
Extracellular Succinate: A Physiological Messenger and a Pathological Trigger

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
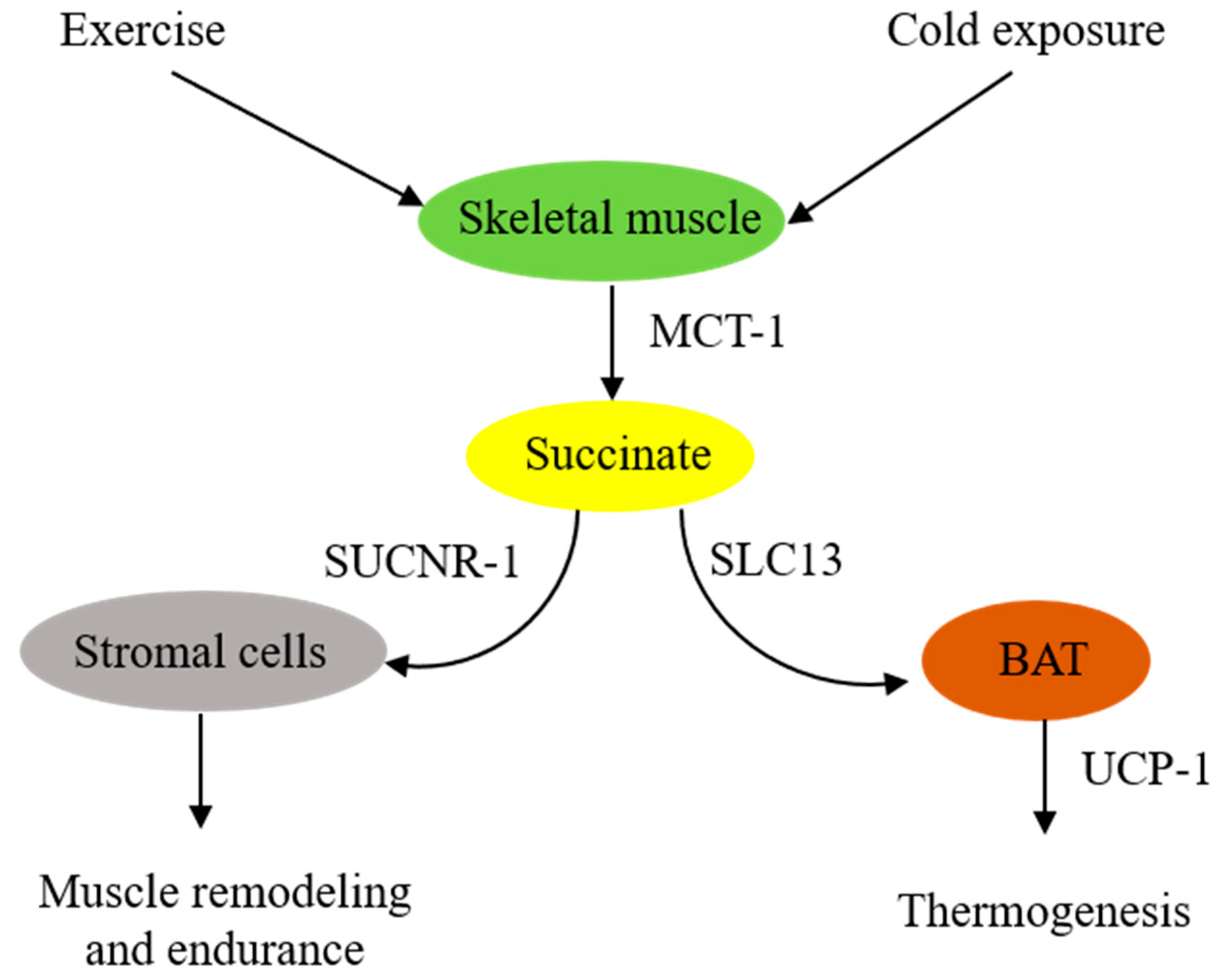
2. Skeletal Muscle-Derived Extracellular Succinate Confers Physiological Adaptation to Exercise and Cold Exposure
2.1. Extracellular Succinate Promotes Muscle Remodeling
2.2. Extracellular Succinate Upregulates Adipose Tissue Energy Expenditure and Thermogenesis
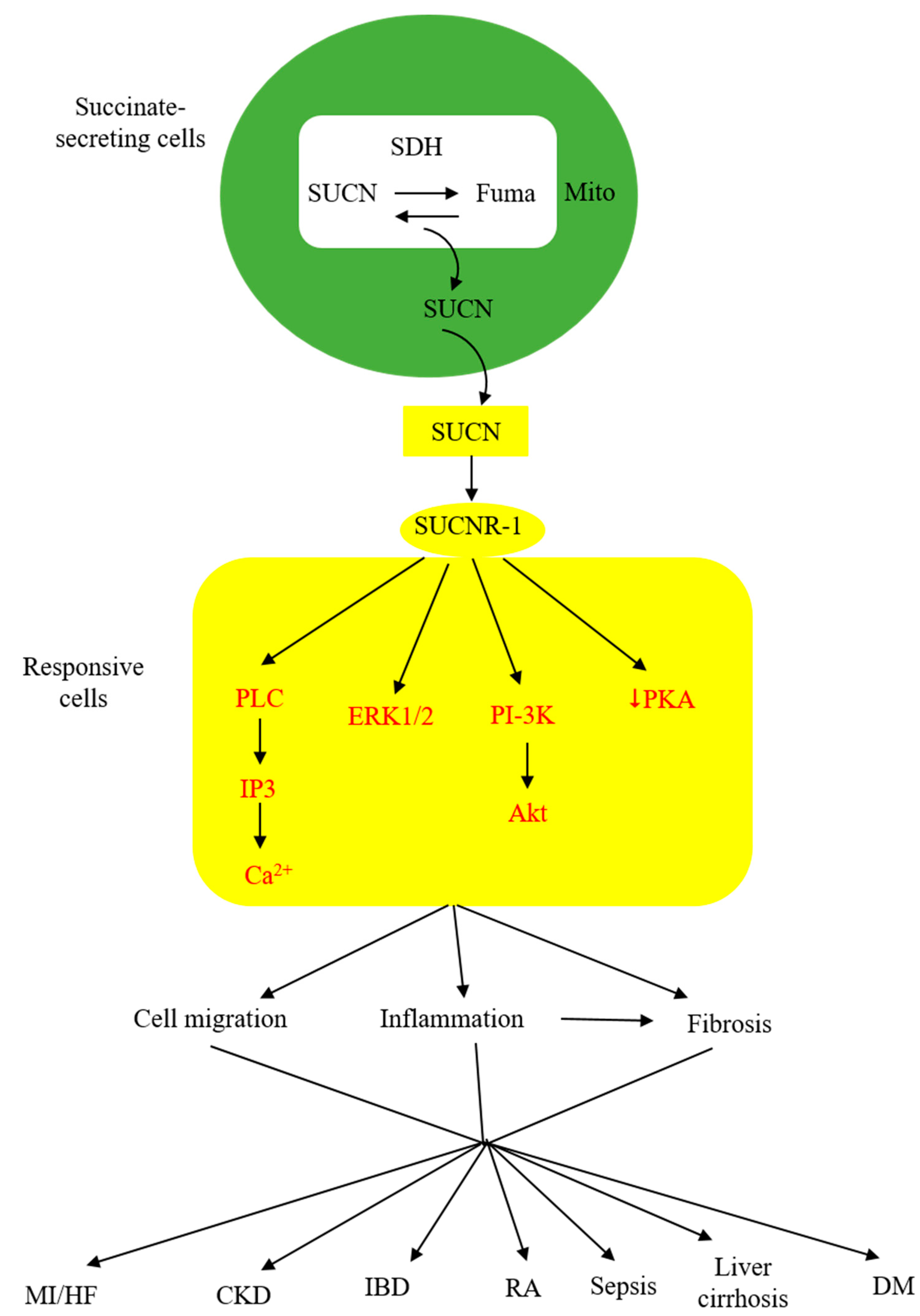
3. Extracellular Succinate Mediates Diverse Pathophysiological Processes via SUCNR-1
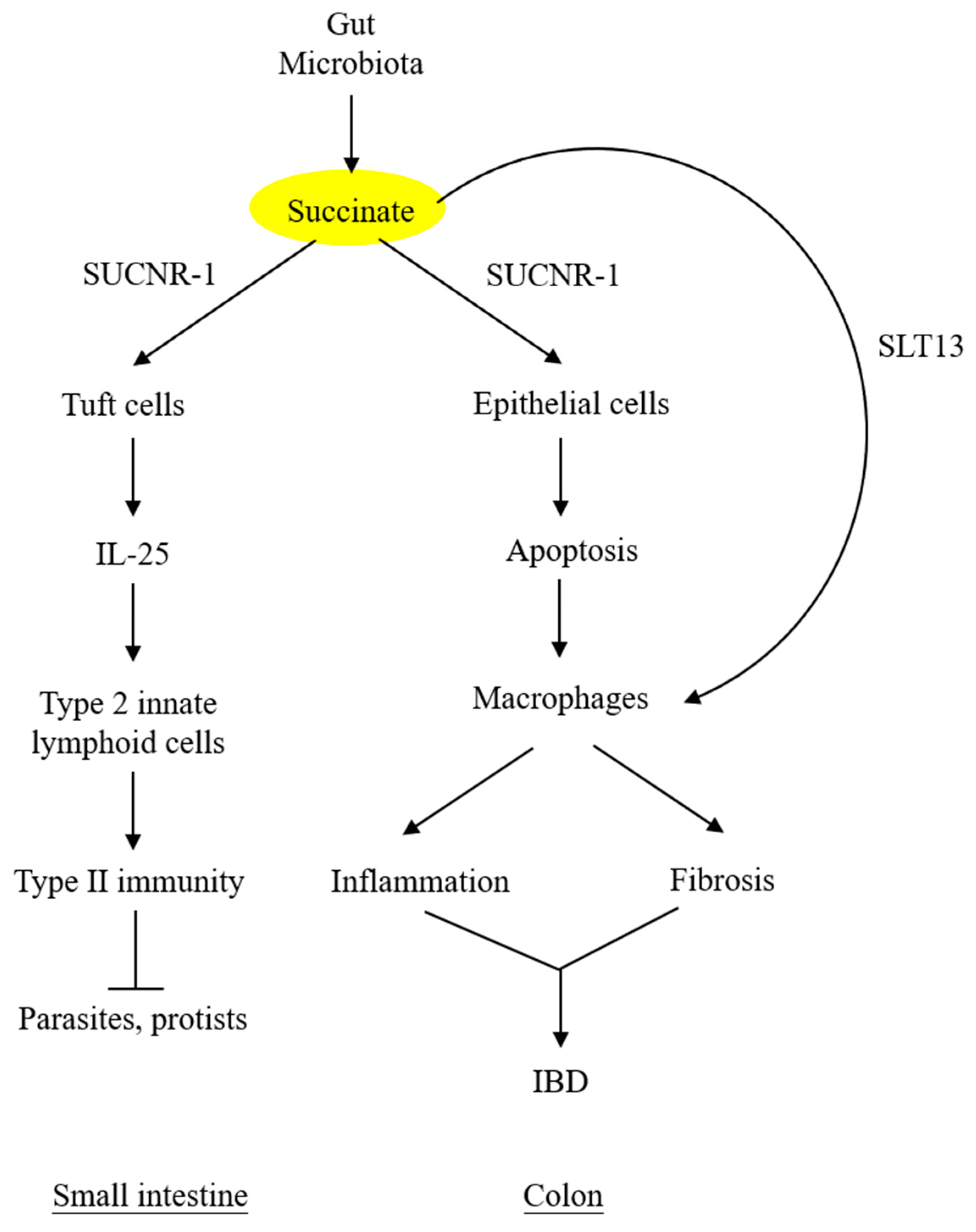
4. Succinate Elicits Intestinal Immunity and Triggers Colon Inflammation
5. Ischemia-Reperfusion (I/R)-Derived Succinate Contributes to Myocardial Infarction (MI)
6. Extracellular Succinate Aggravates Inflammatory Responses through Macrophage Activation
6.1. Activated M1 Macrophages Secrete Succinate to Enhance Inflammatory Response and Exacerbate Inflammatory Disorders
6.2. Adipose Tissues Release Succinate to Recruit Macrophages and Aggravate Inflammation in Obesity
6.3. Succinate-SUCNR-1 Axis Is Involved in Macrophage M2 Polarization and Anti-Inflammatory Actions
6.4. Succinate Controls Inflammatory Response through Cell–Cell Interaction
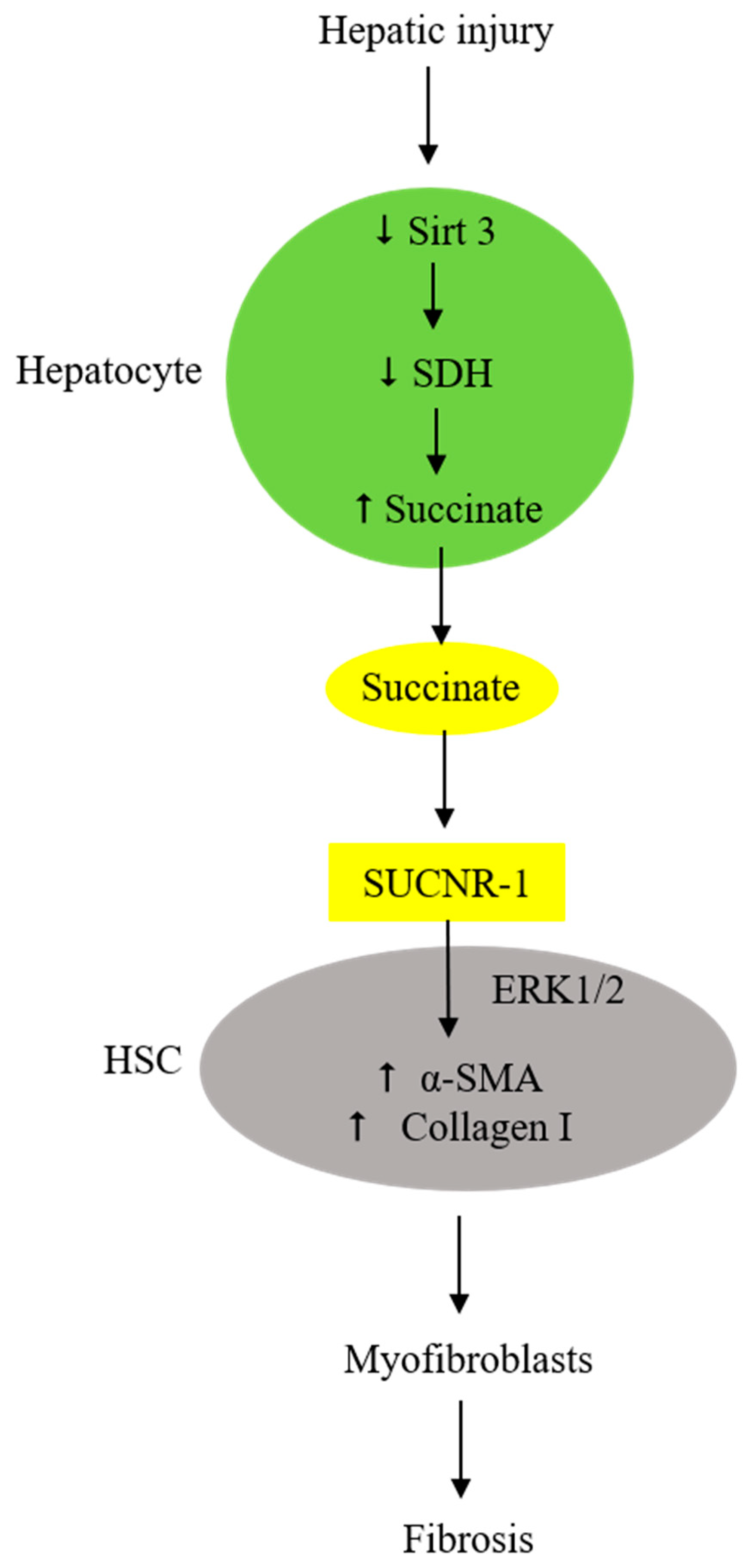
7. Injured Hepatocytes Secrete Succinate to Activate Hepatic Stellate Cells (HSC) and Induce Fibrosis
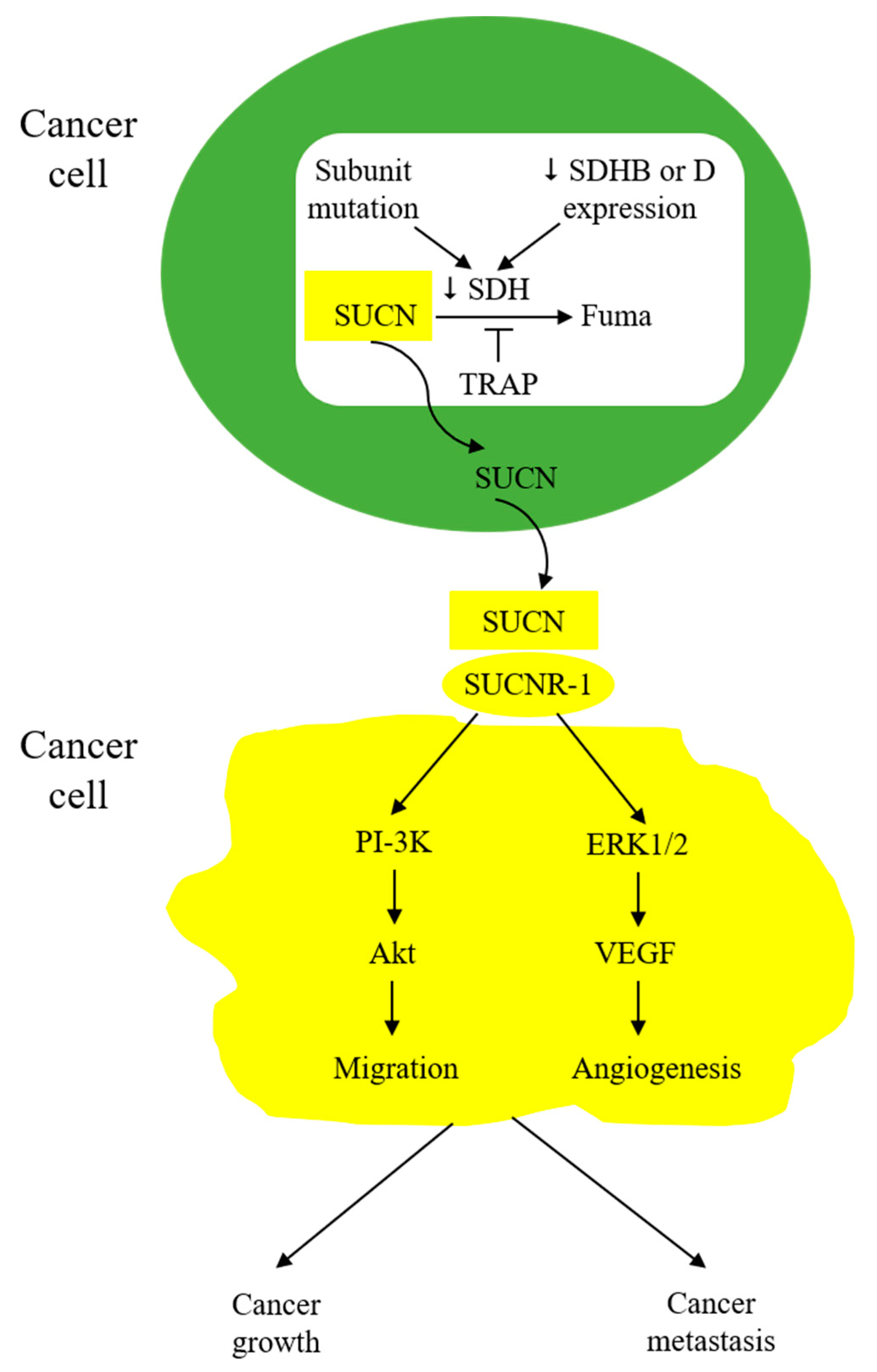
8. Cancer Cell-Derived Succinate Acts as a Messenger in Tumor Microenvironment to Educate Stromal Cells and Promote Cancer Progression
9. Succinate-SUCNR1 Axis as a Therapeutic Target
10. Blood Levels of Succinate in Health and Disease
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Losman, J.A.; Koivunen, P.; Kaelin, W.G., Jr. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat. Rev. Cancer 2020, 20, 710–726. [Google Scholar] [CrossRef]
- Smith, E.H.; Janknecht, R.; Maher, L.J., 3rd. Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum. Mol. Genet. 2007, 16, 3136–3148. [Google Scholar] [CrossRef] [Green Version]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, R.K.; Luchtel, R.A.; Machha, V.; Tischer, A.; Zou, Y.; Pradhan, K.; Ashai, N.; Ramachandra, N.; Albanese, J.M.; Yang, J.I.; et al. Functional succinate dehydrogenase deficiency is a common adverse feature of clear cell renal cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2106947118. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.; An, J.; Rao, A. DNA methylation and hydroxymethylation in hematologic differentiation and transformation. Curr. Opin. Cell Biol. 2015, 37, 91–101. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P.; O’Neill, L.A.J. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784. [Google Scholar] [CrossRef] [Green Version]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta 2016, 1857, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- Hochachka, P.W.; Dressendorfer, R.H. Succinate accumulation in man during exercise. Eur. J. Appl. Physiol. Occup. Physiol. 1976, 35, 235–242. [Google Scholar] [CrossRef]
- Schranner, D.; Kastenmüller, G.; Schönfelder, M.; Römisch-Margl, W.; Wackerhage, H. Metabolite Concentration Changes in Humans after a Bout of Exercise: A Systematic Review of Exercise Metabolomics Studies. Sport. Med. Open 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, A.; Bozi, L.H.M.; Yaghi, O.K.; Mills, E.L.; Xiao, H.; Nicholson, H.E.; Paschini, M.; Paulo, J.A.; Garrity, R.; Laznik-Bogoslavski, D.; et al. pH-Gated Succinate Secretion Regulates Muscle Remodeling in Response to Exercise. Cell 2020, 183, 62–75.e17. [Google Scholar] [CrossRef]
- Yankovskaya, V.; Horsefield, R.; Törnroth, S.; Luna-Chavez, C.; Miyoshi, H.; Léger, C.; Byrne, B.; Cecchini, G.; Iwata, S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science 2003, 299, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rustin, P.; Munnich, A.; Rötig, A. Succinate dehydrogenase and human diseases: New insights into a well-known enzyme. Eur. J. Hum. Genet. 2002, 10, 289–291. [Google Scholar] [CrossRef] [Green Version]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Haller, R.G.; Henriksson, K.G.; Jorfeldt, L.; Hultman, E.; Wibom, R.; Sahlin, K.; Areskog, N.H.; Gunder, M.; Ayyad, K.; Blomqvist, C.G.; et al. Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J. Clin. Invest. 1991, 88, 1197–1206. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.E.; Henriksson, K.G.; Lewis, S.F.; Haller, R.G.; Kennaway, N.G. Mitochondrial myopathy with succinate dehydrogenase and aconitase deficiency. Abnormalities of several iron-sulfur proteins. J. Clin. Investig. 1993, 92, 2660–2666. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Xu, Y.Q.; Yuan, Y.X.; Xu, P.W.; Zhang, C.; Li, F.; Wang, L.N.; Yin, C.; Zhang, L.; Cai, X.C.; et al. Succinate induces skeletal muscle fiber remodeling via SUNCR1 signaling. EMBO Rep. 2019, 20, e47892. [Google Scholar] [CrossRef]
- Xu, G.; Yuan, Y.; Luo, P.; Yang, J.; Zhou, J.; Zhu, C.; Jiang, Q.; Shu, G. Acute Succinate Administration Increases Oxidative Phosphorylation and Skeletal Muscle Explosive Strength via SUCNR1. Front. Vet. Sci. 2022, 8, 808863. [Google Scholar] [CrossRef]
- Himms-Hagen, J. Brown adipose tissue thermogenesis: Interdisciplinary studies. FASEB J. 1990, 4, 2890–2898. [Google Scholar] [CrossRef]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. (Lausanne) 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, E.L.; Pierce, K.A.; Jedrychowski, M.P.; Garrity, R.; Winther, S.; Vidoni, S.; Yoneshiro, T.; Spinelli, J.B.; Lu, G.Z.; Kazak, L.; et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 2018, 560, 102–106. [Google Scholar] [CrossRef]
- Mills, E.L.; Harmon, C.; Jedrychowski, M.P.; Xiao, H.; Garrity, R.; Tran, N.V.; Bradshaw, G.A.; Fu, A.; Szpyt, J.; Reddy, A.; et al. UCP1 governs liver extracellular succinate and inflammatory pathogenesis. Nat. Metab. 2021, 3, 604–617. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Wittenberger, T.; Schaller, H.C.; Hellebrand, S. An expressed sequence tag (EST) data mining strategy succeeding in the discovery of new G-protein coupled receptors. J. Mol. Biol. 2001, 307, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, N.S.; Communi, D.; Hannedouche, S.; Boeynaems, J.M. The fate of P2Y-related orphan receptors: GPR80/99 and GPR91 are receptors of dicarboxylic acids. Purinergic Signal 2004, 1, 17–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toma, I.; Kang, J.J.; Sipos, A.; Vargas, S.; Bansal, E.; Hanner, F.; Meer, E.; Peti-Peterdi, J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Investig. 2008, 118, 2526–2534. [Google Scholar] [CrossRef] [Green Version]
- Khamaysi, A.; Anbtawee-Jomaa, S.; Fremder, M.; Eini-Rider, H.; Shimshilashvili, L.; Aharon, S.; Aizenshtein, E.; Shlomi, T.; Noguchi, A.; Springer, D.; et al. Systemic Succinate Homeostasis and Local Succinate Signaling Affect Blood Pressure and Modify Risks for Calcium Oxalate Lithogenesis. J. Am. Soc. Nephrol. 2019, 30, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Robben, J.H.; Fenton, R.A.; Vargas, S.L.; Schweer, H.; Peti-Peterdi, J.; Deen, P.M.; Milligan, G. Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int. 2009, 76, 1258–1267. [Google Scholar] [CrossRef] [Green Version]
- Trauelsen, M.; Hiron, T.K.; Lin, D.; Petersen, J.E.; Breton, B.; Husted, A.S.; Hjorth, S.A.; Inoue, A.; Frimurer, T.M.; Bouvier, M.; et al. Extracellular succinate hyperpolarizes M2 macrophages through SUCNR1/GPR91-mediated Gq signaling. Cell Rep. 2021, 35, 109246. [Google Scholar] [CrossRef]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwärzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Hakak, Y.; Lehmann-Bruinsma, K.; Phillips, S.; Le, T.; Liaw, C.; Connolly, D.T.; Behan, D.P. The role of the GPR91 ligand succinate in hematopoiesis. J. Leukoc. Biol. 2009, 85, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Högberg, C.; Gidlöf, O.; Tan, C.; Svensson, S.; Nilsson-Öhman, J.; Erlinge, D.; Olde, B. Succinate independently stimulates full platelet activation via cAMP and phosphoinositide 3-kinase-β signaling. J. Thromb. Haemost. 2011, 9, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Krzak, G.; Willis, C.M.; Smith, J.A.; Pluchino, S.; Peruzzotti-Jametti, L. Succinate Receptor 1: An Emerging Regulator of Myeloid Cell Function in Inflammation. Trends Immunol. 2021, 42, 45–58. [Google Scholar] [CrossRef]
- Gilissen, J.; Jouret, F.; Pirotte, B.; Hanson, J. Insight into SUCNR1 (GPR91) structure and function. Pharmacol. Ther. 2016, 159, 56–65. [Google Scholar] [CrossRef]
- de Castro Fonseca, M.; Aguiar, C.J.; da Rocha Franco, J.A.; Gingold, R.N.; Leite, M.F. GPR91: Expanding the frontiers of Krebs cycle intermediates. Cell Commun. Signal 2016, 14, 3. [Google Scholar] [CrossRef] [Green Version]
- Connors, J.; Dawe, N.; Van Limbergen, J. The Role of Succinate in the Regulation of Intestinal Inflammation. Nutrients 2018, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Reichardt, N.; Duncan, S.H.; Young, P.; Belenguer, A.; McWilliam Leitch, C.; Scott, K.P.; Flint, H.J.; Louis, P. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 2014, 8, 1323–1335. [Google Scholar] [CrossRef] [Green Version]
- Fremder, M.; Kim, S.W.; Khamaysi, A.; Shimshilashvili, L.; Eini-Rider, H.; Park, I.S.; Hadad, U.; Cheon, J.H.; Ohana, E. A transepithelial pathway delivers succinate to macrophages, thus perpetuating their pro-inflammatory metabolic state. Cell Rep. 2021, 36, 109521. [Google Scholar] [CrossRef]
- De Vadder, F.; Kovatcheva-Datchary, P.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-Produced Succinate Improves Glucose Homeostasis via Intestinal Gluconeogenesis. Cell Metab. 2016, 24, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Vily-Petit, J.; Soty-Roca, M.; Silva, M.; Raffin, M.; Gautier-Stein, A.; Rajas, F.; Mithieux, G. Intestinal gluconeogenesis prevents obesity-linked liver steatosis and non-alcoholic fatty liver disease. Gut 2020, 69, 2193–2202. [Google Scholar] [CrossRef] [PubMed]
- Nadjsombati, M.S.; McGinty, J.W.; Lyons-Cohen, M.R.; Jaffe, J.B.; DiPeso, L.; Schneider, C.; Miller, C.N.; Pollack, J.L.; Nagana Gowda, G.A.; Fontana, M.F.; et al. Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immun. 2018, 49, 33–41.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, W.; Ren, W.; Ohmoto, M.; Urban, J.F., Jr.; Matsumoto, I.; Margolskee, R.F.; Jiang, P. Activation of intestinal tuft cell-expressed Sucnr1 triggers type 2 immunity in the mouse small intestine. Proc. Natl. Acad. Sci. USA 2018, 115, 5552–5557. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.; O’Leary, C.E.; von Moltke, J.; Liang, H.E.; Ang, Q.Y.; Turnbaugh, P.J.; Radhakrishnan, S.; Pellizzon, M.; Ma, A.; Locksley, R.M. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell 2018, 174, 271–284.e14. [Google Scholar] [CrossRef] [Green Version]
- Jakobsdottir, G.; Xu, J.; Molin, G.; Ahrné, S.; Nyman, M. High-fat diet reduces the formation of butyrate, but increases succinate, inflammation, liver fat and cholesterol in rats, while dietary fibre counteracts these effects. PLoS ONE 2013, 8, e80476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macias-Ceja, D.C.; Ortiz-Masiá, D.; Salvador, P.; Gisbert-Ferrándiz, L.; Hernández, C.; Hausmann, M.; Rogler, G.; Esplugues, J.V.; Hinojosa, J.; Alós, R.; et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal. Immunol. 2019, 12, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zheng, Y.; Zhao, Y.; Zhang, Y.; Li, H.; Zhang, A.; Wang, X.; Wang, W.; Hou, Y.; Wang, J. Succinate/IL-1β Signaling Axis Promotes the Inflammatory Progression of Endothelial and Exacerbates Atherosclerosis. Front. Immunol. 2022, 13, 817572. [Google Scholar] [CrossRef]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, Y.T.; Miller, J.H.; Day, M.M.; Munger, J.C.; Brookes, P.S. Accumulation of Succinate in Cardiac Ischemia Primarily Occurs via Canonical Krebs Cycle Activity. Cell Rep. 2018, 23, 2617–2628. [Google Scholar] [CrossRef] [Green Version]
- Prag, H.A.; Gruszczyk, A.V.; Huang, M.M.; Beach, T.E.; Young, T.; Tronci, L.; Nikitopoulou, E.; Mulvey, J.F.; Ascione, R.; Hadjihambi, A.; et al. Mechanism of succinate efflux upon reperfusion of the ischaemic heart. Cardiovasc. Res. 2021, 117, 1188–1201. [Google Scholar] [CrossRef]
- Kohlhauer, M.; Dawkins, S.; Costa, A.S.H.; Lee, R.; Young, T.; Pell, V.R.; Choudhury, R.P.; Banning, A.P.; Kharbanda, R.K.; Oxford Acute Myocardial Infarction (OxAMI) Study; et al. Metabolomic Profiling in Acute ST-Segment-Elevation Myocardial Infarction Identifies Succinate as an Early Marker of Human Ischemia-Reperfusion Injury. J. Am. Heart Assoc. 2018, 7, e007546. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Luo, H.; Zhou, X.; Cheng, C.Y.; Lin, L.; Liu, B.L.; Liu, K.; Li, P.; Yang, H. Succinate-induced neuronal mitochondrial fission and hexokinase II malfunction in ischemic stroke: Therapeutical effects of kaempferol. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2307–2318. [Google Scholar] [CrossRef] [PubMed]
- Correa, P.R.; Kruglov, E.A.; Thompson, M.; Leite, M.F.; Dranoff, J.A.; Nathanson, M.H. Succinate is a paracrine signal for liver damage. J. Hepatol. 2007, 47, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.T.; Li, L.Z.; Yang, Y.L.; Yin, X.; Liu, Q.; Zhang, L.; Liu, K.; Liu, B.; Li, J.; Qi, L.W. Succinate induces aberrant mitochondrial fission in cardiomyocytes through GPR91 signaling. Cell Death Dis. 2018, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, C.J.; Rocha-Franco, J.A.; Sousa, P.A.; Santos, A.K.; Ladeira, M.; Rocha-Resende, C.; Ladeira, L.O.; Resende, R.R.; Botoni, F.A.; Barrouin Melo, M.; et al. Succinate causes pathological cardiomyocyte hypertrophy through GPR91 activation. Cell Commun. Signal 2014, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Prados, J.C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarique, A.A.; Logan, J.; Thomas, E.; Holt, P.G.; Sly, P.D.; Fantino, E. Phenotypic, functional, and plasticity features of classical and alternatively activated human macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 53, 676–688. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [Green Version]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapetanovic, R.; Afroz, S.F.; Ramnath, D.; Lawrence, G.M.; Okada, T.; Curson, J.E.; de Bruin, J.; Fairlie, D.P.; Schroder, K.; St John, J.C.; et al. Lipopolysaccharide promotes Drp1-dependent mitochondrial fission and associated inflammatory responses in macrophages. Immunol. Cell Biol. 2020, 98, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 2019, 67, 1047–1061. [Google Scholar] [CrossRef] [Green Version]
- van Diepen, J.A.; Robben, J.H.; Hooiveld, G.J.; Carmone, C.; Alsady, M.; Boutens, L.; Bekkenkamp-Grovenstein, M.; Hijmans, A.; Engelke, U.F.H.; Wevers, R.A.; et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia 2017, 60, 1304–1313. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.Y.; Huang, T.W.; Hsieh, Y.T.; Wang, Y.F.; Yen, C.C.; Lee, G.L.; Yeh, C.C.; Peng, Y.J.; Kuo, Y.Y.; Wen, H.T.; et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol. Cell. 2020, 77, 213–227.e5. [Google Scholar] [CrossRef]
- Keiran, N.; Ceperuelo-Mallafré, V.; Calvo, E.; Hernández-Alvarez, M.I.; Ejarque, M.; Núñez-Roa, C.; Horrillo, D.; Maymó-Masip, E.; Rodríguez, M.M.; Fradera, R.; et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat. Immunol. 2019, 20, 581–592. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Vicario, N.; Costa, A.S.H.; Kwok, C.K.; Leonardi, T.; Booty, L.M.; Bicci, I.; Balzarotti, B.; Volpe, G.; et al. Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell 2018, 22, 355–368.e13. [Google Scholar] [CrossRef] [Green Version]
- Vasandan, A.B.; Jahnavi, S.; Shashank, C.; Prasad, P.; Kumar, A.; Prasanna, S.J. Human Mesenchymal stem cells program macrophage plasticity by altering their metabolic status via a PGE2-dependent mechanism. Sci. Rep. 2016, 6, 38308. [Google Scholar] [CrossRef] [Green Version]
- Yañez, R.; Oviedo, A.; Aldea, M.; Bueren, J.A.; Lamana, M.L. Prostaglandin E2 plays a key role in the immunosuppressive properties of adipose and bone marrow tissue-derived mesenchymal stromal cells. Exp. Cell Res. 2010, 316, 3109–3123. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, S.; Quattrini, A.; Brambilla, E.; Gritti, A.; Salani, G.; Dina, G.; Galli, R.; Del Carro, U.; Amadio, S.; Bergami, A.; et al. Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature 2003, 422, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, S.; Zanotti, L.; Rossi, B.; Brambilla, E.; Ottoboni, L.; Salani, G.; Martinello, M.; Cattalini, A.; Bergami, A.; Furlan, R.; et al. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature 2005, 436, 266–271. [Google Scholar] [CrossRef]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Barron, L. Macrophages: Master regulators of inflammation and fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Park, S.Y.; Le, C.T.; Sung, K.Y.; Choi, D.H.; Cho, E.H. Succinate induces hepatic fibrogenesis by promoting activation, proliferation, and migration, and inhibiting apoptosis of hepatic stellate cells. Biochem. Biophys. Res. Commun. 2018, 496, 673–678. [Google Scholar] [CrossRef]
- Li, Y.H.; Woo, S.H.; Choi, D.H.; Cho, E.H. Succinate causes α-SMA production through GPR91 activation in hepatic stellate cells. Biochem. Biophys. Res. Commun. 2015, 463, 853–858. [Google Scholar] [CrossRef]
- Liu, X.J.; Xie, L.; Du, K.; Liu, C.; Zhang, N.P.; Gu, C.J.; Wang, Y.; Abdelmalek, M.F.; Dong, W.Y.; Liu, X.P.; et al. Succinate-GPR-91 receptor signalling is responsible for nonalcoholic steatohepatitis-associated fibrosis: Effects of DHA supplementation. Liver Int. 2020, 40, 830–843. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Choi, D.H.; Lee, E.H.; Seo, S.R.; Lee, S.; Cho, E.H. Sirtuin 3 (SIRT3) Regulates α-Smooth Muscle Actin (α-SMA) Production through the Succinate Dehydrogenase-G Protein-coupled Receptor 91 (GPR91) Pathway in Hepatic Stellate Cells. J. Biol. Chem. 2016, 291, 10277–10292. [Google Scholar] [CrossRef] [Green Version]
- Finley, L.W.; Haas, W.; Desquiret-Dumas, V.; Wallace, D.C.; Procaccio, V.; Gygi, S.P.; Haigis, M.C. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE 2011, 6, e23295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haigis, M.C.; Deng, C.X.; Finley, L.W.; Kim, H.S.; Gius, D. SIRT3 is a mitochondrial tumor suppressor: A scientific tale that connects aberrant cellular ROS, the Warburg effect, and carcinogenesis. Cancer Res. 2012, 72, 2468–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mu, X.; Zhao, T.; Xu, C.; Shi, W.; Geng, B.; Shen, J.; Zhang, C.; Pan, J.; Yang, J.; Hu, S.; et al. Oncometabolite succinate promotes angiogenesis by upregulating VEGF expression through GPR91-mediated STAT3 and ERK activation. Oncotarget 2017, 8, 13174–13185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, P.L.; Wu, W.H.; Hu, T.H.; Chen, C.W.; Cheng, H.C.; Li, C.F.; Tsai, W.H.; Tsai, H.J.; Hsieh, M.C.; Chuang, J.H.; et al. Decreased succinate dehydrogenase B in human hepatocellular carcinoma accelerates tumor malignancy by inducing the Warburg effect. Sci. Rep. 2018, 8, 3081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspuria, P.P.; Lunt, S.Y.; Väremo, L.; Vergnes, L.; Gozo, M.; Beach, J.A.; Salumbides, B.; Reue, K.; Wiedemeyer, W.R.; Nielsen, J.; et al. Succinate dehydrogenase inhibition leads to epithelial-mesenchymal transition and reprogrammed carbon metabolism. Cancer Metab. 2014, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, T.; Zhang, S.; Zhou, J.; Wang, Y.; Di, W. Succinate dehydrogenase subunit B inhibits the AMPK-HIF-1α pathway in human ovarian cancer in vitro. J. Ovarian Res. 2014, 7, 115. [Google Scholar] [CrossRef] [Green Version]
- Cervera, A.M.; Apostolova, N.; Crespo, F.L.; Mata, M.; McCreath, K.J. Cells silenced for SDHB expression display characteristic features of the tumor phenotype. Cancer Res. 2008, 68, 4058–4067. [Google Scholar] [CrossRef] [Green Version]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.; Douglas, F.; George, E.; Sköldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Kerlan, V.; Plouin, P.F.; Rötig, A.; Jeunemaitre, X. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J. Clin. Endocrinol. Metab. 2002, 87, 4771–4774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Crespin, M.; Nau, V.; Khau Van Kien, P.; Corvol, P.; Plouin, P.F.; Jeunemaitre, X.; et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003, 63, 5615–5621. [Google Scholar]
- King, K.S.; Prodanov, T.; Kantorovich, V.; Fojo, T.; Hewitt, J.K.; Zacharin, M.; Wesley, R.; Lodish, M.; Raygada, M.; Gimenez-Roqueplo, A.P.; et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: Significant link to SDHB mutations. J. Clin. Oncol. 2011, 29, 4137–4142. [Google Scholar] [CrossRef] [Green Version]
- Matlac, D.M.; Hadrava Vanova, K.; Bechmann, N.; Richter, S.; Folberth, J.; Ghayee, H.K.; Ge, G.B.; Abunimer, L.; Wesley, R.; Aherrahrou, R.; et al. Succinate Mediates Tumorigenic Effects via Succinate Receptor 1: Potential for New Targeted Treatment Strategies in Succinate Dehydrogenase Deficient Paragangliomas. Front. Endocrinol. (Lausanne) 2021, 12, 589451. [Google Scholar] [CrossRef]
- Bardella, C.; Pollard, P.J.; Tomlinson, I. SDH mutations in cancer. Biochim. Biophys. Acta 2011, 1807, 1432–1443. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.H.; Plescia, J.; Dohi, T.; Rosa, J.; Doxsey, S.J.; Altieri, D.C. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell 2007, 131, 257–270. [Google Scholar] [CrossRef]
- Lisanti, S.; Garlick, D.S.; Bryant, K.G.; Tavecchio, M.; Mills, G.B.; Lu, Y.; Kossenkov, A.V.; Showe, L.C.; Languino, L.R.; Altieri, D.C. Transgenic Expression of the Mitochondrial Chaperone TNFR-associated Protein 1 (TRAP1) Accelerates Prostate Cancer Development. J. Biol. Chem. 2016, 291, 25247–25254. [Google Scholar] [CrossRef] [Green Version]
- Sciacovelli, M.; Guzzo, G.; Morello, V.; Frezza, C.; Zheng, L.; Nannini, N.; Calabrese, F.; Laudiero, G.; Esposito, F.; Landriscina, M.; et al. The mitochondrial chaperone TRAP1 promotes neoplastic growth by inhibiting succinate dehydrogenase. Cell Metab. 2013, 17, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Mak, P.; Leav, I.; Pursell, B.; Bae, D.; Yang, X.; Taglienti, C.A.; Gouvin, L.M.; Sharma, V.M.; Mercurio, A.M. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: Implications for Gleason grading. Cancer Cell 2010, 17, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.C.; Wu, J.Y.; Wu, K.K. Cancer-derived extracellular succinate: A driver of cancer metastasis. J. Biomed. Sci. 2022, 29, 93. [Google Scholar] [CrossRef]
- D’Anna, F.; Van Dyck, L.; Xiong, J.; Zhao, H.; Berrens, R.V.; Qian, J.; Bieniasz-Krzywiec, P.; Chandra, V.; Schoonjans, L.; Matthews, J.; et al. DNA methylation repels binding of hypoxia-inducible transcription factors to maintain tumor immunotolerance. Genome Biol. 2020, 21, 182. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouhi, P.; Lee, S.L.; Cao, Z.; Hedlund, E.M.; Jensen, L.D.; Cao, Y. Pathological angiogenesis facilitates tumor cell dissemination and metastasis. Cell Cycle 2010, 9, 913–917. [Google Scholar] [CrossRef] [Green Version]
- Kula-Alwar, D.; Prag, H.A.; Krieg, T. Targeting Succinate Metabolism in Ischemia/Reperfusion Injury. Circulation 2019, 140, 1968–1970. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [Green Version]
- Harber, K.J.; de Goede, K.E.; Verberk, S.G.S.; Meinster, E.; de Vries, H.E.; van Weeghel, M.; de Winther, M.P.J.; Van den Bossche, J. Succinate Is an Inflammation-Induced Immunoregulatory Metabolite in Macrophages. Metabolites 2020, 10, 372. [Google Scholar] [CrossRef]
- Malaisse, W.J.; Nadi, A.B.; Ladriere, L.; Zhang, T.M. Protective effects of succinic acid dimethyl ester infusion in experimental endotoxemia. Nutrition 1997, 13, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.L.; Ladrière, L.; Vincent, J.L.; Malaisse, W.J. Prolongation of survival time by infusion of succinic acid dimethyl ester in a caecal ligation and perforation model of sepsis. Horm. Metab. Res. 2000, 32, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Protti, A.; Singer, M. Bench-to-bedside review: Potential strategies to protect or reverse mitochondrial dysfunction in sepsis-induced organ failure. Crit. Care 2006, 10, 228. [Google Scholar] [CrossRef] [Green Version]
- Jalloh, I.; Helmy, A.; Howe, D.J.; Shannon, R.J.; Grice, P.; Mason, A.; Gallagher, C.N.; Stovell, M.G.; van der Heide, S.; Murphy, M.P.; et al. Focally perfused succinate potentiates brain metabolism in head injury patients. J. Cereb. Blood Flow Metab. 2017, 37, 2626–2638. [Google Scholar] [CrossRef] [PubMed]
- Czibik, G.; Steeples, V.; Yavari, A.; Ashrafian, H. Citric acid cycle intermediates in cardioprotection. Circ. Cardiovasc. Genet. 2014, 7, 711–719. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, J.M.; Venkatachalam, M.A.; Roeser, N.F.; Nissim, I. Mitochondrial dysfunction during hypoxia/reoxygenation and its correction by anaerobic metabolism of citric acid cycle intermediates. Proc. Natl. Acad. Sci. USA 2000, 97, 2826–2831. [Google Scholar] [CrossRef]
- Ariza, A.C.; Deen, P.M.; Robben, J.H. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. (Lausanne) 2012, 3, 22. [Google Scholar] [CrossRef] [Green Version]
- Velcicky, J.; Wilcken, R.; Cotesta, S.; Janser, P.; Schlapbach, A.; Wagner, T.; Piechon, P.; Villard, F.; Bouhelal, R.; Piller, F.; et al. Discovery and Optimization of Novel SUCNR1 Inhibitors: Design of Zwitterionic Derivatives with a Salt Bridge for the Improvement of Oral Exposure. J. Med. Chem. 2020, 63, 9856–9875. [Google Scholar] [CrossRef]
- Haffke, M.; Fehlmann, D.; Rummel, G.; Boivineau, J.; Duckely, M.; Gommermann, N.; Cotesta, S.; Sirockin, F.; Freuler, F.; Littlewood-Evans, A.; et al. Structural basis of species-selective antagonist binding to the succinate receptor. Nature 2019, 574, 581–585. [Google Scholar] [CrossRef]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The human serum metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [Green Version]
- Rautureau, G.J.P.; Morio, B.; Guibert, S.; Lefevre, C.; Perrier, J.; Alves, A.; Chauvin, M.A.; Pinteur, C.; Monet, M.A.; Godet, M.; et al. Dietary obesity in mice is associated with lipid deposition and metabolic shifts in the lungs sharing features with the liver. Sci. Rep. 2021, 11, 8712. [Google Scholar] [CrossRef] [PubMed]
- Tong, W.; Hannou, S.A.; Wang, Y.; Astapova, I.; Sargsyan, A.; Monn, R.; Thiriveedi, V.; Li, D.; McCann, J.R.; Rawls, J.F.; et al. The intestine is a major contributor to circulating succinate in mice. FASEB J. 2022, 36, e22546. [Google Scholar] [CrossRef]
- Beloborodova, N.; Pautova, A.; Sergeev, A.; Fedotcheva, N. Serum Levels of Mitochondrial and Microbial Metabolites Reflect Mitochondrial Dysfunction in Different Stages of Sepsis. Metabolites 2019, 9, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadagopan, N.; Li, W.; Roberds, S.L.; Major, T.; Preston, G.M.; Yu, Y.; Tones, M.A. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am. J. Hypertens. 2007, 20, 1209–1215. [Google Scholar] [CrossRef] [PubMed]
- Osuna-Prieto, F.J.; Martinez-Tellez, B.; Ortiz-Alvarez, L.; Di, X.; Jurado-Fasoli, L.; Xu, H.; Ceperuelo-Mallafré, V.; Núñez-Roa, C.; Kohler, I.; Segura-Carretero, A.; et al. Elevated plasma succinate levels are linked to higher cardiovascular disease risk factors in young adults. Cardiovasc. Diabetol. 2021, 20, 151. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Sharp, W.W.; Fang, Y.H.; Han, M.; Zhang, H.J.; Hong, Z.; Banathy, A.; Morrow, E.; Ryan, J.J.; Archer, S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: Therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. 2014, 28, 316–326. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.B.; Subrayan, S.; Lim, S.Y.; Yellon, D.M.; Davidson, S.M.; Hausenloy, D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010, 121, 2012–2022. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, K.K. Extracellular Succinate: A Physiological Messenger and a Pathological Trigger. Int. J. Mol. Sci. 2023, 24, 11165. https://doi.org/10.3390/ijms241311165
Wu KK. Extracellular Succinate: A Physiological Messenger and a Pathological Trigger. International Journal of Molecular Sciences. 2023; 24(13):11165. https://doi.org/10.3390/ijms241311165
Chicago/Turabian StyleWu, Kenneth K. 2023. "Extracellular Succinate: A Physiological Messenger and a Pathological Trigger" International Journal of Molecular Sciences 24, no. 13: 11165. https://doi.org/10.3390/ijms241311165