
Glucose Deprivation Induces Cancer Cell Death through Failure of ROS Regulation
 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
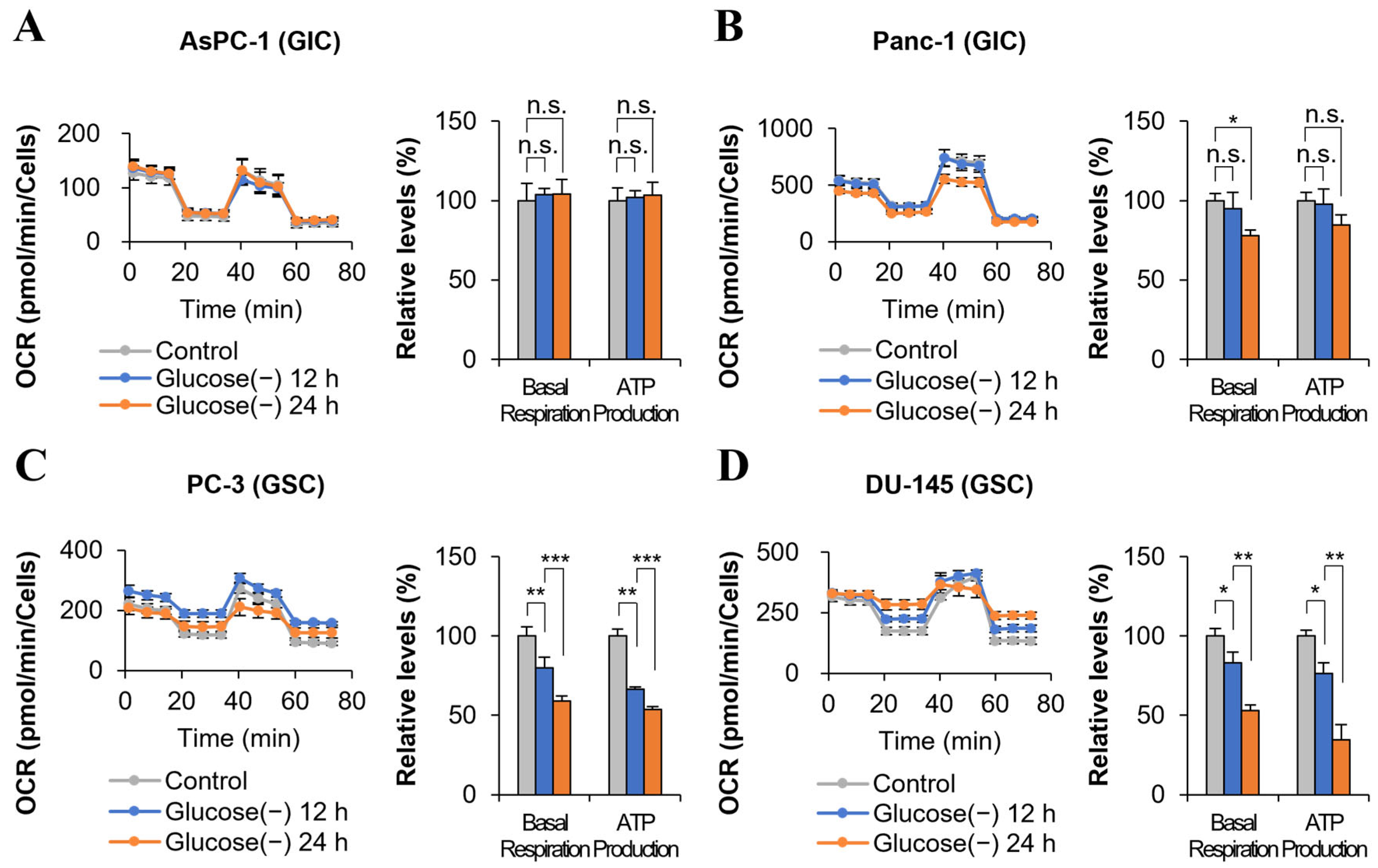
2.1. ATP Production Decreases in Some Cancer Cells after 24 h of Glucose Deprivation
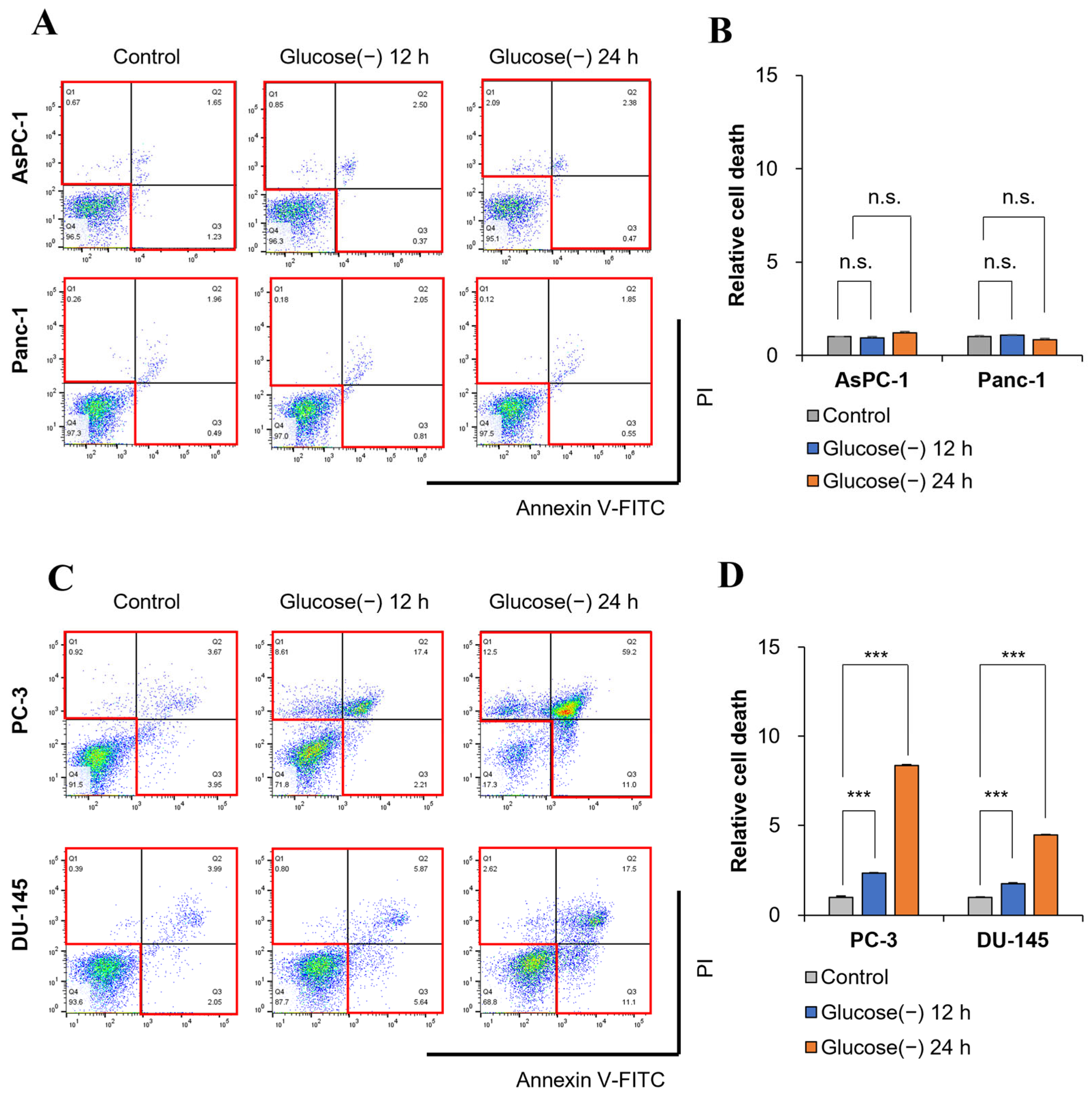
2.2. Glucose Deprivation Induces Cell Death in GSC within 12 h
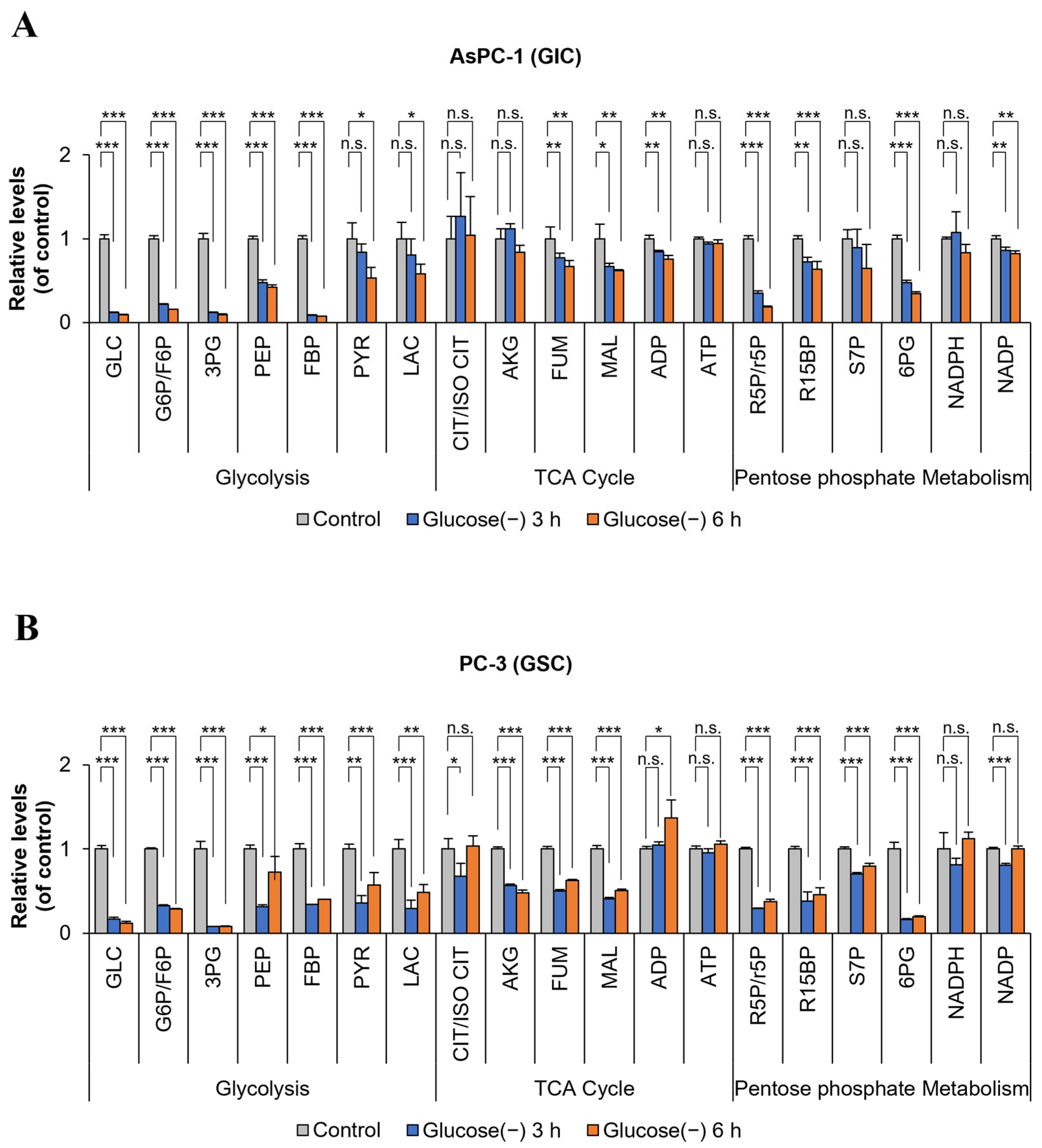
2.3. ATP Levels in GSC and GIC do Not Change Markedly after 6 h of Glucose Deprivation
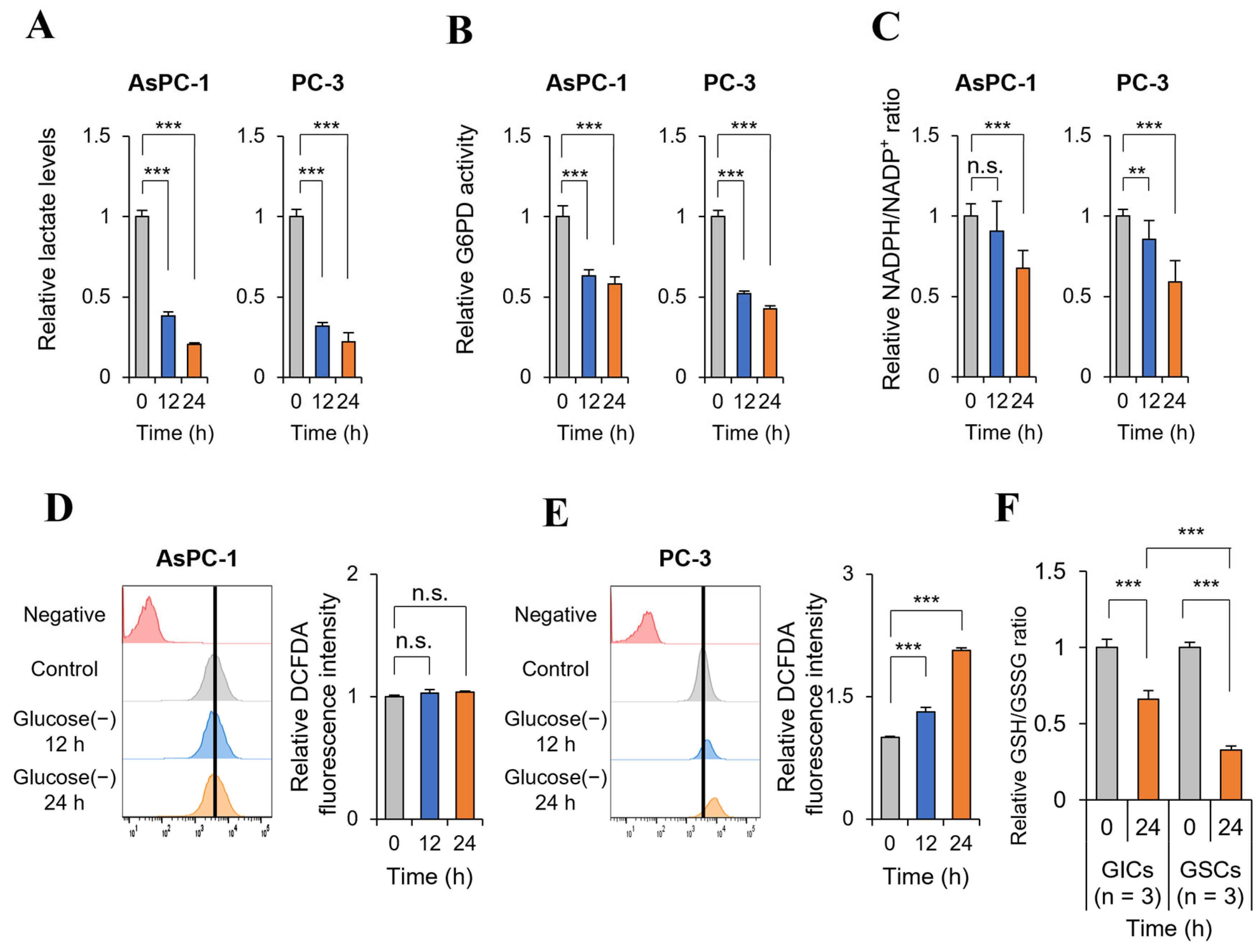
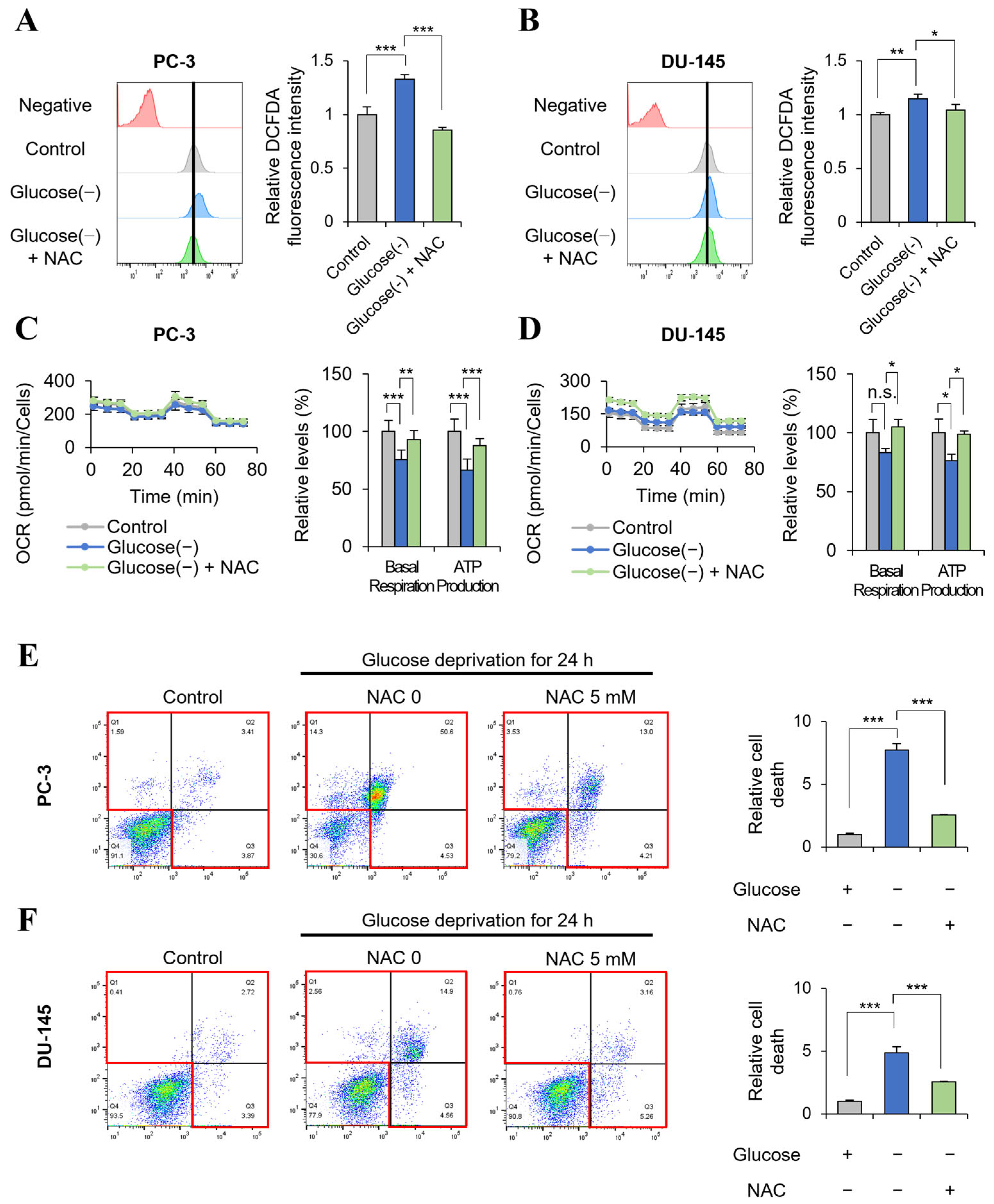
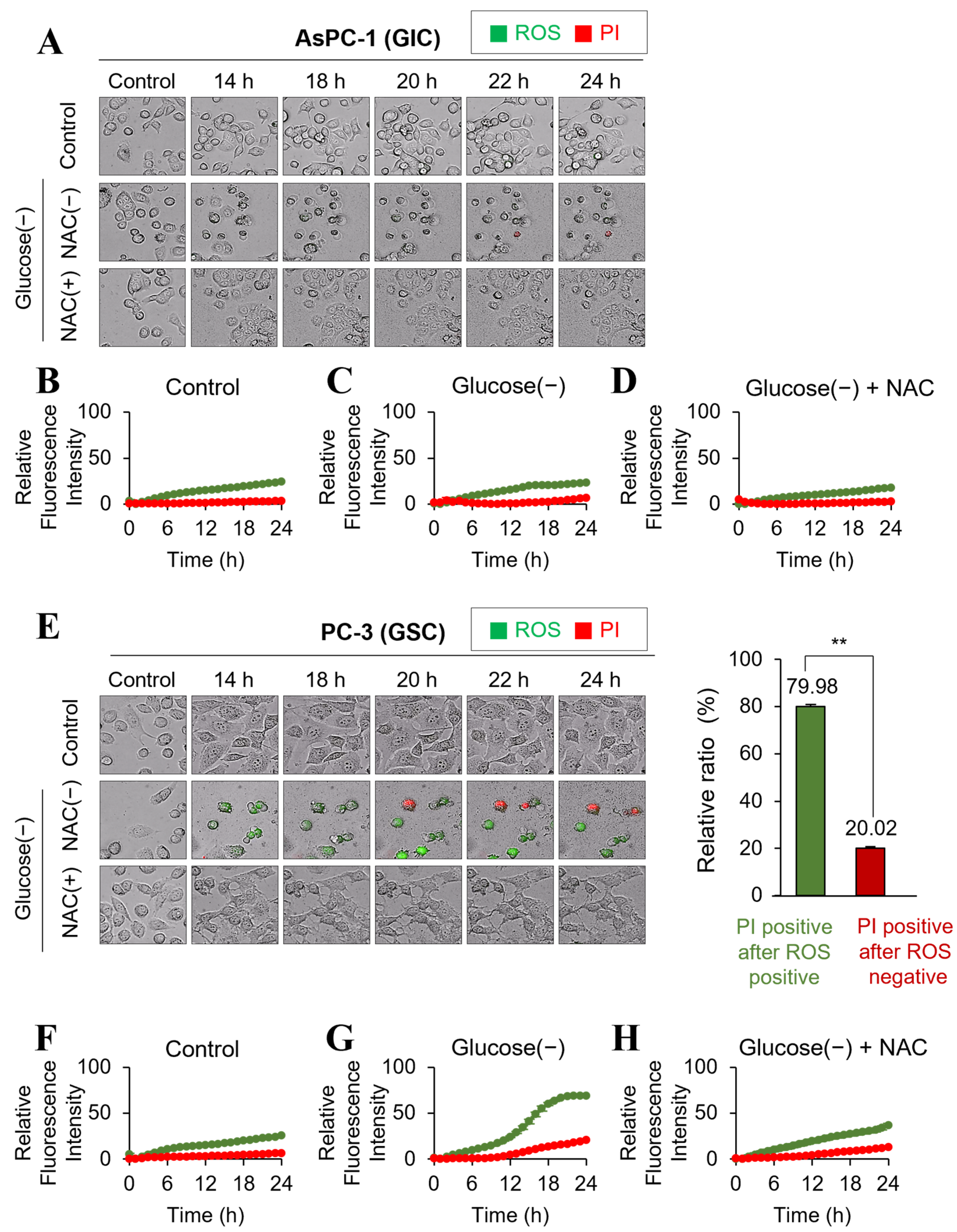
2.4. Glucose Deprivation Induces Cell Death Only in GSC with a Significant Increase in ROS Production
2.5. Glucose Deprivation-Induced GSC Cell Death Is Rescued by the Antioxidant NAC
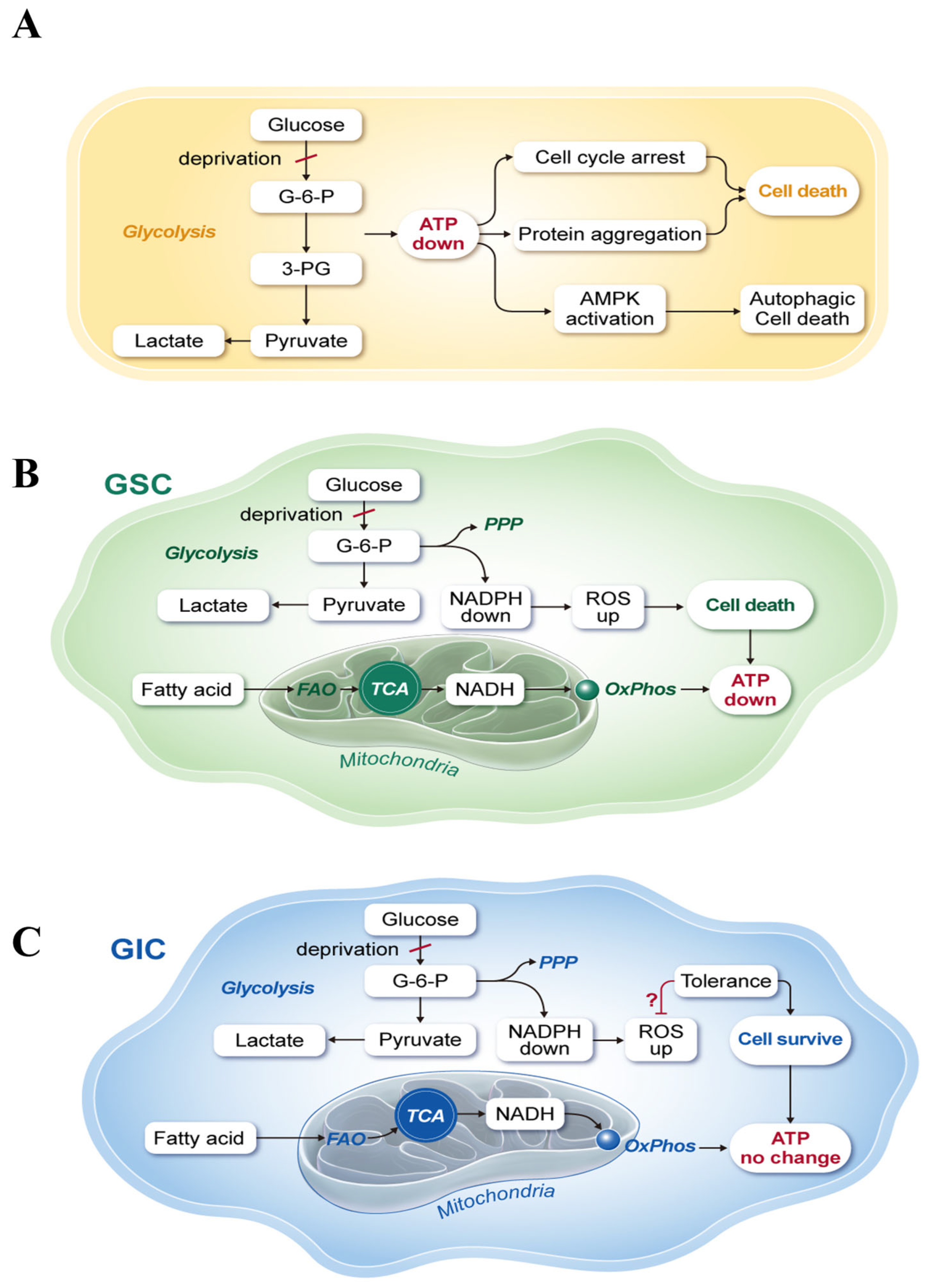
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Seahorse Mito Stress Test Assay
4.3. FITC Annexin V and Propidium Iodide (PI) Cell Death Detection
4.4. Reactive Oxygen Species (ROS) Measurement
4.5. Lactate Measurement
4.6. ATP Measurement
4.7. NADPH Assay
4.8. GSH/GSSG Ratio
4.9. LC-MS/MS
4.10. Real-Time Live Cell Imaging and High Content Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, H.; Woo, S.M.; Jang, H.; Kang, M.; Kim, S.Y. Cancer depends on fatty acids for ATP production: A possible link between cancer and obesity. Semin. Cancer Biol. 2022, 86, 347–357. [Google Scholar] [CrossRef]
- Lee, J.S.; Oh, S.J.; Choi, H.J.; Kang, J.H.; Lee, S.H.; Ha, J.S.; Woo, S.M.; Jang, H.; Lee, H.; Kim, S.Y. ATP Production Relies on Fatty Acid Oxidation Rather than Glycolysis in Pancreatic Ductal Adenocarcinoma. Cancers 2020, 12, 2477. [Google Scholar] [CrossRef]
- Ganapathy-Kanniappan, S.; Geschwind, J.F. Tumor glycolysis as a target for cancer therapy: Progress and prospects. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Puschel, F.; Leon-Annicchiarico, C.L.; O’Connor, H.; Martin, S.J.; Palou-Gramon, D.; Lucendo, E.; Munoz-Pinedo, C. Glucose Deprivation Induces ATF4-Mediated Apoptosis through TRAIL Death Receptors. Mol. Cell. Biol. 2017, 37, e00479-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro-Maldonado, A.; Tait, S.W.; Ramirez-Peinado, S.; Ricci, J.E.; Fabregat, I.; Green, D.R.; Munoz-Pinedo, C. Glucose deprivation induces an atypical form of apoptosis mediated by caspase-8 in Bax-, Bak-deficient cells. Cell Death Differ. 2010, 17, 1335–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karabiyik, C.; Vicinanza, M.; Son, S.M.; Rubinsztein, D.C. Glucose starvation induces autophagy via ULK1-mediated activation of PIKfyve in an AMPK-dependent manner. Dev. Cell 2021, 56, 1961–1975.e5. [Google Scholar] [CrossRef] [PubMed]
- Graham, N.A.; Tahmasian, M.; Kohli, B.; Komisopoulou, E.; Zhu, M.; Vivanco, I.; Teitell, M.A.; Wu, H.; Ribas, A.; Lo, R.S.; et al. Glucose deprivation activates a metabolic and signaling amplification loop leading to cell death. Mol. Syst. Biol. 2012, 8, 589. [Google Scholar] [CrossRef]
- Sweet, S.; Singh, G. Accumulation of human promyelocytic leukemic (HL-60) cells at two energetic cell cycle checkpoints. Cancer Res. 1995, 55, 5164–5167. [Google Scholar]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sanchez-Perez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Aldini, G.; Altomare, A.; Baron, G.; Vistoli, G.; Carini, M.; Borsani, L.; Sergio, F. N-Acetylcysteine as an antioxidant and disulphide breaking agent: The reasons why. Free Radic. Res. 2018, 52, 751–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O.; Posener, K.; Negelein, E. Über den stoffwechsel der carcinomzelle. Naturwissenschaften 1924, 12, 1131–1137. [Google Scholar] [CrossRef]
- Eagle, H. Amino acid metabolism in mammalian cell cultures. Science 1959, 130, 432–437. [Google Scholar] [CrossRef]
- Puck, T.T.; Cieciura, S.J.; Robinson, A. Genetics of somatic mammalian cells. III. Long-term cultivation of euploid cells from human and animal subjects. J. Exp. Med. 1958, 108, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends. Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 2019, 1, 404–415. [Google Scholar] [CrossRef]
- Peng, F.; Liao, M.; Qin, R.; Zhu, S.; Peng, C.; Fu, L.; Chen, Y.; Han, B. Regulated cell death (RCD) in cancer: Key pathways and targeted therapies. Signal Transduct. Target. Ther. 2022, 7, 286. [Google Scholar] [CrossRef]
- Wilkie-Grantham, R.P.; Matsuzawa, S.; Reed, J.C. Novel phosphorylation and ubiquitination sites regulate reactive oxygen species-dependent degradation of anti-apoptotic c-FLIP protein. J. Biol. Chem. 2013, 288, 12777–12790. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef] [PubMed]
- Kodama, R.; Kato, M.; Furuta, S.; Ueno, S.; Zhang, Y.; Matsuno, K.; Yabe-Nishimura, C.; Tanaka, E.; Kamata, T. ROS-generating oxidases Nox1 and Nox4 contribute to oncogenic Ras-induced premature senescence. Genes Cells 2013, 18, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Choi, A.; Jun, Y.; Kim, N.; Yook, J.I.; Kim, S.Y.; Lee, S.; Kang, S.W. Glutathione peroxidase-1 regulates adhesion and metastasis of triple-negative breast cancer cells via FAK signaling. Redox Biol. 2020, 29, 101391. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 721–733. [Google Scholar] [CrossRef]
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.H.; Lee, S.H.; Hong, D.; Lee, J.S.; Ahn, H.S.; Ahn, J.H.; Seong, T.W.; Lee, C.H.; Jang, H.; Hong, K.M.; et al. Aldehyde dehydrogenase is used by cancer cells for energy metabolism. Exp. Mol. Med. 2016, 48, e272. [Google Scholar] [CrossRef] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Bensaude, O. Increased thermal aggregation of proteins in ATP-depleted mammalian cells. Eur. J. Biochem. 1994, 220, 239–246. [Google Scholar] [CrossRef]
- Song, C.F.; Hu, Y.H.; Mang, Z.G.; Ye, Z.; Chen, H.D.; Jing, D.S.; Fan, G.X.; Ji, S.R.; Yu, X.J.; Xu, X.W.; et al. Hernandezine induces autophagic cell death in human pancreatic cancer cells via activation of the ROS/AMPK signaling pathway. Acta Pharmacol. Sin. 2023, 44, 865–876. [Google Scholar] [CrossRef]
- Izyumov, D.S.; Avetisyan, A.V.; Pletjushkina, O.Y.; Sakharov, D.V.; Wirtz, K.W.; Chernyak, B.V.; Skulachev, V.P. "Wages of fear": Transient threefold decrease in intracellular ATP level imposes apoptosis. Biochim. Biophys. Acta 2004, 1658, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar] [PubMed]
- Ju, H.Q.; Lin, J.F.; Tian, T.; Xie, D.; Xu, R.H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef] [PubMed]
- Kuehne, A.; Emmert, H.; Soehle, J.; Winnefeld, M.; Fischer, F.; Wenck, H.; Gallinat, S.; Terstegen, L.; Lucius, R.; Hildebrand, J.; et al. Acute Activation of Oxidative Pentose Phosphate Pathway as First-Line Response to Oxidative Stress in Human Skin Cells. Mol. Cell 2015, 59, 359–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manegold, C. Pemetrexed (Alimta, MTA, multitargeted antifolate, LY231514) for malignant pleural mesothelioma. Semin. Oncol. 2003, 30 (Suppl. S10), 32–36. [Google Scholar] [CrossRef]
- Bae, S.H.; Kim, J.H.; Park, T.H.; Lee, K.; Lee, B.I.; Jang, H. BMS794833 inhibits macrophage efferocytosis by directly binding to MERTK and inhibiting its activity. Exp. Mol. Med. 2022, 54, 1450–1460. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, M.; Kang, J.H.; Sim, I.A.; Seong, D.Y.; Han, S.; Jang, H.; Lee, H.; Kang, S.W.; Kim, S.-Y. Glucose Deprivation Induces Cancer Cell Death through Failure of ROS Regulation. Int. J. Mol. Sci. 2023, 24, 11969. https://doi.org/10.3390/ijms241511969
Kang M, Kang JH, Sim IA, Seong DY, Han S, Jang H, Lee H, Kang SW, Kim S-Y. Glucose Deprivation Induces Cancer Cell Death through Failure of ROS Regulation. International Journal of Molecular Sciences. 2023; 24(15):11969. https://doi.org/10.3390/ijms241511969
Chicago/Turabian StyleKang, Mingyu, Joon H. Kang, In A. Sim, Do Y. Seong, Suji Han, Hyonchol Jang, Ho Lee, Sang W. Kang, and Soo-Youl Kim. 2023. "Glucose Deprivation Induces Cancer Cell Death through Failure of ROS Regulation" International Journal of Molecular Sciences 24, no. 15: 11969. https://doi.org/10.3390/ijms241511969