
Exploring Transcriptional Regulation of Hyperaccumulation in Sedum plumbizincicola through Integrated Transcriptome Analysis and CRISPR/Cas9 Technology
Abstract
:1. Introduction
2. Results
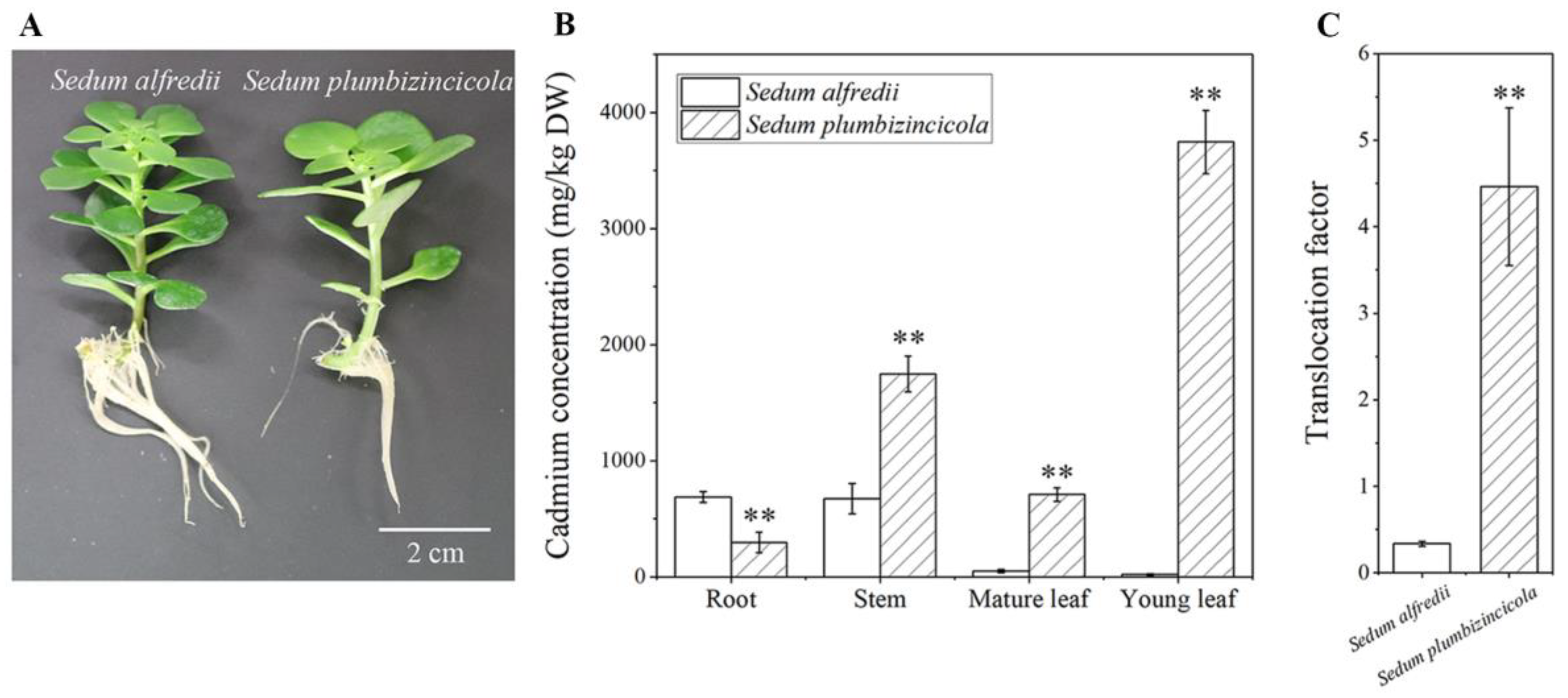
2.1. Cd Accumulation Differences between S. plumbizincicola and S. alfredii (NHE)
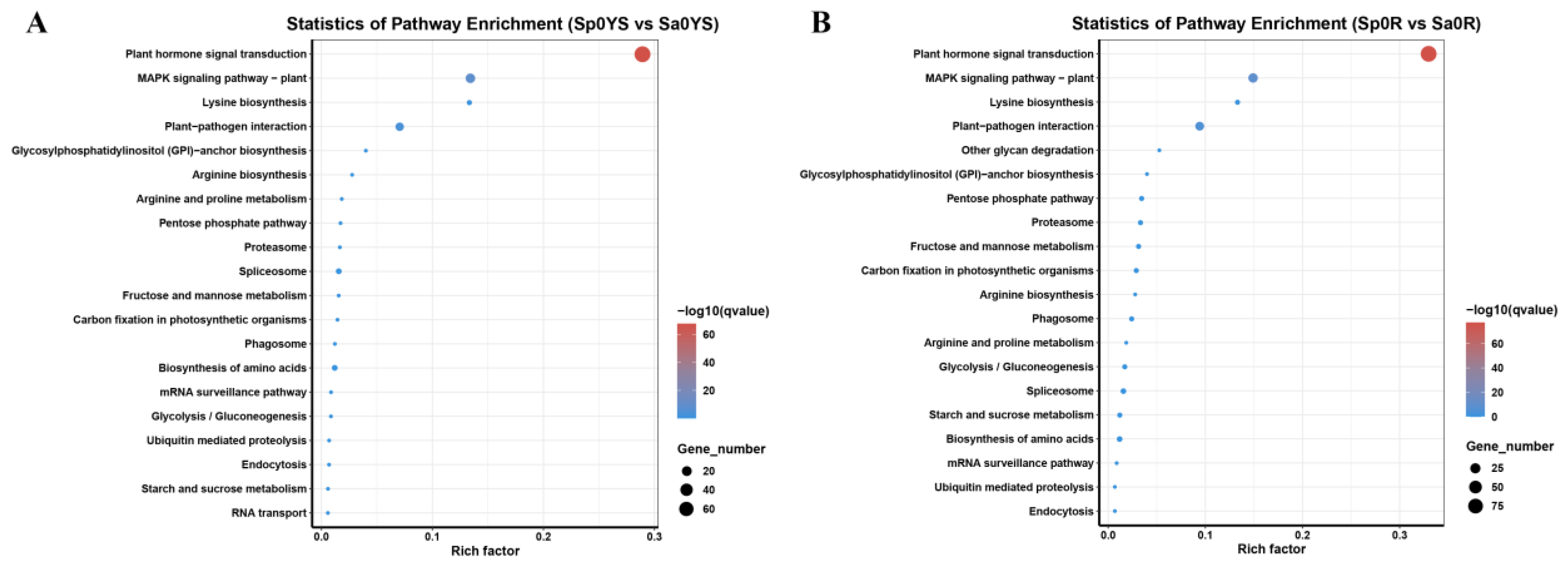
2.2. RNA-Seq Analysis on S. plumbizincicola and S. alfredii under Cadmium Treatment
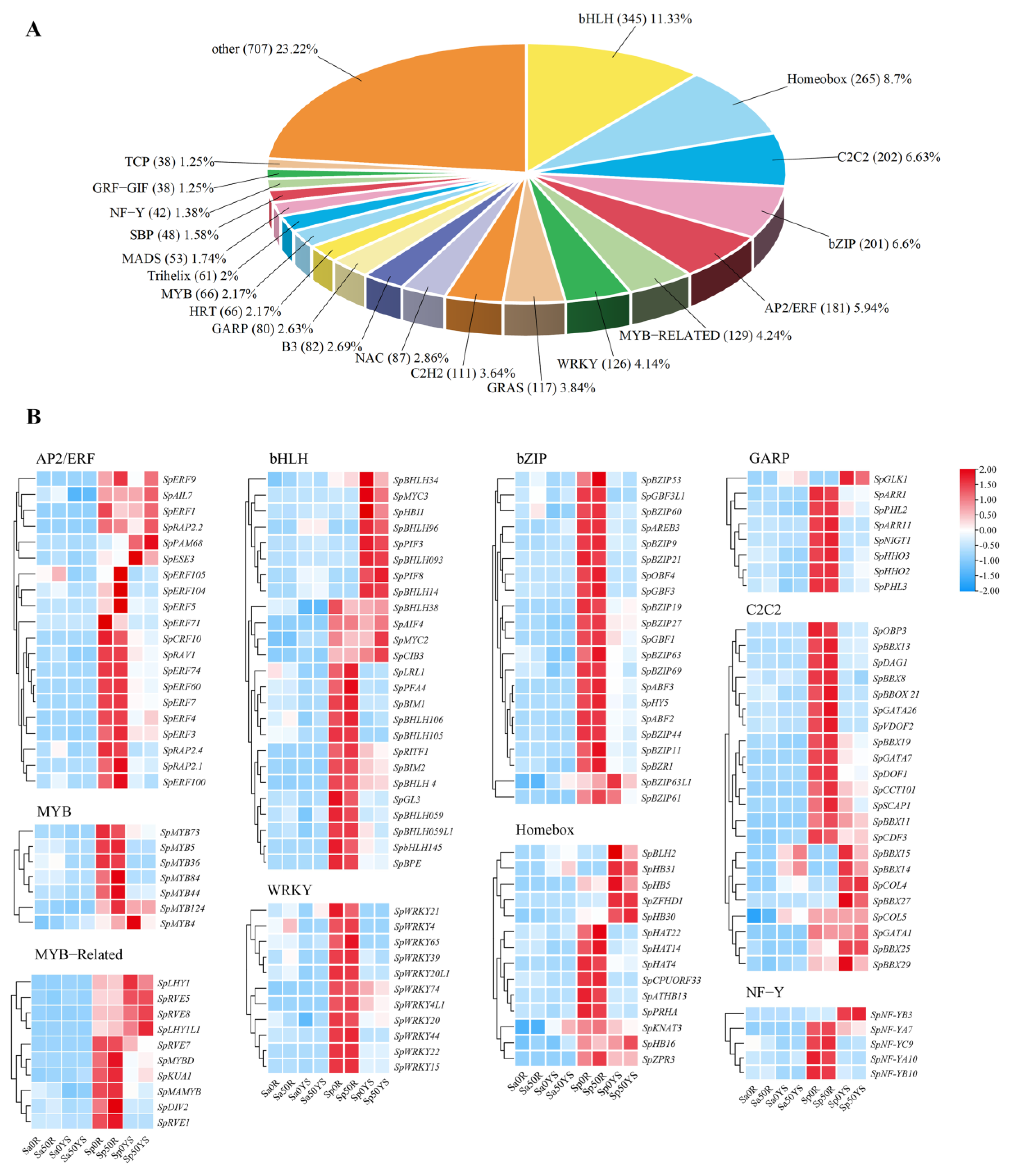
2.3. Expression of Transcriptional Regulatory Genes
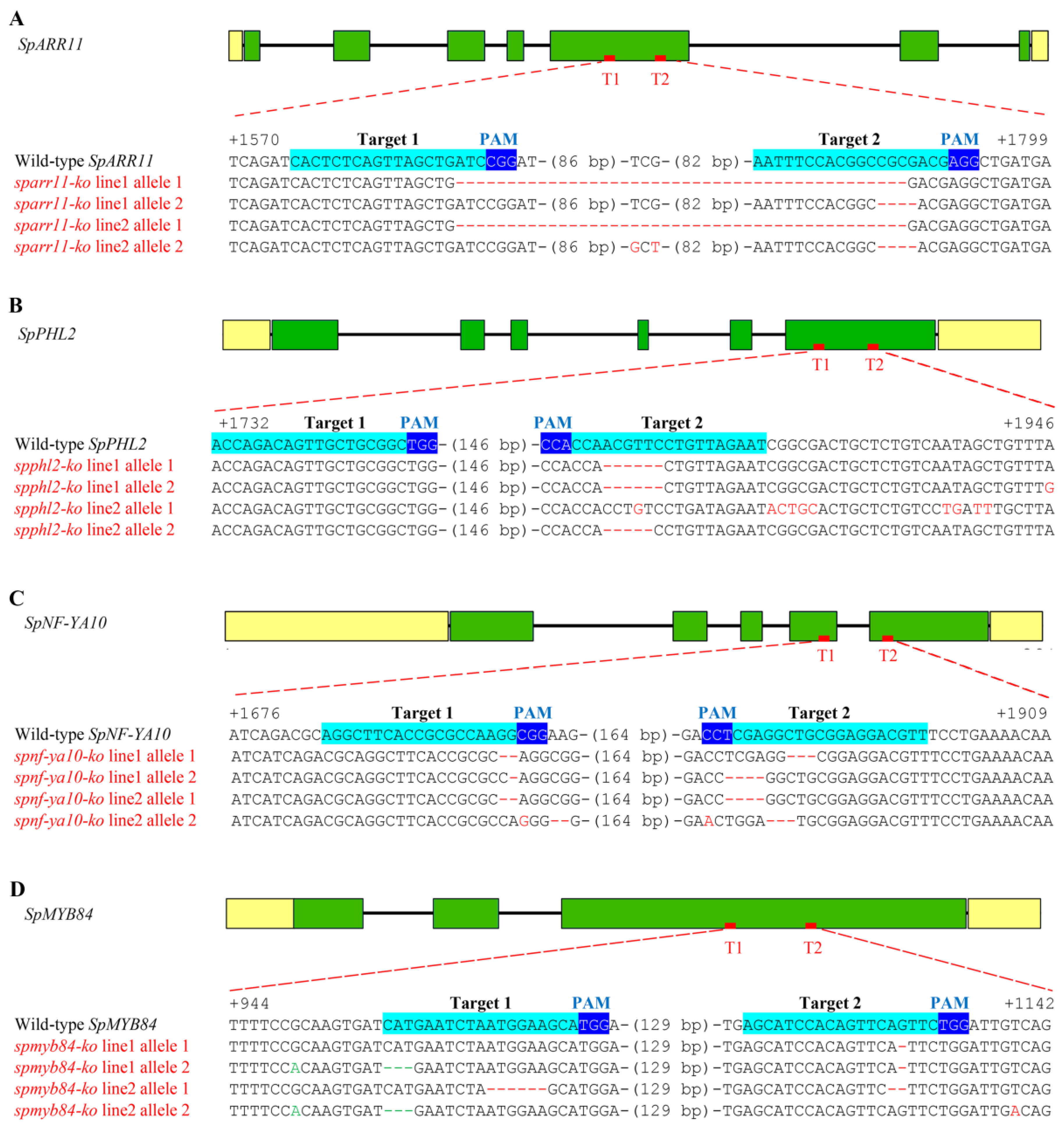
2.4. Creating Knockout Mutants of Four Transcription Factor Genes Using CRISPR/Cas9 System
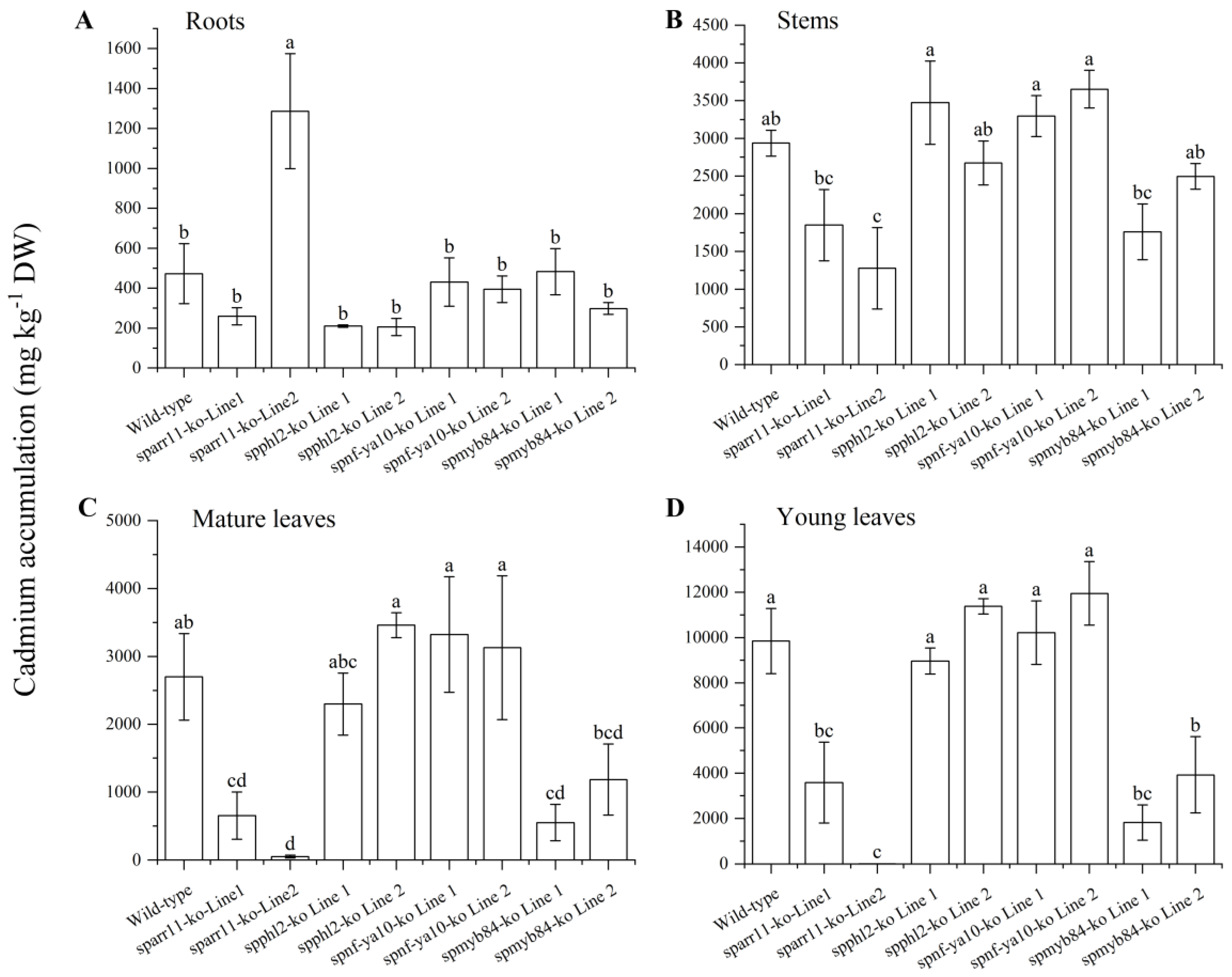
2.5. Cd Accumulation Analysis of the Transcription Factor Genes Knockout Mutants
3. Discussion
4. Materials and Methods
4.1. Plant Material and Growth Condition
4.2. Determination of Cd Content in Plants
4.3. Transcriptome Sequencing
4.4. Transcriptome Analysis
4.5. Transmembrane, Subcellular and Motif Prediction of Hyperaccumulation-Related Candidate Genes
4.6. Transcription Factor Genes Cloning and Knockout Vector Construction
4.7. Plant Transformations
4.8. Determination of Cadmium Content in Transgenic Knockout Plants
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
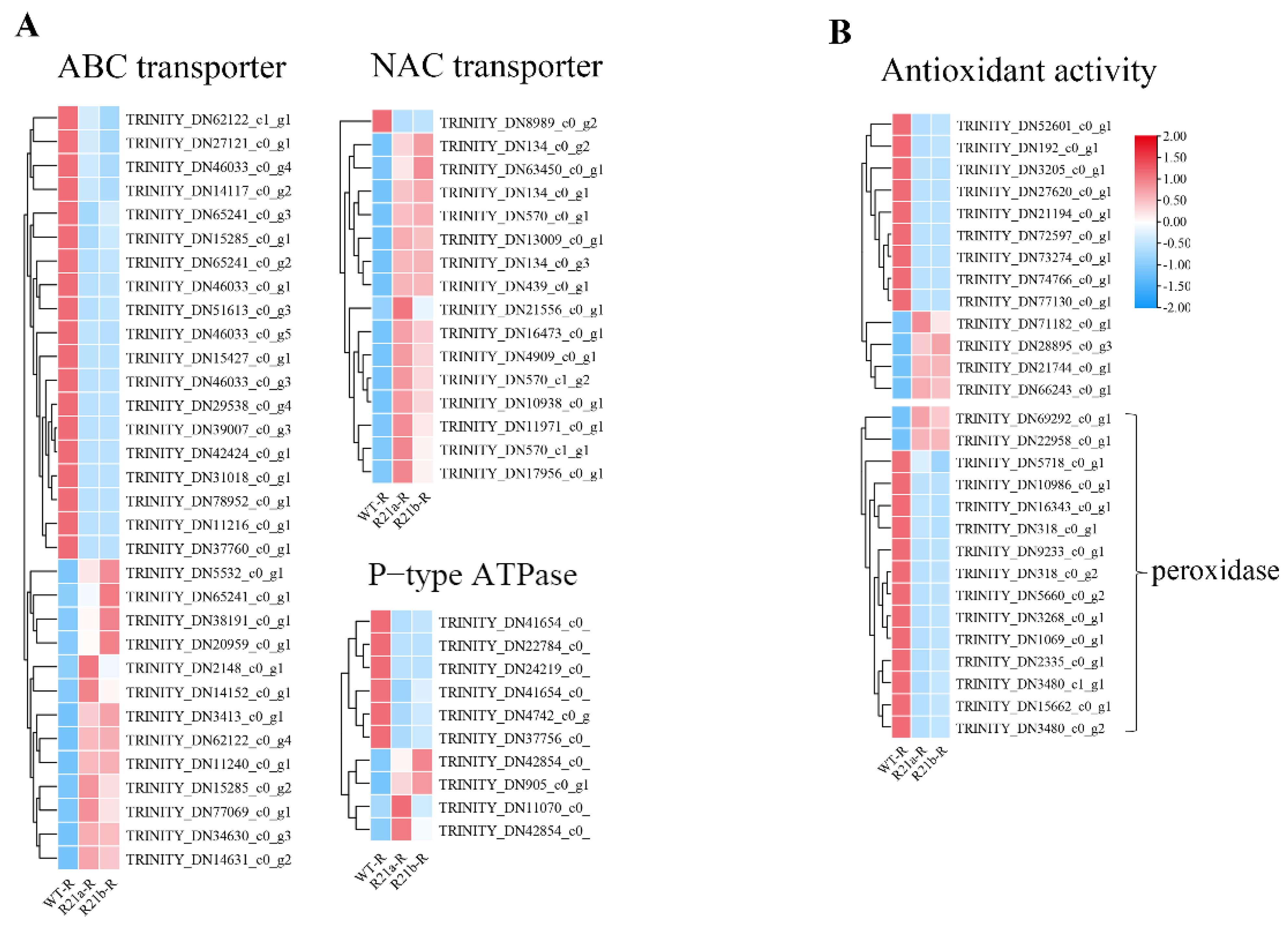
Appendix A.1. Transcriptome Analysis of Knockout Mutants from SpARR11 and SpMYB84
Appendix A.1.1. Transcriptome Sequencing
Appendix A.1.2. Transcriptome Analysis
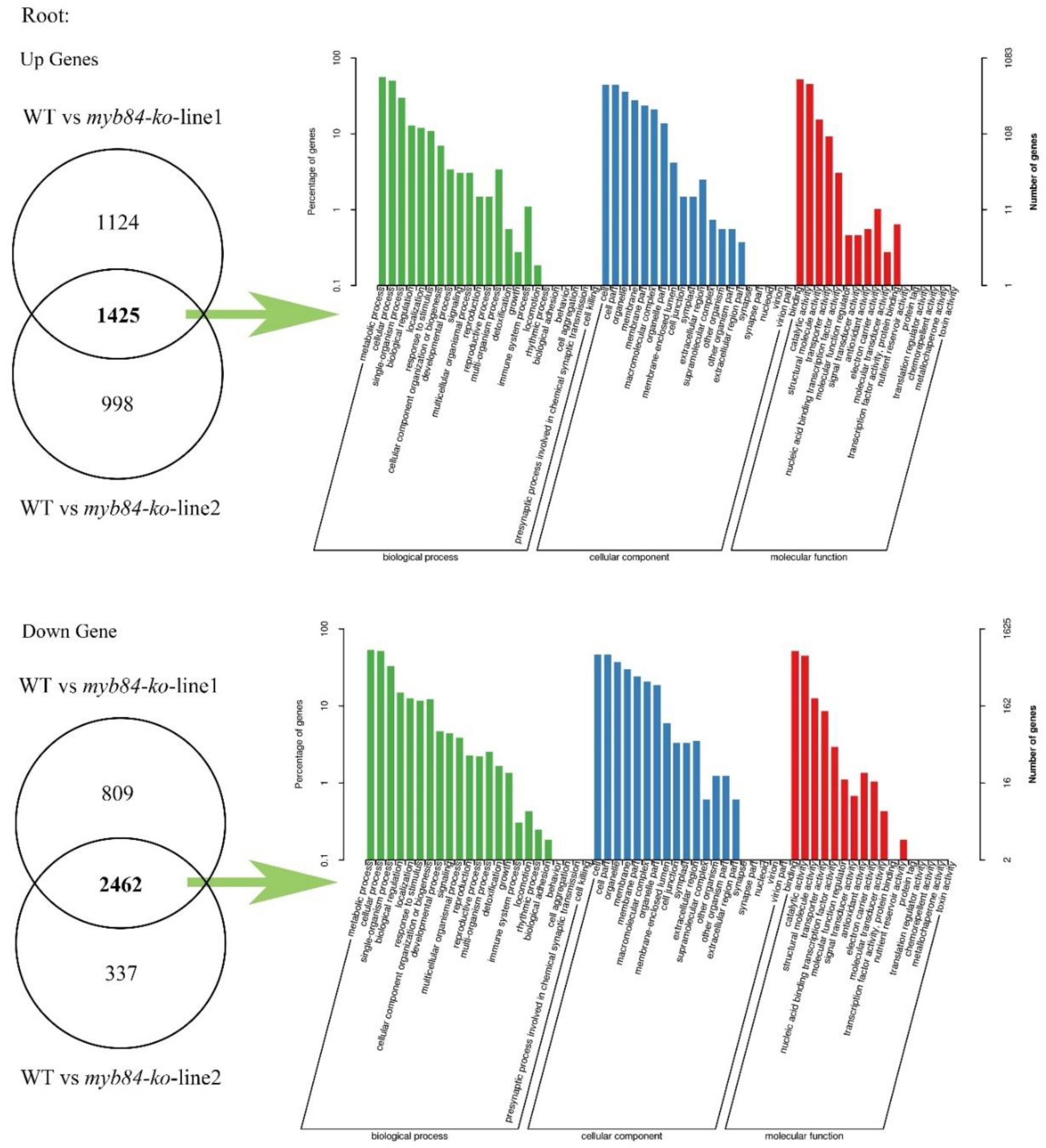
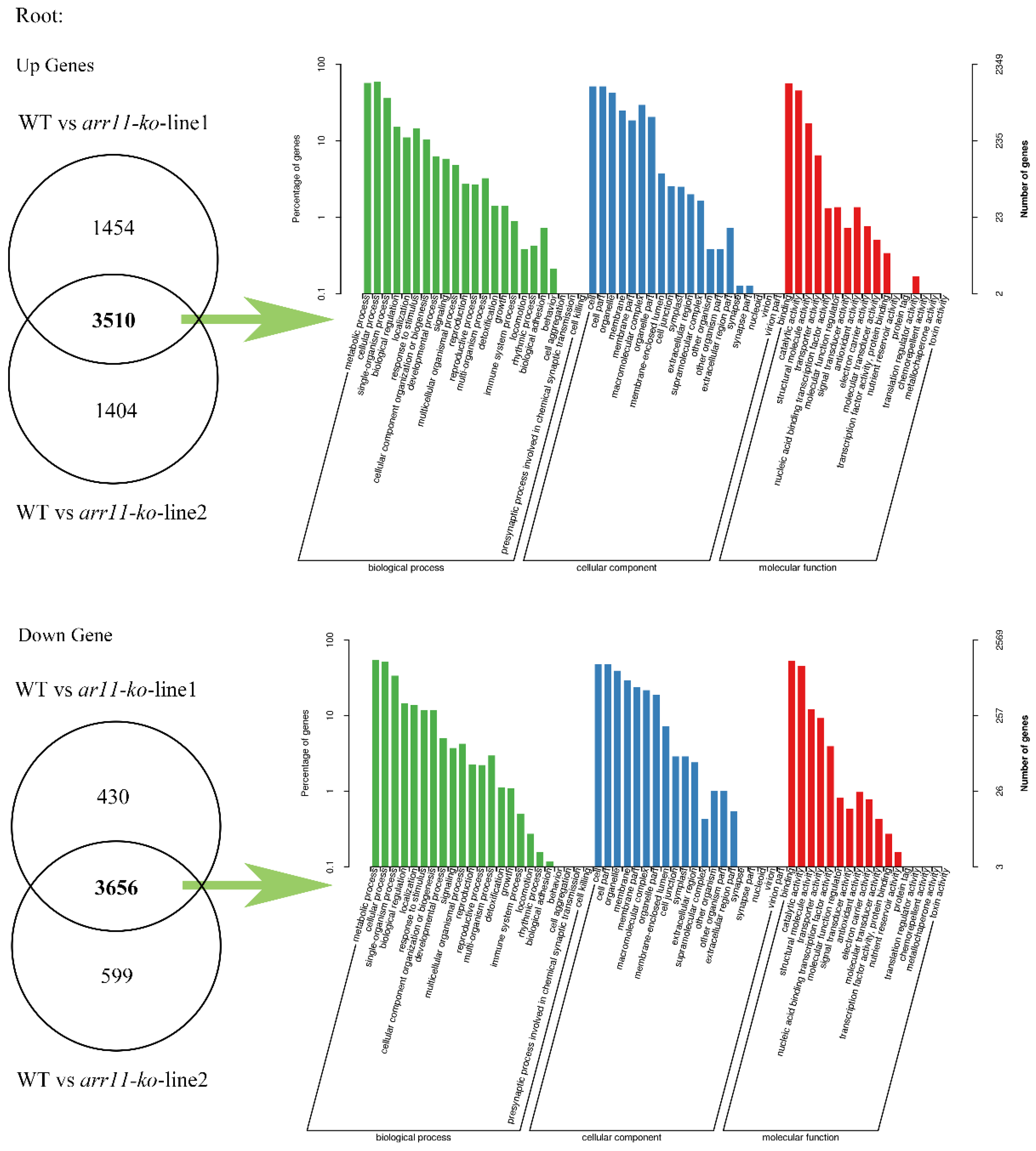
Appendix A.1.3. Genes Knockout Mutants Transcriptome Differential Gene Analysis




References
- Genchi, G.; Sinicropi, M.S.; Lauria, G.; Carocci, A.; Catalano, A. The effects of cadmium toxicity. Int. J. Environ. Res. Public Health 2020, 17, 3782. [Google Scholar] [CrossRef]
- Meyer, C.; Verbruggen, N. The use of the model species Arabidopsis halleri towards phytoextraction of cadmium polluted soils. New Biotechnol. 2012, 30, 9–14. [Google Scholar] [CrossRef]
- Manara, A.; Elisa, F.; Fasani, E.; Furini, A.; DalCorso, G. Evolution of the metal hyperaccumulation and hypertolerance traits. Plant Cell Environ. 2020, 43, 2969–2986. [Google Scholar] [CrossRef]
- Weber, M.; Trampczynska, A.; Clemens, S. Comparative transcriptome analysis of toxic metal responses in Arabidopsis thaliana and the Cd2+-hypertolerant facultative metallophyte Arabidopsis halleri. Plant Cell Environ. 2006, 29, 950–963. [Google Scholar] [CrossRef]
- Hanikenne, M.; Talke, I.N.; Haydon, M.J.; Lanz, C.; Nolte, A.; Motte, P.; Kroymann, J.; Weigel, D.; Kramer, U. Evolution of metal hyperaccumulation required cis-regulatory changes and triplication of HMA4. Nature 2008, 453, 391–395. [Google Scholar] [CrossRef]
- Milner, M.J.; Mitani-Ueno, N.; Yamaji, N.; Yokosho, K.; Craft, E.; Fei, Z.; Ebbs, S.; Zambrano, M.C.; Ma, J.F.; Kochian, L.V. Root and shoot transcriptome analysis of two ecotypes of Noccaea caerulescens uncovers the role of NcNramp1 in Cd hyperaccumulation. Plant J. 2014, 78, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Ueno, D.; Milner, M.J.; Yamaji, N.; Yokosho, K.; Koyama, E.; Clemencia, Z.M.; Kaskie, M.; Ebbs, S.; Kochian, L.V.; Ma, J.F. Elevated expression of TcHMA3 plays a key role in the extreme Cd tolerance in a Cd-hyperaccumulating ecotype of Thlaspi caerulescens. Plant J. 2011, 66, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Van de Mortel, J.E.; Almar Villanueva, L.; Schat, H.; Kwekkeboom, J.; Coughlan, S.; Moerland, P.D.; Ver Loren van Themaat, E.; Koornneef, M.; Aarts, M.G. Large expression differences in genes for iron and zinc homeostasis, stress response, and lignin biosynthesis distinguish roots of Arabidopsis thaliana and the related metal hyperaccumulator Thlaspi caerulescens. Plant Physiol. 2006, 142, 1127–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, J.S.; Wang, Y.J.; Ge, D.; Ma, H.L.; Zhang, Y.J.; Gong, J.M. A pivotal role of cell wall in cadmium accumulation in the Crassulaceae hyperaccumulator Sedum plumbizincicola. Mol. Plant 2017, 10, 771–774. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhao, H.; Wu, L.; Liu, A.; Zhao, F.J.; Xu, W. Heavy metal ATPase 3 (HMA3) confers cadmium hypertolerance on the cadmium/zinc hyperaccumulator Sedum plumbizincicola. New Phytol. 2017, 215, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wang, L.; Zhao, F.J.; Wu, L.; Liu, A.; Xu, W. SpHMA1 is a chloroplast cadmium exporter protecting photochemical reactions in the Cd hyperaccumulator Sedum plumbizincicola. Plant Cell Environ. 2019, 42, 1112–1124. [Google Scholar] [CrossRef]
- Li, Z.; Wu, L.; Hu, P.; Luo, Y.; Zhang, H.; Christie, P. Repeated phytoextraction of four metal-contaminated soils using the cadmium/zinc hyperaccumulator Sedum plumbizincicola. Environ. Pollut. 2014, 189, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Mitra, M.; Banerjee, S.; Roy, S. MYB4 transcription factor, a member of R2R3-subfamily of MYB domain protein, regulates cadmium tolerance via enhanced protection against oxidative damage and increases expression of PCS1 and MT1C in Arabidopsis. Plant Sci. 2020, 297, 110501. [Google Scholar] [CrossRef]
- Sheng, Y.; Yan, X.; Huang, Y.; Han, Y.; Zhang, C.; Ren, Y.; Fan, T.; Xiao, F.; Liu, Y.; Cao, S. The WRKY transcription factor, WRKY13, activates PDR8 expression to positively regulate cadmium tolerance in Arabidopsis. Plant Cell Environ. 2019, 42, 891–903. [Google Scholar] [CrossRef]
- Hu, S.; Yu, Y.; Chen, Q.; Mu, G.; Shen, Z.; Zheng, L. OsMYB45 plays an important role in rice resistance to cadmium stress. Plant Sci. 2017, 264, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Meisel, L.; Lam, E. Switching of gene expression: Analysis of the factors that spatially and temporally regulate plant gene expression. Genet. Eng. 1997, 19, 183–199. [Google Scholar] [CrossRef]
- Hehl, R. From experiment-driven database analyses to database-driven experiments in Arabidopsis thaliana transcription factor research. Plant Sci. 2017, 262, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, B.; Ding, W.; Liu, X.; Yang, D.L.; Wei, P.; Cao, F.; Zhu, S.; Zhang, F.; Mao, Y.; et al. Efficient genome editing in plants using a CRISPR/Cas system. Cell Res. 2013, 23, 1229–1232. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Zhang, H.; Xu, N.; Zhang, B.; Gou, F.; Zhu, J.K. Application of the CRISPR-Cas system for efficient genome engineering in plants. Mol. Plant 2013, 6, 2008–2011. [Google Scholar] [CrossRef] [Green Version]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Hussain, D.; Haydon, M.J.; Wang, Y.; Wong, E.; Sherson, S.M.; Young, J.; Camakaris, J.; Harper, J.F.; Cobbett, C.S. P-type ATPase heavy metal transporters with roles in essential zinc homeostasis in Arabidopsis. Plant Cell 2004, 16, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.; Ma, X.; Luo, S.; Gao, J.; Yang, X.; Feng, Y. SaZIP4, an uptake transporter of Zn/Cd hyperaccumulator Sedum alfredii Hance. Environ. Exp. Bot. 2018, 155, 107–117. [Google Scholar] [CrossRef]
- Hall, J.L. Cellular mechanisms for heavy metal detoxification and tolerance. J. Exp. Bot. 2002, 53, 1–11. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, M.; Tian, S.; Lu, L.; Shohag, M.J.I.; Yang, X. Metallothionein 2 (SaMT2) from Sedum alfredii Hance confers increased cd tolerance and accumulation in yeast and tobacco. PLoS ONE 2014, 9, e102750. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.S.; Ding, G.; Meng, S.; Yi, H.Y.; Gong, J.M. Enhanced metal tolerance correlates with heterotypic variation in SpMTL, a metallothionein-like protein from the hyperaccumulator Sedum plumbizincicola. Plant Cell Environ. 2017, 40, 1368–1378. [Google Scholar] [CrossRef]
- Zheng, P.; Cao, L.; Zhang, C.; Pan, W.; Wang, W.; Yu, X.; Li, Y.; Fan, T.; Miao, M.; Tang, X.; et al. MYB43 as a novel substrate for CRL4PRL1 E3 ligases negatively regulates cadmium tolerance through transcriptional inhibition of HMAs in Arabidopsis. New Phytol. 2022, 234, 884–901. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, R.; Ju, Q.; Li, W.; Tran, L.P.; Xu, J. The R2R3-MYB transcription factor MYB49 regulates cadmium accumulation. Plant Physiol. 2019, 180, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Fan, T.; Zhu, X.; Wu, X.; Ouyang, J.; Jiang, L.; Cao, S. WRKY12 represses GSH1 expression to negatively regulate cadmium tolerance in Arabidopsis. Plant Mol. Biol. 2019, 99, 149–159. [Google Scholar] [CrossRef]
- Chen, S.; Yu, M.; Li, H.; Wang, Y.; Lu, Z.; Zhang, Y.; Liu, M.; Qiao, G.; Wu, L.; Han, X.; et al. SaHsfA4c from Sedum alfredii Hance enhances cadmium tolerance by regulating ROS-scavenger activities and heat shock proteins expression. Front. Plant Sci. 2020, 11, 142. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Kisko, M.; Dubos, C.; Lacombe, B.; Berthomieu, P.; Krouk, G.; Rouached, H. TransDetect identifies a new regulatory module controlling phosphate accumulation. Plant Physiol. 2017, 175, 916–926. [Google Scholar] [CrossRef] [Green Version]
- Falconieri, G.S.; Bertini, L.; Bizzarri, E.; Proietti, S.; Caruso, C. Plant defense: ARR11 response regulator as a potential player in Arabidopsis. Front. Plant Sci. 2022, 13, 995178. [Google Scholar] [CrossRef]
- Wu, L.H.; Liu, Y.J.; Zhou, S.B.; Guo, F.G.; Bi, D.; Guo, X.H.; Baker, A.J.M.; Smith, J.A.C.; Luo, Y.M. Sedum plumbizincicola X.H. Guo et S.B. Zhou ex L.H. Wu (Crassulaceae): A new species from Zhejiang Province, China. Plant Syst. Evol. 2013, 299, 487–498. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Xing, H.L.; Dong, L.; Wang, Z.P.; Zhang, H.Y.; Han, C.Y.; Liu, B.; Wang, X.C.; Chen, Q.J. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 2014, 14, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.P.; Xing, H.L.; Dong, L.; Zhang, H.Y.; Han, C.Y.; Wang, X.C.; Chen, Q.J. Egg cell-specific promoter-controlled CRISPR/Cas9 efficiently generates homozygous mutants for multiple target genes in Arabidopsis in a single generation. Genome Biol. 2015, 16, 144. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Best-Hit-A.th 1 | Gene Name | Gene Family | Motif 2 | Sa0YS 3 | Sa0R 3 | Sp0YS 3 | Sp0R 3 |
|---|---|---|---|---|---|---|---|---|
| Sp-R_1-10k_transcript/44752 | AT2G41130.1 | BHLH106 | bHLH | HLH | 6.83 | 25.97 | 10.49 | 77.64 |
| Sp-R_1-10k_transcript/39703 | AT4G02590.2 | BHLH059 | bHLH | HLH, Tom22 | 6.36 | 13.10 | 14.80 | 42.77 |
| Sp-R_1-10k_transcript/42007 | AT1G42990.1 | BZIP60 | bZIP | bZIP_1, bZIP_2, | 2.56 | 7.45 | 8.61 | 25.85 |
| Sp-R_1-10k_transcript/40522 | AT1G75390.1 | BZIP44 | bZIP | bZIP_1, bZIP_2, | 0.69 | 3.04 | 23.96 | 82.14 |
| Sp-R_1-10k_transcript/35867 | AT3G16857.2 | ARR11 | GARP | Response_reg, Myb_DNA-binding, | 0.17 | 0.63 | 0.63 | 23.22 |
| Sp-R_1-10k_transcript/39786 | AT4G13640.1 | PHL3 | GARP | Myb_CC_LHEQLE, Myb_DNA-binding, | 5.44 | 17.36 | 17.51 | 76.14 |
| Sp-R_1-10k_transcript/38907 | AT3G24120.1 | PHL2 | GARP | Myb_CC_LHEQLE, Myb_DNA-binding, | 4.68 | 6.82 | 13.02 | 23.05 |
| Sp-R_1-10k_transcript/17174 | AT1G14350.1 | MYB124 | MYB | Myb_DNA-binding, Myb_DNA-bind_6, | 4.05 | 5.51 | 14.93 | 13.85 |
| Sp-R_1-10k_transcript/38968 | AT3G49690.1 | MYB84 | MYB | Myb_DNA-binding, Myb_DNA-bind_6 | 0.05 | 11.72 | 0.15 | 64.96 |
| Sp-R_1-10k_transcript/39578 | AT5G57620.1 | MYB36 | MYB | Myb_DNA-binding, Myb_DNA-bind_6, | 0.15 | 8.69 | 0.19 | 37.67 |
| Sp-R_1-10k_transcript/38848 | AT5G06510.1 | NF-YA10 | NF-Y | CBFB_NFYA | 1.10 | 3.14 | 2.39 | 23.14 |
| Sp-R_1-10k_transcript/40870 | AT1G29280.1 | WRKY65 | WRKY | WRKY | 4.50 | 12.28 | 2.51 | 37.59 |
| Sp-R_1-10k_transcript/21995 | AT2G37260.1 | WRKY44 | WRKY | WRKY, FLYWCH | 0.61 | 4.15 | 1.59 | 62.10 |
| Sp-R_1-10k_transcript/26282 | AT2G30590.1 | WRKY21 | WRKY | WRKY, Plant_zn_clust | 9.34 | 13.06 | 9.79 | 35.90 |
| Sp-R_1-10k_transcript/41387 | AT2G23320.1 | WRKY15 | WRKY | WRKY, Plant_zn_clust | 4.89 | 7.71 | 21.96 | 90.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Mo, Y.; Han, L.; Sun, Z.; Xu, W. Exploring Transcriptional Regulation of Hyperaccumulation in Sedum plumbizincicola through Integrated Transcriptome Analysis and CRISPR/Cas9 Technology. Int. J. Mol. Sci. 2023, 24, 11845. https://doi.org/10.3390/ijms241411845
Zhang Y, Mo Y, Han L, Sun Z, Xu W. Exploring Transcriptional Regulation of Hyperaccumulation in Sedum plumbizincicola through Integrated Transcriptome Analysis and CRISPR/Cas9 Technology. International Journal of Molecular Sciences. 2023; 24(14):11845. https://doi.org/10.3390/ijms241411845
Chicago/Turabian StyleZhang, Yixin, Yanlan Mo, Liyuan Han, Zhenyuan Sun, and Wenzhong Xu. 2023. "Exploring Transcriptional Regulation of Hyperaccumulation in Sedum plumbizincicola through Integrated Transcriptome Analysis and CRISPR/Cas9 Technology" International Journal of Molecular Sciences 24, no. 14: 11845. https://doi.org/10.3390/ijms241411845