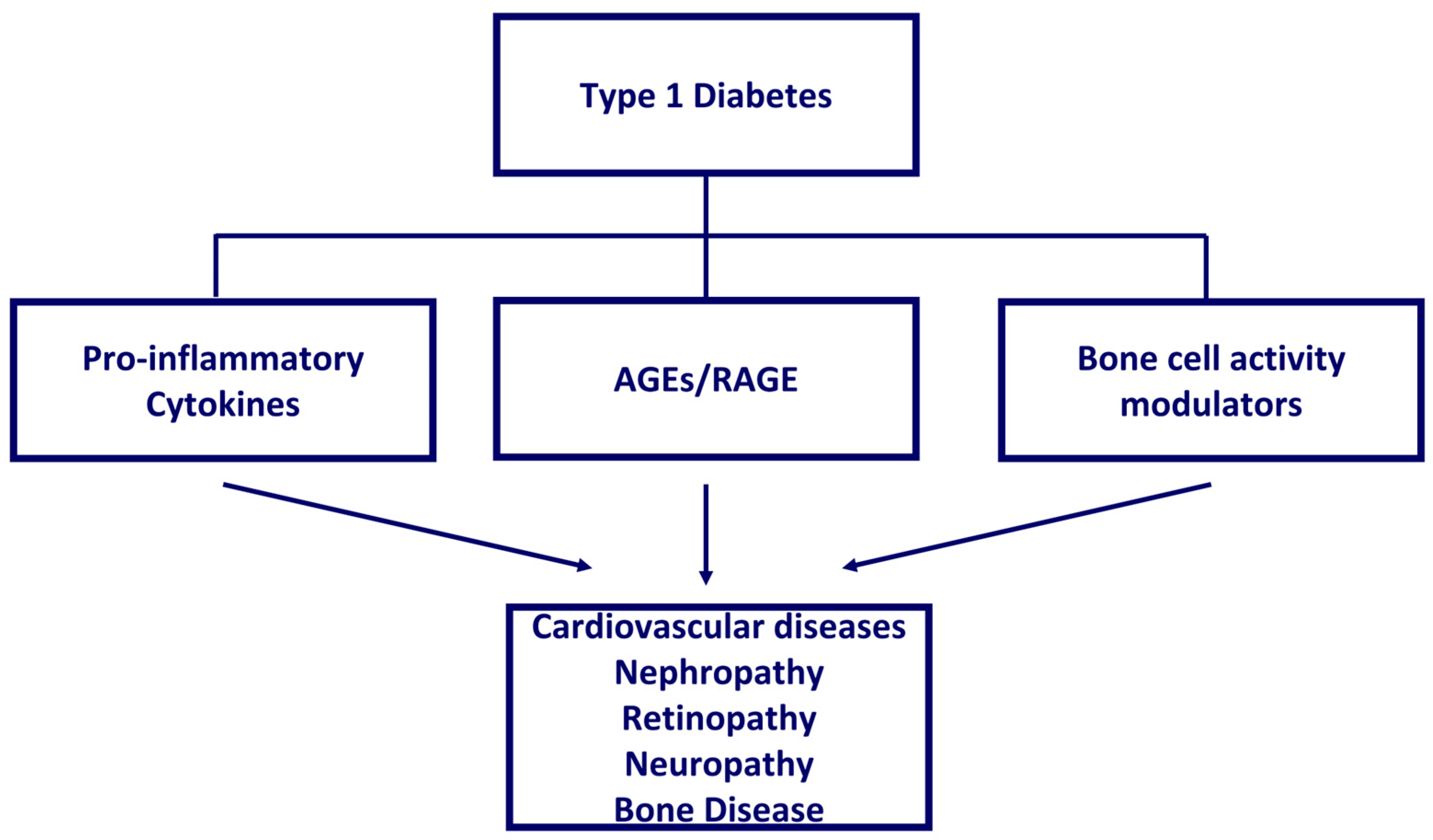{kind=link}
| Advantages of CGM (rtCGM/FGM) | Use of rtCGM | Use of FGM | Advantages and Use of Insulin Pumps (CSII) | Advantages of AID Systems | Use of AID Systems (SAP, LGS, PLGS) |
|---|---|---|---|---|---|
| Accuracy and precision data Automatic setting of date, time Direct information on the diary tool, pre- and postprandial averages, standard deviation (SD) Alarms, reminders Trend indicators Prediction of HbA1c Bolus calculator Detection of ketonemia Data download Features of the specific software for a given instrument Pump connection Application for viewing data on smartphones connectivity | Recommended in the following conditions: - Asymptomatic, recurrent, severe hypoglycemia ≥2/year - Inadequate metabolic control and/or poor QoL - Need for simple and predictive alarms Others: > 10 SMBG/day agophobia | Recommended in the following conditions: - Lower risk of hypoglycemia - Inadequate metabolic control and/or poor QoL - Motivation to check the readings of the device several times a day Others: > 10 SMBG/day agophobia | Provide insulin delivery in two ways: - basal insulin delivery in a steady, measured, and continuous dose - surge (“bolus”) doses at mealtime Recommended in the following conditions: Inadequate metabolic control (elevated HbA1c, persistently above the desirable target and/or glycemic instability) despite intensive and optimized use of MDI High insulin sensitivity Risk of recurrent, nocturnal, or severe hypoglycemia Lifestyle impairment with MDI Newborn and preschool children Compliance of children, parents, and/or caregivers with technological devices | Increase in TIR, especially in the night hours, without an increased risk of hypoglycemia | Recommended in the following conditions: Frequent episodes of disabling and/or unrecognized severe hypoglycemia despite optimization of insulin pump therapy and SMBG Age < 6 years School age with frequent hypoglycemia, glycemic instability, or suboptimal glycemia Need for extremely frequent glycemic self-monitoring (>10/day) Compliance of children, parents, and/or caregivers with technological devices Not recommended if: No improvement in the risk of hypoglycemia and metabolic control, poor compliance, inability to wear the sensor continuously or to manage the “lag time”, interpret the trend arrows, perform the calibration correctly |
| Pump Insulin Closed-Loop Term | Medtronic 670G Rapid Acting Auto Mode | Medtronic 780G Rapid Acting Smart Guard | Tandem t:slim X2 Ultra Rapid and Rapid Acting Control IQ |
|---|---|---|---|
| Algorithm | Treat-to-target proportional integral derivative with insulin feedback | Treat-to-target proportional integral derivative with insulin feedback; added fuzzy logic component | Treat-to-range predictive control |
| Set-up | TDD, weight, basal rates, ICR, ISF, active insulin time 7 days of manual mode | TDD, weight, basal rates, ICR, ISF, active insulin time 7 days of manual mode | TDD, weight, basal rates, ICR, ISF |
| Automated corrections used to supplement basal delivery | Based on total daily insulin dose last 2–6 days | Based on total daily insulin dose last 2–6 days | Based on pre-programmed basal rates |
| Automated insulin delivery | No | Yes, if glucose >120 mg/dL, and at maximum “auto basal” delivery | Yes (max 1/hour), if glucose predicted to be >180 mg/dL, delivers 60% of calculated dose |
| Glucose target | 120 mg/dL Change to 150 mg/dL for set duration (30 min–12 h) | 120 mg/dL Change to 150 mg/dL for set duration (30 min–24 h) | 110–150 mg/dL Exercise range 140–160 mg/dL (manual start/stop only) Sleep range 110–120 mg/dL, prevents autocorrections |
| Adjustable setting | I:C ratio, active insulin time, glucose target | I:C ratio, active insulin time, “BG target” (algorithm target) | Basal rates, I:C ratio, ISF, glucose target range |
| No adjustable setting | Basal rates, ISF (automatically calculated and adapted) | Basal rates, ISF (automatically calculated and adapted) | Active insulin time (set at 5 h) |
| Exercise mode | No | Yes | Yes |
| Boost mode | No | No | No |
| Sick day rules | Recommended to revert to open loop for illness and/or ketone | Recommended to revert to open loop for illness and/or ketone | Recommended to revert to open loop for illness and/or ketone |
| Automatically reverts to open loop if… | Prolonged hyperglycemia, max/min insulin delivery, loss of CGM data, sensor integrity concerns, lack of calibrations. | Prolonged hyperglycemia, max/min insulin delivery, loss of CGM data, sensor integrity concerns, lack of calibrations. | Prolonged hyperglycemia (670 G only), max/min insulin delivery, loss of CGM data, sensor integrity concerns, lack of calibrations. |
| Meal bolus | Strenghten I:C ratios 10–25% Pre-meal boluses recommended for optimal outcomes | Bolus calculator automatically provides sensor glucose level for calculation or BG if performed in past 12 min Pre-meal boluses recommended | Bolus calculator automatically pro vides sensor glucose level for calcula tion If sensor glucose < 110, system will prompt to reduce carb bolus Pre-meal boluses recommended |
| Hypo treatment | Consider treating hypoglycemia with fewer carbohydrates (5 g CHO) depending on recent insulin delivery since insulin will likely have decreased or suspended | Consider treating hypoglycemia with fewer carbohydrates (5 g CHO) depending on recent insulin delivery since insulin will likely have decreased or suspended | Consider treating hypoglycemia with fewer carbohydrates (5 g CHO) depending on recent insulin delivery since insulin will likely have decreased or suspended |
| System optimization | Fingerstick BG check required. Enter into pump to give correction bolus If insulin sensitivity fluctuates widely overnight (e.g., young children), setting a temp target to prevent hypoglycemia No need to change/edit recommended bolus doses (algorithm has been likely giving more insulin in background) Extended/combo bolus function not available | Use of temp target will turn off automated corrections If insulin sensitivity fluctuates widely overnight (e.g., young children), setting a temp target to prevent hypoglycemia No need to change/edit recommended bolus doses (algorithm has been likely giving more insulin in background) Extended/combo bolus function not available | Use exercise activity, considering that auto-corrections will still occur Set sleep activity schedule overnight for tighter target Adjust doses for individuals with shorter active insulin time Extended bolus available, up to 2 h |
| Type of sensor | Guardian 3 | Guardian 3 | Dexcom G6 |
| Calibrations | 3–4 per day (before meals and at bedtime). Avoid when glucose fluctuates widely (e.g., after eating, after treating hypo, during exercise) | 3–4 per day (before meals and at bedtime). Avoid when glucose fluctuates widely (e.g., after eating, after treating hypo, during exercise) | Rarely required (factory calibrated) |
| Sensor life | 7 days | 7 days | 10 days |
| Remote monitoring | No | Yes, Carelink Connect app | Yes, Dexcom G6 Follow app (CGM data) |
| Upload/Data sharing | Manual downloading | Yes, MiniMed mobile app (pump and CGM data) | Yes, t:connect mobile app |
| Remote boluses | No, fingerstick BG check needed | Yes | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbano, F.; Farella, I.; Brunetti, G.; Faienza, M.F. Pediatric Type 1 Diabetes: Mechanisms and Impact of Technologies on Comorbidities and Life Expectancy. Int. J. Mol. Sci. 2023, 24, 11980. https://doi.org/10.3390/ijms241511980
Urbano F, Farella I, Brunetti G, Faienza MF. Pediatric Type 1 Diabetes: Mechanisms and Impact of Technologies on Comorbidities and Life Expectancy. International Journal of Molecular Sciences. 2023; 24(15):11980. https://doi.org/10.3390/ijms241511980
Chicago/Turabian StyleUrbano, Flavia, Ilaria Farella, Giacomina Brunetti, and Maria Felicia Faienza. 2023. "Pediatric Type 1 Diabetes: Mechanisms and Impact of Technologies on Comorbidities and Life Expectancy" International Journal of Molecular Sciences 24, no. 15: 11980. https://doi.org/10.3390/ijms241511980