
Whole Genome Sequencing Revealed Inherited Rare Oligogenic Variants Contributing to Schizophrenia and Major Depressive Disorder in Two Families

Abstract
:1. Introduction
2. Results
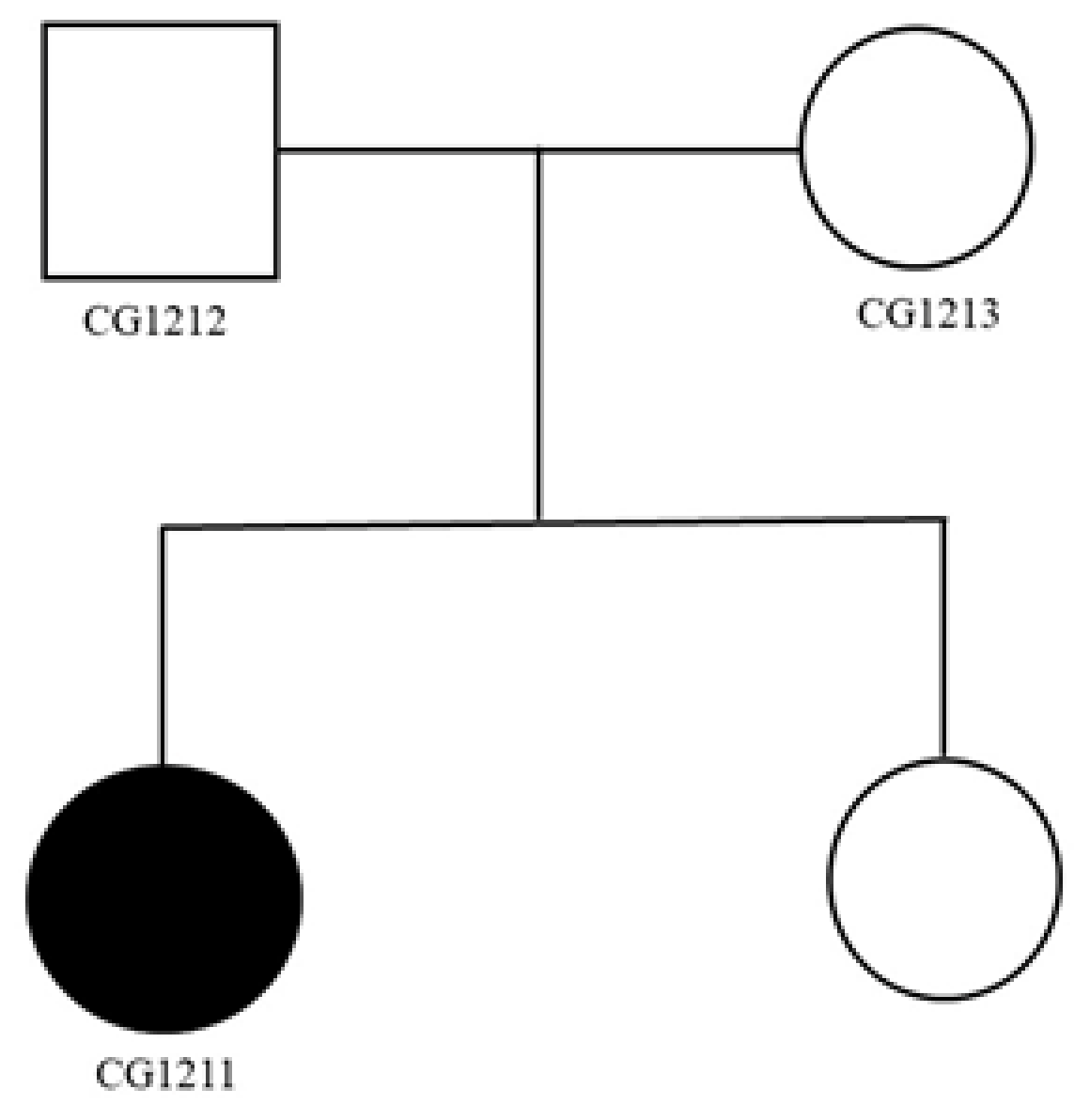
2.1. Clinical Reports of Family 1
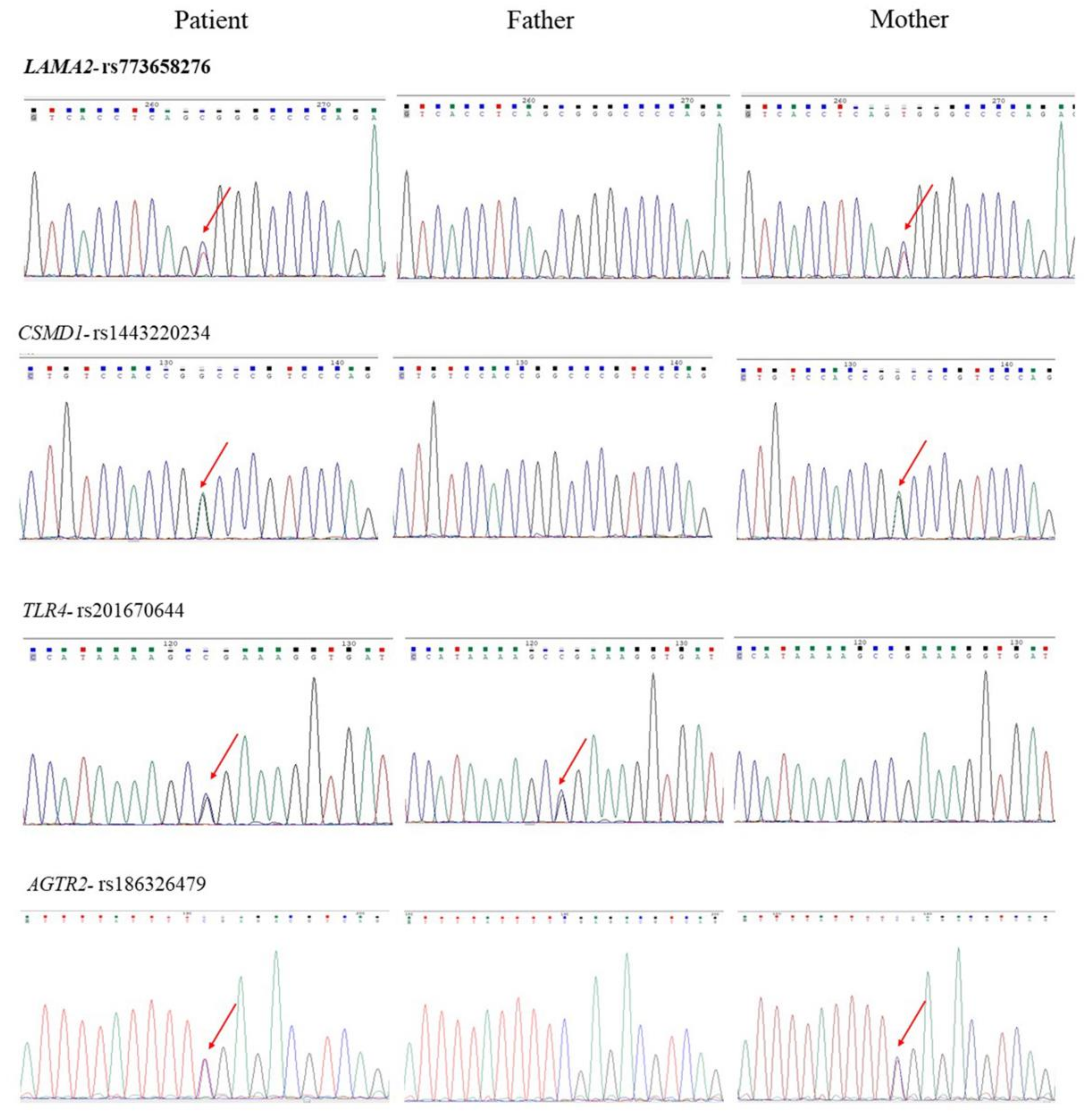
2.2. Genetic Findings of Family 1
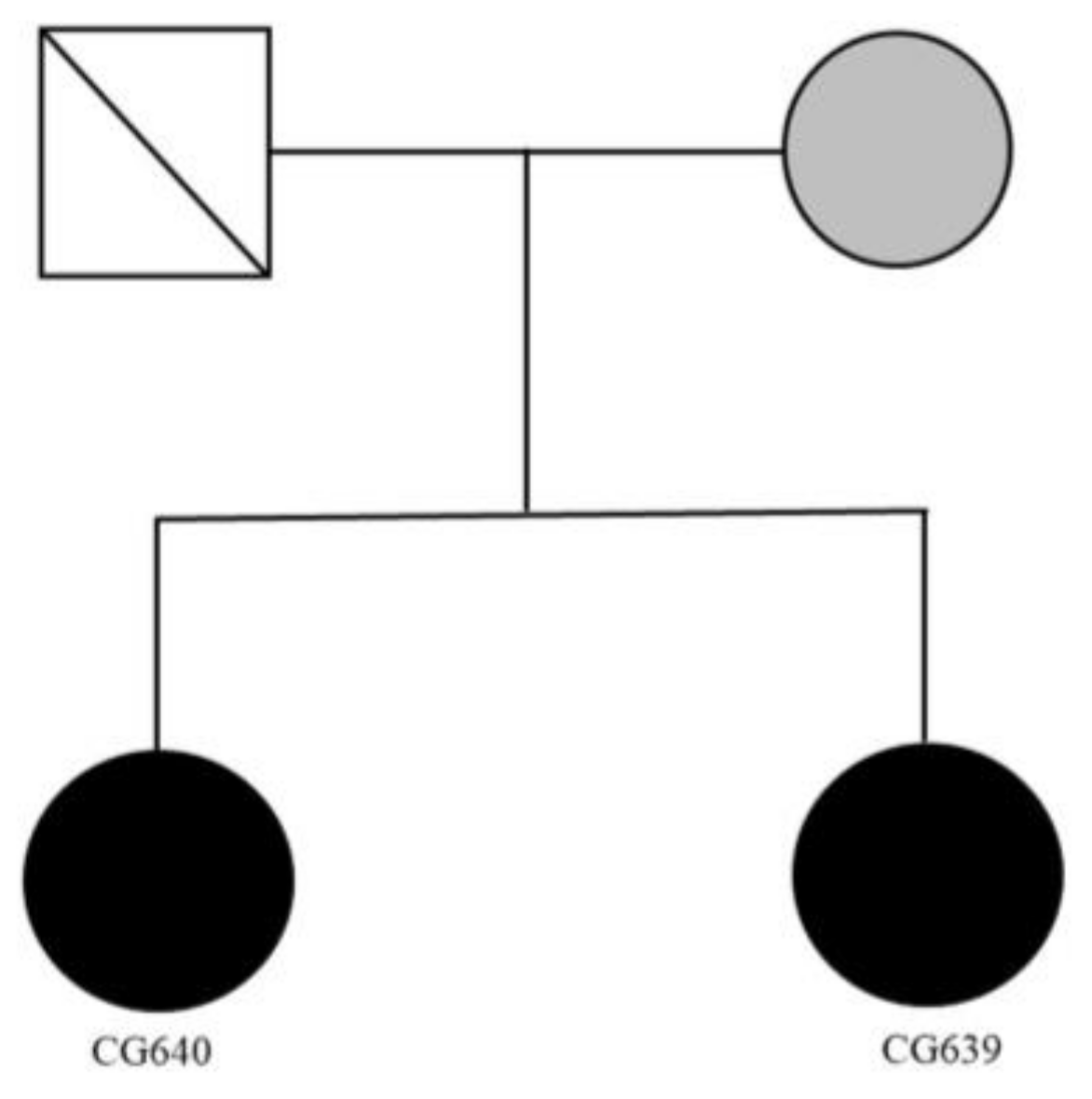
2.3. Clinical Reports of Family 2
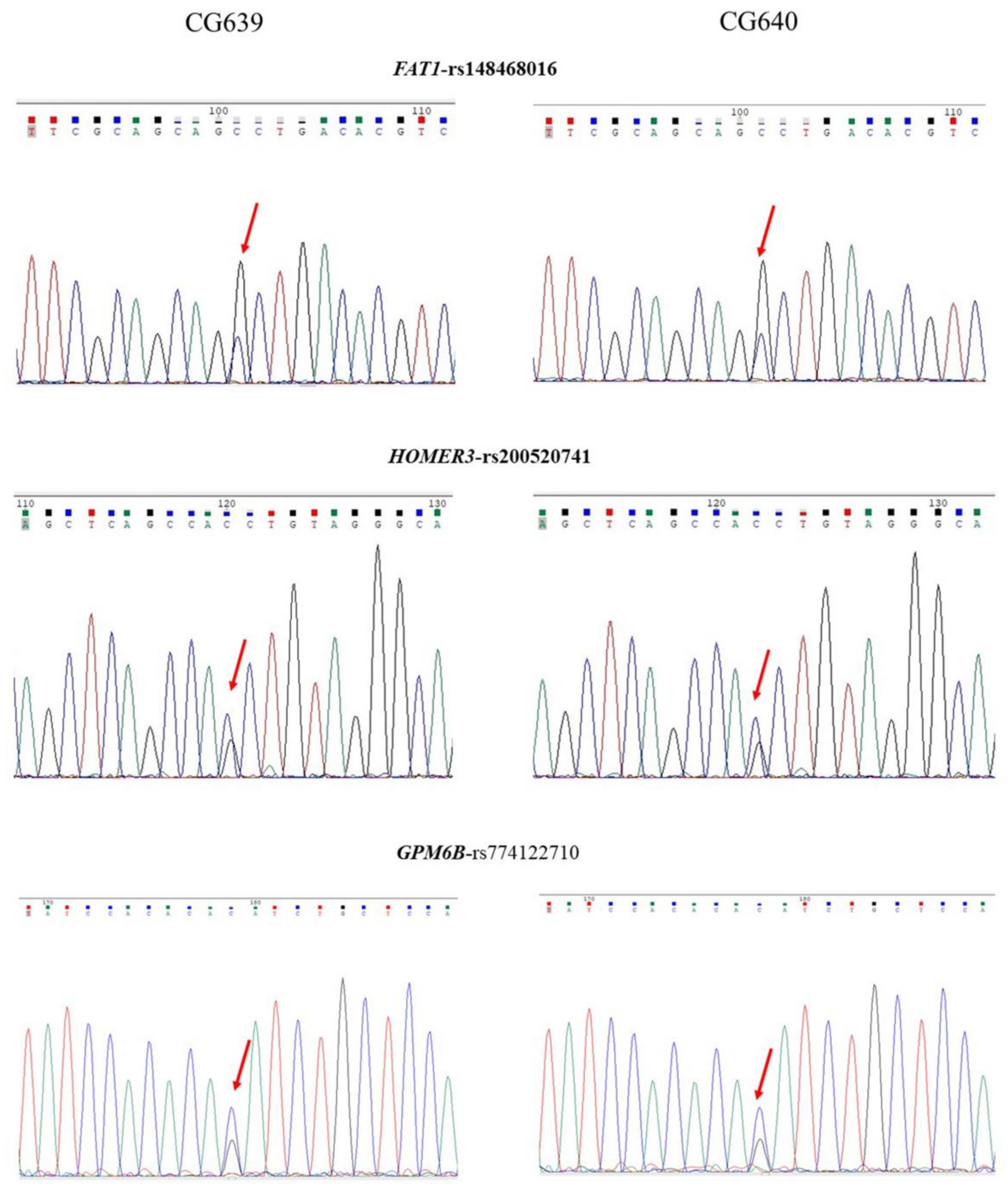
2.4. Genetic Findings of Family 2
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Whole-Genome Sequencing (WGS) Analysis
4.3. Bioinformatics Analysis
4.4. Sanger Sequencing
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sullivan, P.F.; Kendler, K.S.; Neale, M.C. Schizophrenia as a complex trait: Evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 2003, 60, 1187–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, P.F.; Neale, M.C.; Kendler, K.S. Genetic epidemiology of major depression: Review and meta-analysis. Am. J. Psychiatry 2000, 157, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.S.; Coombes, B.J. Genetic contributions to bipolar disorder: Current status and future directions. Psychol. Med. 2021, 51, 2156–2167. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Takahashi, A.; Kamatani, Y.; Okahisa, Y.; Kunugi, H.; Mori, N.; Sasaki, T.; Ohmori, T.; Okamoto, Y.; Kawasaki, H.; et al. A genome-wide association study identifies two novel susceptibility loci and trans population polygenicity associated with bipolar disorder. Mol. Psychiatry 2018, 23, 639–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattarinussi, G.; Delvecchio, G.; Sambataro, F.; Brambilla, P. The effect of polygenic risk scores for major depressive disorder, bipolar disorder and schizophrenia on morphological brain measures: A systematic review of the evidence. J. Affect. Disord. 2022, 310, 213–222. [Google Scholar] [CrossRef]
- Flint, J. The genetic basis of major depressive disorder. Mol. Psychiatry 2023, in press. [Google Scholar] [CrossRef]
- Thalamuthu, A.; Mills, N.T.; Berger, K.; Minnerup, H.; Grotegerd, D.; Dannlowski, U.; Meinert, S.; Opel, N.; Repple, J.; Gruber, M.; et al. Genome-wide interaction study with major depression identifies novel variants associated with cognitive function. Mol. Psychiatry 2022, 27, 1111–1119. [Google Scholar] [CrossRef]
- Legge, S.E.; Santoro, M.L.; Periyasamy, S.; Okewole, A.; Arsalan, A.; Kowalec, K. Genetic architecture of schizophrenia: A review of major advancements. Psychol. Med. 2021, 51, 2168–2177. [Google Scholar] [CrossRef]
- Hall, J.; Bray, N.J. Schizophrenia Genomics: Convergence on Synaptic Development, Adult Synaptic Plasticity, or Both? Biol. Psychiatry 2022, 91, 709–717. [Google Scholar] [CrossRef]
- MacIntyre, D.J.; Blackwood, D.H.R.; Porteous, D.J.; Pickard, B.S.; Muir, W.J. Chromosomal abnormalities and mental illness. Mol. Psychiatry 2003, 8, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Blackwood, D.H.R.; Thiagarajah, T.; Malloy, P.; Pickard, B.S.; Muir, W.J. Chromosome abnormalities, mental retardation and the search for genes in bipolar disorder and schizophrenia. Neurotox. Res. 2008, 14, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Jouan, L.; Gauthier, J.; Dion, P.A.; Rouleau, G.A. Rare variants in complex traits: Novel identification strategies and the role of de novo mutations. Hum. Hered. 2012, 74, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, D.; Sebat, J. CNVs: Harbingers of a rare variant revolution in psychiatric genetics. Cell 2012, 148, 1223–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifu, S.C.; Kohn, B.; Vlasie, A.; Patrichi, B.-E. Genetics of schizophrenia (Review). Exp. Ther. Med. 2020, 20, 3462–3468. [Google Scholar] [CrossRef]
- Rees, E.; Kirov, G. Copy number variation and neuropsychiatric illness. Curr. Opin. Genet. Dev. 2021, 68, 57–63. [Google Scholar] [CrossRef]
- Kato, T. Whole genome/exome sequencing in mood and psychotic disorders. Psychiatry Clin. Neurosci. 2015, 69, 65–76. [Google Scholar] [CrossRef]
- Glahn, D.C.; Nimgaonkar, V.L.; Raventós, H.; Contreras, J.; McIntosh, A.M.; Thomson, P.A.; Jablensky, A.; McCarthy, N.S.; Charlesworth, J.C.; Blackburn, N.B.; et al. Rediscovering the value of families for psychiatric genetics research. Mol. Psychiatry 2019, 24, 523–535. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Kato, H.; Kimura, H.; Kushima, I.; Takahashi, N.; Aleksic, B.; Ozaki, N. The genetic architecture of schizophrenia: Review of large-scale genetic studies. J. Hum. Genet. 2023, 68, 175–182. [Google Scholar] [CrossRef]
- Havdahl, A.; Niarchou, M.; Starnawska, A.; Uddin, M.; van der Merwe, C.; Warrier, V. Genetic contributions to autism spectrum disorder. Psychol. Med. 2021, 51, 2260–2273. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Guo, R.; Xu, W.; Liu, X.; Zhao, C.; Guo, Q.; Xu, W.; Ni, X.; Hao, C.; et al. Genetic diagnostic yields of 354 Chinese ASD children with rare mutations by a pipeline of genomic tests. Front. Genet. 2023, 14, 1108440. [Google Scholar] [CrossRef]
- Nakamura, T.; Takata, A. The molecular pathology of schizophrenia: An overview of existing knowledge and new directions for future research. Mol. Psychiatry 2023, in press. [Google Scholar] [CrossRef]
- Morris, E.; Inglis, A.; Austin, J. Psychiatric genetic counseling for people with copy number variants associated with psychiatric conditions. Clin. Genet. 2022, 102, 369–378. [Google Scholar] [CrossRef]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Chen, C.-H.; Huang, A.; Huang, Y.-S.; Fang, T.-H. Identification of a Rare Novel KMT2C Mutation That Presents with Schizophrenia in a Multiplex Family. J. Pers. Med. 2021, 11, 1254. [Google Scholar] [CrossRef]
- Chen, C.-H.; Cheng, M.-C.; Hu, T.-M.; Ping, L.-Y.; Kushima, I.; Aleksic, B. Identification of rare mutations of the vasoactive intestinal peptide receptor 2 gene in schizophrenia. Psychiatr. Genet. 2022, 32, 125–130. [Google Scholar] [CrossRef]
- Chen, C.-H.; Huang, Y.-S.; Fang, T.-H. Identification of a novel nonsense homozygous mutation of LINS1 gene in two sisters with intellectual disability, schizophrenia, and anxiety. Biomed. J. 2021, 44, 748–751. [Google Scholar] [CrossRef]
- Chen, C.-H.; Huang, Y.-S.; Liao, D.-L.; Huang, C.-Y.; Lin, C.-H.; Fang, T.-H. Identification of Rare Mutations of Two Presynaptic Cytomatrix Genes BSN and PCLO in Schizophrenia and Bipolar Disorder. J. Pers. Med. 2021, 11, 1057. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Fang, T.-H.; Kung, B.; Chen, C.-H. Two Genetic Mechanisms in Two Siblings with Intellectual Disability, Autism Spectrum Disorder, and Psychosis. J. Pers. Med. 2022, 12, 1013. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-H.; Cheng, M.-C.; Hu, T.-M.; Ping, L.-Y. Chromosomal Microarray Analysis as First-Tier Genetic Test for Schizophrenia. Front. Genet. 2021, 12, 620496. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.-C.; Chien, W.-H.; Huang, Y.-S.; Fang, T.-H.; Chen, C.-H. Translational Study of Copy Number Variations in Schizophrenia. Int. J. Mol. Sci. 2021, 23, 457. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, A.; Foley, A.R.; Zambon, A.A.; Bönnemann, C.G.; Muntoni, F. LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness. Front. Mol. Neurosci. 2020, 13, 123. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-Y.; Yang, J.-J.; Li, S.-Y.; Lee, I.-C. A Wide Spectrum of Genetic Disorders Causing Severe Childhood Epilepsy in Taiwan: A Case Series of Ultrarare Genetic Cause and Novel Mutation Analysis in a Pilot Study. J. Pers. Med. 2020, 10, 281. [Google Scholar] [CrossRef]
- Accogli, A.; Severino, M.; Riva, A.; Madia, F.; Balagura, G.; Iacomino, M.; Carlini, B.; Baldassari, S.; Giacomini, T.; Croci, C.; et al. Targeted re-sequencing in malformations of cortical development: Genotype-phenotype correlations. Seizure 2020, 80, 145–152. [Google Scholar] [CrossRef]
- Nouri, Z.; Sarmadi, A.; Narrei, S.; Sehhati, M.; Tabatabaiefar, M.A. Whole exome sequencing identified a novel LAMA2 frameshift variant causing merosin-deficient congenital muscular dystrophy in a patient with cardiomyopathy, and autism-like behavior. Neuromuscul. Disord. 2022, 32, 776–784. [Google Scholar] [CrossRef]
- Xu, B.; Ionita-Laza, I.; Roos, J.L.; Boone, B.; Woodrick, S.; Sun, Y.; Levy, S.; Gogos, J.A.; Karayiorgou, M. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 2012, 44, 1365–1369. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.-Q.; Zhou, Z.-Y.; He, X.; Wang, H.; Guo, X.-L.; Hao, C.-J.; Guo, Y.; Zhen, X.-C.; Li, W. Dysbindin deficiency in sandy mice causes reduction of snapin and displays behaviors related to schizophrenia. Schizophr. Res. 2008, 106, 218–228. [Google Scholar] [CrossRef]
- Hu, H.; Wang, X.; Li, C.; Li, Y.; Hao, J.; Zhou, Y.; Yang, X.; Chen, P.; Shen, X.; Zhang, S. Loss of Dysbindin Implicates Synaptic Vesicle Replenishment Dysregulation as a Potential Pathogenic Mechanism in Schizophrenia. Neuroscience 2021, 452, 138–152. [Google Scholar] [CrossRef]
- Dickman, D.K.; Tong, A.; Davis, G.W. Snapin is critical for presynaptic homeostatic plasticity. J. Neurosci. 2012, 32, 8716–8724. [Google Scholar] [CrossRef] [Green Version]
- Ermis Akyuz, E.; Bell, S.M. The Diverse Role of CUB and Sushi Multiple Domains 1 (CSMD1) in Human Diseases. Genes 2022, 13, 2332. [Google Scholar] [CrossRef] [PubMed]
- Håvik, B.; Le Hellard, S.; Rietschel, M.; Lybæk, H.; Djurovic, S.; Mattheisen, M.; Mühleisen, T.W.; Degenhardt, F.; Priebe, L.; Maier, W.; et al. The complement control-related genes CSMD1 and CSMD2 associate to schizophrenia. Biol. Psychiatry 2011, 70, 35–42. [Google Scholar] [CrossRef] [PubMed]
- The Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium. Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 2011, 43, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Fatima, A.; Farooq, M.; Abdullah, U.; Tariq, M.; Mustafa, T.; Iqbal, M.; Tommerup, N.; Mahmood Baig, S. Genome-Wide Supported Risk Variants in MIR137, CACNA1C, CSMD1, DRD2, and GRM3 Contribute to Schizophrenia Susceptibility in Pakistani Population. Psychiatry Investig. 2017, 14, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Liu, F.; Xu, X.; Bai, Y. Replicated association between the European GWAS locus rs10503253 at CSMD1 and schizophrenia in Asian population. Neurosci. Lett. 2017, 647, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cheng, Z.; Wang, J.; Jin, C.; Yuan, J.; Wang, G.; Zhang, F.; Zhao, X. No association between the rs10503253 polymorphism in the CSMD1 gene and schizophrenia in a Han Chinese population. BMC Psychiatry 2016, 16, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotan, A.; Fenckova, M.; Bralten, J.; Alttoa, A.; Dixson, L.; Williams, R.W.; van der Voet, M. Neuroinformatic analyses of common and distinct genetic components associated with major neuropsychiatric disorders. Front. Neurosci. 2014, 8, 331. [Google Scholar] [CrossRef]
- Chen, X.; Long, F.; Cai, B.; Chen, X.; Qin, L.; Chen, G. A Novel Relationship for Schizophrenia, Bipolar, and Major Depressive Disorder. Part 8: A Hint from Chromosome 8 High Density Association Screen. Mol. Neurobiol. 2017, 54, 5868–5882. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Alonso, A.; Ekelund, J.; Sarin, A.-P.; Miettunen, J.; Veijola, J.; Järvelin, M.-R.; Hennah, W. Genome-Wide Association Study of Psychosis Proneness in the Finnish Population. Schizophr. Bull. 2017, 43, 1304–1314. [Google Scholar] [CrossRef]
- Woo, H.J.; Yu, C.; Kumar, K.; Reifman, J. Large-scale interaction effects reveal missing heritability in schizophrenia, bipolar disorder and posttraumatic stress disorder. Transl. Psychiatry 2017, 7, e1089. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Fu, X.; Tang, Z.; Li, C.; Xu, Y.; Zhang, F.; Zhou, D.; Zhu, C. Altered expression of the CSMD1 gene in the peripheral blood of schizophrenia patients. BMC Psychiatry 2019, 19, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd El Gayed, E.M.; Rizk, M.S.; Ramadan, A.N.; Bayomy, N.R. mRNA Expression of the CUB and Sushi Multiple Domains 1 (CSMD1) and Its Serum Protein Level as Predictors for Psychosis in the Familial High-Risk Children and Young Adults. ACS Omega 2021, 6, 24128–24138. [Google Scholar] [CrossRef] [PubMed]
- Hatzimanolis, A.; Foteli, S.; Stefanatou, P.; Ntigrintaki, A.-A.; Ralli, I.; Kollias, K.; Nikolaou, C.; Gazouli, M.; Stefanis, N.C. Deregulation of complement components C4A and CSMD1 peripheral expression in first-episode psychosis and links to cognitive ability. Eur. Arch. Psychiatry Clin. Neurosci. 2022, 272, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Dou, M.; Su, W.; Jiang, Z.; Duan, Q.; Cao, B.; Chen, Y. Identifying novel proteins underlying schizophrenia via integrating pQTLs of the plasma, CSF, and brain with GWAS summary data. BMC Med. 2022, 20, 474. [Google Scholar] [CrossRef]
- Venkatasubramanian, G.; Debnath, M. The TRIPS (Toll-like receptors in immuno-inflammatory pathogenesis) Hypothesis: A novel postulate to understand schizophrenia. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 44, 301–311. [Google Scholar] [CrossRef]
- Garcia Bueno, B.; Caso, J.R.; Madrigal, J.L.; Leza, J.C. Innate immune receptor Toll-like receptor 4 signalling in neuropsychiatric diseases. Neurosci. Biobehav. Rev. 2016, 64, 134–147. [Google Scholar] [CrossRef]
- Carnac, T. Schizophrenia Hypothesis: Autonomic Nervous System Dysregulation of Fetal and Adult Immune Tolerance. Front. Syst. Neurosci. 2022, 16, 844383. [Google Scholar] [CrossRef]
- McKernan, D.P.; Dennison, U.; Gaszner, G.; Cryan, J.F.; Dinan, T.G. Enhanced peripheral toll-like receptor responses in psychosis: Further evidence of a pro-inflammatory phenotype. Transl. Psychiatry 2011, 1, e36. [Google Scholar] [CrossRef] [Green Version]
- García-Bueno, B.; Gassó, P.; MacDowell, K.S.; Callado, L.F.; Mas, S.; Bernardo, M.; Lafuente, A.; Meana, J.J.; Leza, J.C. Evidence of activation of the Toll-like receptor-4 proinflammatory pathway in patients with schizophrenia. J. Psychiatry Neurosci. 2016, 41, E46–E55. [Google Scholar] [CrossRef] [Green Version]
- Balaji, R.; Subbanna, M.; Shivakumar, V.; Abdul, F.; Venkatasubramanian, G.; Debnath, M. Pattern of expression of Toll like receptor (TLR)-3 and -4 genes in drug-naïve and antipsychotic treated patients diagnosed with schizophrenia. Psychiatry Res. 2020, 285, 112727. [Google Scholar] [CrossRef]
- Gurung, J.; Bera, N.K.; Lama, M.; Singh, B. Association of TLR-4 896A/G, TLR-4 1196C/T, and TLR-9 C/T polymorphism with schizophrenia in Indian Bengalee patient. Indian J. Psychiatry 2022, 64, 579–587. [Google Scholar] [CrossRef]
- Mostafa, M.; Elwasify, M.; Fathy, A.A.; Abdelsalam, M. Toll-Like Receptor 4 Gene Polymorphisms and Susceptibility to Schizophrenia: A Case-Control Study. Immunol. Investig. 2022, 51, 2009–2024. [Google Scholar] [CrossRef]
- Park, H.S.; Jung, H.Y.; Park, E.Y.; Kim, J.; Lee, W.J.; Bae, Y.S. Cutting edge: Direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J. Immunol. 2004, 173, 3589–3593. [Google Scholar] [CrossRef] [Green Version]
- Ershova, E.S.; Shmarina, G.V.; Martynov, A.V.; Zakharova, N.V.; Veiko, R.V.; Umriukhin, P.E.; Kostyuk, G.P.; Kutsev, S.I.; Veiko, N.N.; Kostyuk, S.V. NADPH-oxidase 4 gene over-expression in peripheral blood lymphocytes of the schizophrenia patients. PLoS ONE 2022, 17, e0269130. [Google Scholar] [CrossRef]
- Fyhrquist, F.; Saijonmaa, O. Renin-angiotensin system revisited. J. Intern. Med. 2008, 264, 224–236. [Google Scholar] [CrossRef]
- Turner, A.J.; Nalivaeva, N.N. Angiotensin-converting enzyme 2 (ACE2): Two decades of revelations and re-evaluation. Peptides 2022, 151, 170766. [Google Scholar] [CrossRef]
- Junho, C.V.C.; Trentin-Sonoda, M.; Panico, K.; Dos Santos, R.S.N.; Abrahão, M.V.; Vernier, I.C.S.; Fürstenau, C.R.; Carneiro-Ramos, M.S. Cardiorenal syndrome: Long road between kidney and heart. Heart Fail. Rev. 2022, 27, 2137–2153. [Google Scholar] [CrossRef]
- Pedreanez, A.; Mosquera, J.; Munoz, N.; Robalino, J.; Tene, D. Diabetes, heart damage, and angiotensin II. What is the relationship link between them? A minireview. Endocr. Regul. 2022, 56, 55–65. [Google Scholar] [CrossRef]
- Kagan, M.; Pleniceanu, O.; Vivante, A. The genetic basis of congenital anomalies of the kidney and urinary tract. Pediatr. Nephrol. 2022, 37, 2231–2243. [Google Scholar] [CrossRef]
- Gribouval, O.; Morinière, V.; Pawtowski, A.; Arrondel, C.; Sallinen, S.-L.; Saloranta, C.; Clericuzio, C.; Viot, G.; Tantau, J.; Blesson, S.; et al. Spectrum of mutations in the renin-angiotensin system genes in autosomal recessive renal tubular dysgenesis. Hum. Mutat. 2012, 33, 316–326. [Google Scholar] [CrossRef]
- Gubler, M.-C. Renal tubular dysgenesis. Pediatr. Nephrol. 2014, 29, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Szczepanska-Sadowska, E.; Wsol, A.; Cudnoch-Jedrzejewska, A.; Czarzasta, K.; Żera, T. Multiple Aspects of Inappropriate Action of Renin-Angiotensin, Vasopressin, and Oxytocin Systems in Neuropsychiatric and Neurodegenerative Diseases. J. Clin. Med. 2022, 11, 908. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.; Kuruppu, S.; Rajapakse, N.W. Chronic Renin-Angiotensin System Activation Induced Neuroinflammation: Common Mechanisms Underlying Hypertension and Dementia? J. Alzheimer’s Dis. 2022, 85, 943–955. [Google Scholar] [CrossRef]
- Wu, H.; Sun, Q.; Yuan, S.; Wang, J.; Li, F.; Gao, H.; Chen, X.; Yang, R.; Xu, J. AT1 Receptors: Their Actions from Hypertension to Cognitive Impairment. Cardiovasc. Toxicol. 2022, 22, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, V.S.; Beachem, M.A.; Edwards, P.S.; Ladd, S.; Miller, K.E.; de Mollerat, X.; Clarkson, K.; DuPont, B.; Schwartz, C.E.; Stevenson, R.E.; et al. AGTR2 mutations in X-linked mental retardation. Science 2002, 296, 2401–2403. [Google Scholar] [CrossRef] [PubMed]
- Ylisaukko-oja, T.; Rehnström, K.; Vanhala, R.; Tengström, C.; Lähdetie, J.; Järvelä, I. Identification of two AGTR2 mutations in male patients with non-syndromic mental retardation. Hum. Genet. 2004, 114, 211–213. [Google Scholar] [CrossRef]
- Takeshita, E.; Nakagawa, E.; Nakatani, K.; Sasaki, M.; Goto, Y.-I. Novel AGTR2 missense mutation in a Japanese boy with severe mental retardation, pervasive developmental disorder, and epilepsy. Brain Dev. 2012, 34, 776–779. [Google Scholar] [CrossRef]
- Bienvenu, T.; Poirier, K.; Van Esch, H.; Hamel, B.; Moraine, C.; Fryns, J.P.; Ropers, H.H.; Beldjord, C.; Yntema, H.G.; Chelly, J. Rare polymorphic variants of the AGTR2 gene in boys with non-specific mental retardation. J. Med. Genet. 2003, 40, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, J.; Dähmlow, S.; Guse, M.; Hetzer, R.; Regitz-Zagrosek, V. The assertion that a G21V mutation in AGTR2 causes mental retardation is not supported by other studies. Hum. Genet. 2004, 114, 396–397. [Google Scholar] [CrossRef]
- Huang, D.; Sun, W.; Strom, C.M. Sequence variations in AGTR2 are unlikely to be associated with X-linked mental retardation. Am. J. Med. Genet. Part A 2005, 139, 243–244. [Google Scholar] [CrossRef]
- Piton, A.; Redin, C.; Mandel, J.-L. XLID-causing mutations and associated genes challenged in light of data from large-scale human exome sequencing. Am. J. Hum. Genet. 2013, 93, 368–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Yi, J.H.; Lee, S.; Park, C.-H.; Ryu, J.H.; Shin, K.S.; Kang, S.J. Defective neurogenesis and schizophrenia-like behavior in PARP-1-deficient mice. Cell Death Dis. 2019, 10, 943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.M.; Hoff, J.D.; Zeese, M.L.; Corfas, G. Poly (ADP-Ribose) Polymerase 1 Regulates Cajal-Retzius Cell Development and Neural Precursor Cell Adhesion. Front. Cell Dev. Biol. 2021, 9, 693595. [Google Scholar] [CrossRef] [PubMed]
- Usui, N.; Berto, S.; Konishi, A.; Kondo, M.; Konopka, G.; Matsuzaki, H.; Shimada, S. Zbtb16 regulates social cognitive behaviors and neocortical development. Transl. Psychiatry 2021, 11, 242. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.; Liang, X.; Chen, J.; Hong, J.; Li, L.; He, Q.; Cai, X. History and progression of Fat cadherins in health and disease. OncoTargets Ther. 2016, 9, 7337–7343. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Gong, Y.; Liang, X. Role of FAT1 in health and disease (Review). Oncol. Lett. 2021, 21, 398. [Google Scholar] [CrossRef]
- Blair, I.P.; Chetcuti, A.F.; Badenhop, R.F.; Scimone, A.; Moses, M.J.; Adams, L.J.; Craddock, N.; Green, E.; Kirov, G.; Owen, M.J.; et al. Positional cloning, association analysis and expression studies provide convergent evidence that the cadherin gene FAT contains a bipolar disorder susceptibility allele. Mol. Psychiatry 2006, 11, 372–383. [Google Scholar] [CrossRef] [Green Version]
- Abou Jamra, R.; Becker, T.; Georgi, A.; Feulner, T.; Schumacher, J.; Stromaier, J.; Schirmbeck, F.; Schulze, T.G.; Propping, P.; Rietschel, M.; et al. Genetic variation of the FAT gene at 4q35 is associated with bipolar affective disorder. Mol. Psychiatry 2008, 13, 277–284. [Google Scholar] [CrossRef]
- Light, K.J.; Miller, A.L.; Doughty, C.J.; Joyce, P.R.; Olds, R.J.; Kennedy, M.A. FAT and bipolar affective disorder. Mol. Psychiatry 2007, 12, 899–900. [Google Scholar] [CrossRef]
- Frei, J.A.; Brandenburg, C.; Nestor, J.E.; Hodzic, D.M.; Plachez, C.; McNeill, H.; Dykxhoorn, D.M.; Nestor, M.W.; Blatt, G.J.; Lin, Y.-C. Postnatal expression profiles of atypical cadherin FAT1 suggest its role in autism. Biol. Open 2021, 10, bio056457. [Google Scholar] [CrossRef]
- Costa, C.I.S.; da Silva Montenegro, E.M.; Zarrei, M.; de Sá Moreira, E.; Silva, I.M.W.; de Oliveira Scliar, M.; Wang, J.Y.T.; Zachi, E.C.; Branco, E.V.; da Costa, S.S.; et al. Copy number variations in a Brazilian cohort with autism spectrum disorders highlight the contribution of cell adhesion genes. Clin. Genet. 2022, 101, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Hernando-Davalillo, C.; Martín, A.A.S.; Borregan Prats, M.; Ortigoza-Escobar, J.D. De novo 4q35.2 duplication containing FAT1 is associated with autism spectrum disorder. Clin. Genet. 2022, 102, 434–437. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi-Yamaguchi, Y.; Furuichi, T. The Homer family proteins. Genome Biol. 2007, 8, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, P.; Li, X.; Fei, Z.; Poon, W. Scaffold protein Homer 1: Implications for neurological diseases. Neurochem. Int. 2012, 61, 731–738. [Google Scholar] [CrossRef]
- Quadir, S.G.; dos Santos, J.R.B.; Campbell, R.R.; Wroten, M.G.; Singh, N.; Holloway, J.J.; Bal, S.K.; Camarini, R.; Szumlinski, K.K. Homer2 regulates alcohol and stress cross-sensitization. Addict. Biol. 2016, 21, 613–633. [Google Scholar] [CrossRef] [Green Version]
- Castelli, V.; Brancato, A.; Cavallaro, A.; Lavanco, G.; Cannizzaro, C. Homer2 and Alcohol: A Mutual Interaction. Front. Psychiatry 2017, 8, 268. [Google Scholar] [CrossRef] [Green Version]
- Azaiez, H.; Decker, A.R.; Booth, K.T.; Simpson, A.C.; Shearer, A.E.; Huygen, P.L.M.; Bu, F.; Hildebrand, M.S.; Ranum, P.T.; Shibata, S.B.; et al. HOMER2, a stereociliary scaffolding protein, is essential for normal hearing in humans and mice. PLoS Genet. 2015, 11, e1005137. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Wang, Q.; Gu, H.; Zhang, X.; Qi, Y.; Liu, Y. Whole exome sequencing identified a second pathogenic variant in HOMER2 for autosomal dominant non-syndromic deafness. Clin. Genet. 2018, 94, 419–428. [Google Scholar] [CrossRef]
- Wu, Q.; Gong, B.; Jiang, A.; Qin, X. Case report and literature analysis: Autoimmune cerebellar ataxia associated with homer-3 antibodies. Front. Neurol. 2022, 13, 951659. [Google Scholar] [CrossRef]
- Hansen, N.; Radenbach, K.; Rentzsch, K.; Fox, J.; Wiltfang, J.; Bartels, C. Cerebrospinal Fluid Homer-3 Autoantibodies in a Patient with Amnestic Mild Cognitive Impairment. Brain Sci. 2023, 13, 125. [Google Scholar] [CrossRef]
- Han, K.-M.; Han, M.-R.; Kim, A.; Kang, W.; Kang, Y.; Kang, J.; Tae, W.-S.; Cho, Y.; Ham, B.-J. A study combining whole-exome sequencing and structural neuroimaging analysis for major depressive disorder. J. Affect. Disord. 2020, 262, 31–39. [Google Scholar] [CrossRef]
- Yan, Y.; Narayanan, V.; Lagenaur, C. Expression of members of the proteolipid protein gene family in the developing murine central nervous system. J. Comp. Neurol. 1996, 370, 465–478. [Google Scholar] [CrossRef]
- Werner, H.; Dimou, L.; Klugmann, M.; Pfeiffer, S.; Nave, K.-A. Multiple splice isoforms of proteolipid M6B in neurons and oligodendrocytes. Mol. Cell. Neurosci. 2001, 18, 593–605. [Google Scholar] [CrossRef]
- Sanchez-Roige, S.; Fontanillas, P.; Elson, S.L.; Pandit, A.; Schmidt, E.M.; Foerster, J.R.; Abecasis, G.R.; Gray, J.C.; de Wit, H.; Davis, L.K.; et al. Genome-wide association study of delay discounting in 23,217 adult research participants of European ancestry. Nat. Neurosci. 2018, 21, 16–18. [Google Scholar] [CrossRef]
- Fiori, L.M.; Zouk, H.; Himmelman, C.; Turecki, G. X chromosome and suicide. Mol. Psychiatry 2011, 16, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Fuchsova, B.; Alvarez Juliá, A.; Rizavi, H.S.; Frasch, A.C.; Pandey, G.N. Altered expression of neuroplasticity-related genes in the brain of depressed suicides. Neuroscience 2015, 299, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Fratelli, C.; Siqueira, J.; Silva, C.; Ferreira, E.; Silva, I. 5HTTLPR Genetic Variant and Major Depressive Disorder: A Review. Genes 2020, 11, 1260. [Google Scholar] [CrossRef]
- Stein, K.; Maruf, A.A.; Müller, D.J.; Bishop, J.R.; Bousman, C.A. Serotonin Transporter Genetic Variation and Antidepressant Response and Tolerability: A Systematic Review and Meta-Analysis. J. Pers. Med. 2021, 11, 1334. [Google Scholar] [CrossRef]
- Gershon, E.S. Bipolar illness and schizophrenia as oligogenic diseases: Implications for the future. Biol. Psychiatry 2000, 47, 240–244. [Google Scholar] [CrossRef]
- Du, Y.; Li, Z.; Liu, Z.; Zhang, N.; Wang, R.; Li, F.; Zhang, T.; Jiang, Y.; Zhi, X.; Wang, Z.; et al. Nonrandom occurrence of multiple de novo coding variants in a proband indicates the existence of an oligogenic model in autism. Genet. Med. 2020, 22, 170–180. [Google Scholar] [CrossRef]
- Wang, T.; Zhao, P.A.; Eichler, E.E. Rare variants and the oligogenic architecture of autism. Trends Genet. 2022, 38, 895–903. [Google Scholar] [CrossRef] [PubMed]
- John, J.; Bhattacharyya, U.; Yadav, N.; Kukshal, P.; Bhatia, T.; Nimgaonkar, V.L.; Deshpande, S.N.; Thelma, B.K. Multiple rare inherited variants in a four generation schizophrenia family offer leads for complex mode of disease inheritance. Schizophr. Res. 2020, 216, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Elston, R.C.; Zhu, X. The meaning of interaction. Hum. Hered. 2010, 70, 269–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Psychiatric Association; DSM-5 Task Force. Diagnostic and Statistical Manual of Mental Disorders: DSM-5; American Psychiatric Association: Washington, DC, USA, 2013; Volume 5. [Google Scholar]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.-J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name and SNP | Mutation Location | Inheritance | Taiwan Biobank (MAF) | ALFA | SIFT | PolyPhen-2 | Mutation Taster |
|---|---|---|---|---|---|---|---|
| LAMA2 rs773658276 | NC_000006.11:g.129691055C > T NM_000426.3:c.4879C > T NM_000426.3:p.Arg1627Trp | Mother | 0.000330 | 0 | Damaging (0.004) | Probably damaging (0.996) | Disease-causing |
| CSMD1 rs1443220234 | NC_000008.10:g.3141807G > A NM_033225.5:c.4012C > T NM_033225.5:p.Pro1338Ser | Mother | 0 | 0 | Damaging (0.034) | Possibly damaging (0.513) | Disease-causing |
| TLR4 rs201670644 | NC_000009.11:g.120476597C > G NM_003266.3:c.2071C > G NM_003266.3:p.Arg691Gly | Father | 0 | 0.000172 | Damaging (0.000) | Probably damaging (1.000) | Disease-causing |
| AGTR2 rs186326479 | NC_000023.10:g.115304077C > T NM_000686.4:c.544C > T NM_000686.4:p.Arg182X | Mother | 0.005020 | 0.000035 | N/A | N/A | Disease-causing |
| Gene Name and SNP | Mutation Location | Taiwan Biobank (MAF) | ALFA | SIFT (Score) | PolyPhen-2 | Mutation Taster |
|---|---|---|---|---|---|---|
| FAT1 rs148468016 | NC_000004.11:g.187509861G > C NM_005245.3:c.13652C > G NM_005245.3:p.Ala4551Gly | 0.017469 | 0.00029 | Damaging (0.009) | Probably damaging (1.000) | Disease-causing |
| HOMER3 rs200520741 | NC_000019.9:g.19042434C > G NM_001145722.1:c.691G > C NM_001145722.1:p.Val231Leu | 0.001980 | 0.000378 | Damaging (0.027) | Probably damaging (0.998) | Disease-causing |
| GPM6B rs774122710 | NC_000023.10:g.13797959G > C NM_005278.3:c.555C > G NM_005278.3:p.Ile185Met | 0.000670 | 0 | Damaging (0.007) | Probably damaging (0.992) | Disease-causing |
| Gene Name and SNP | Forward Primer Sequences (5′-3′) | Reverse Primer Sequences (5′-3′) | Size (bp) |
|---|---|---|---|
| AGTR2 rs186326479 | CTGGCTCTTTGGACCTGTGATGTG | CATTAAGGCAATCCCAGCTGACCA | 316 |
| CSMD1 rs1443220234 | CGCTGTGCCACCTACTGGAGAACT | GGGTTGTGTGAAAGCGAAATGAGC | 317 |
| LAMA2 rs773658276 | CCCCCATAGAGCTGTTGTGAAA | TACCCTGGTCAGCAGCTCGTTCAT | 375 |
| TLR4 rs201670644 | TCAAGCCAGGATGAGGACTGGGTA | CCTGAGCAGGGTCTTCTCCACCTT | 300 |
| GPM6B rs774122710 | CCTCCCTGAAGTTTCCACCCAGAA | CTGGCTGGGTGTGTTTGGTTTCTC | 320 |
| FAT1 rs148468016 | GGGGAGTTGAGAGTCAGACTTCCG | AGAGAACCCCATGCCCCTTACCCG | 244 |
| HOMER3 rs200520741 | GCGAGGCCCAGGAACCACACTTG | TGGGTTTGAGACAATGCCAGCCTC | 347 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, I.-H.; Huang, Y.-S.; Fang, T.-H.; Chen, C.-H. Whole Genome Sequencing Revealed Inherited Rare Oligogenic Variants Contributing to Schizophrenia and Major Depressive Disorder in Two Families. Int. J. Mol. Sci. 2023, 24, 11777. https://doi.org/10.3390/ijms241411777
Chung I-H, Huang Y-S, Fang T-H, Chen C-H. Whole Genome Sequencing Revealed Inherited Rare Oligogenic Variants Contributing to Schizophrenia and Major Depressive Disorder in Two Families. International Journal of Molecular Sciences. 2023; 24(14):11777. https://doi.org/10.3390/ijms241411777
Chicago/Turabian StyleChung, I-Hang, Yu-Shu Huang, Ting-Hsuan Fang, and Chia-Hsiang Chen. 2023. "Whole Genome Sequencing Revealed Inherited Rare Oligogenic Variants Contributing to Schizophrenia and Major Depressive Disorder in Two Families" International Journal of Molecular Sciences 24, no. 14: 11777. https://doi.org/10.3390/ijms241411777