
Whole Exome Sequencing of 20 Spanish Families: Candidate Genes for Non-Syndromic Pediatric Cataracts
 , , , , , ,
, , , , , ,  and
and
Abstract
:1. Introduction
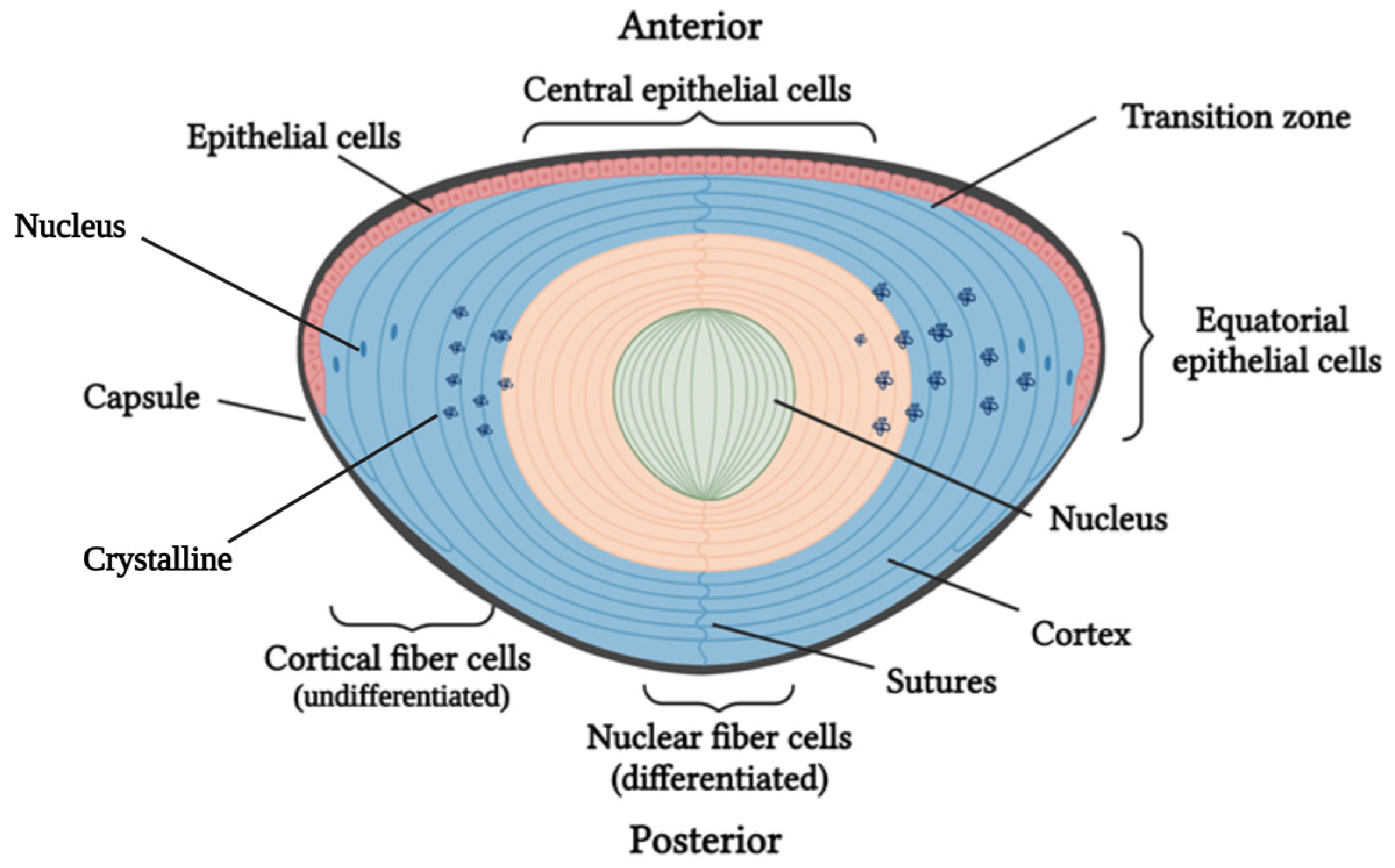
1.1. Cataract Formation: Lens Structure and Function

1.2. Classification of Pediatric Cataracts
1.3. Pediatric Cataracts: Prevalence and Etiology
1.4. Mechanism of Pediatric Cataract Development
1.5. Genetics of Pediatric Cataracts
1.5.1. Crystallin Protein Genes
1.5.2. Membrane Protein Genes
1.5.3. Cytoskeletal Protein Genes
1.5.4. Transcription Factors and Lens Growth Factors
1.5.5. Degradation Proteins or Chaperones
1.5.6. Metabolism-Associated Genes
1.6. Symptoms and Treatment of Pediatric Cataracts
2. Results
2.1. Ophthalmological Examination
2.2. Molecular Genetics
3. Discussion
4. Materials and Methods
4.1. Ophthalmological Studies
4.2. Genetic Studies
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Description |
|---|---|
| PVS1 | Null variant (nonsense, frameshift, canonical ± 1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where loss of function is a known mechanism of disease. |
| PS1 | Same amino acid change as a previously established pathogenic variant regardless of nucleotide change. |
| PS2 | De novo (both maternity and paternity confirmed) in a patient with the disease and no family history. |
| PS3 | Well-established in vitro or in vivo functional studies supporting a damaging effect on the gene or gene product. |
| BS1 | Allele frequency is greater than expected for the disorder. |
| PM1 | Located in a mutational hotspot and/or critical and well-established functional domain (e.g., active site of an enzyme) with no benign variation. |
| PM2 | Absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, and Exome Aggregation Consortium. |
| PM3 | For recessive disorders, detected in trans with a pathogenic variant. |
| PM4 | Protein length changes as a result of in-frame deletions/insertions in a non-repeat region or stop-loss variants. |
| PM5 | Novel missense change at an amino acid residue where a different missense change determined to be pathogenic has previously been observed. |
| PP1 | Co-segregation with disease in multiple affected family members in a gene definitively known to cause the disease. |
| PP2 | Missense variant in a gene that has a low rate of benign missense variation and in which missense variants are a common mechanism of disease. |
| PP3 | Multiple lines of computational evidence support a deleterious effect on the gene or gene product (e.g., conservation, evolutionary, splicing impact). |
| PP5 | Reputable source recently reports variant as pathogenic, but the evidence is not available to the laboratory to perform an independent evaluation. |
| BP4 | Multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc.). |
| BP6 | Reputable source recently reports variant as benign, but the evidence is not available for the laboratory to perform an independent evaluation. |
| Evidence Type | Evidence |
|---|---|
| Section 1: Initial Assessment of Genomic Content | |
| Copy number loss content | 1A. Contains protein coding or other known functionally important elements |
| 1B. Does NOT contain protein coding or any known functionally important elements | |
| Section 2: Overlap with Established/Predicted Haploinsufficient (HI) or Established Benign Genes/Genomic Regions (Skip to Section 3 if your copy number loss DOES NOT overlap these types of genes/regions) | |
| Overlap with ESTABLISHED HI genes or genomic regions and consideration of reason for referral | 2A. Complete overlap of an established HI gene/genomic region |
2B. Partial overlap of an established HI genomic region
| |
| 2C. Partial overlap with the 5′ end of an established HI gene (3′ end of the gene not involved); | |
| 2C-1. And coding sequence is involved; | |
| 2C-2. And only the 5′ UTR is involved. | |
| 2D. Partial overlap with the 3′ end of an established HI gene (5′ end of the gene not involved); | |
| 2D-1 And only the 3′ untranslated region is involved; | |
| 2D-2. And only the last exon is involved. Other established pathogenic variants have been reported in this exon; | |
| 2D-3. And only the last exon is involved. No other established pathogenic variants have been reported in this exon; | |
| 2D-4. And it includes other exons in addition to the last exon. Nonsense-mediated decay is expected to occur. | |
| 2E. Both breakpoints are within the same gene (intragenic CNV; gene-level sequence variant). | |
| Overlap with ESTABLISHED benign genes or genomic regions | 2F. Completely contained within an established benign CNV region. |
| 2G. Overlaps an established benign CNV but includes additional genomic material. | |
| Haploinsufficiency Predictors | 2H. Two or more HI predictors suggest that AT LEAST ONE gene in the interval is HI. |
| Section 3: Evaluation of Gene Number | |
| Number of protein-coding RefSeq genes wholly or partially included in the copy number loss | 3A. 0–24 genes |
| 3B. 25–34 genes | |
| 3C. 35+ genes | |
| Section 4: Detailed Evaluation of Genomic Content Using Cases from the Published Literature, Public Databases, and/or Internal Lab Data (Skip to Section 5 if either your CNV overlapped with an established HI gene/region in Section 2 OR there have been no reports associating either the CNV or any genes within the CNV with human phenotypes caused by loss of function (LOF) or copy number loss) | |
| Individual case evidence, de novo occurrences | Reported proband (from the literature, public databases, or internal lab data) has either:
|
| 4A. The reported phenotype is highly specific and relatively unique to the gene or genomic region; | |
| 4B. The reported phenotype is consistent with the gene/genomic region, is highly specific, but not necessarily unique to the gene/genomic region; | |
| 4C. The reported phenotype is consistent with the gene/genomic region but is not highly specific and/or has high genetic heterogeneity. | |
| Individual case evidence, inconsistent phenotype | 4D. The reported phenotype is NOT consistent with what is expected for the gene/genomic region or not consistent in general. |
| Individual case evidence, unknown inheritance | 4E. Reported proband has a highly specific phenotype consistent with the gene/genomic region, but the inheritance of the variant is unknown. |
| Individual case evidence, segregation among similarly affected family members | 4F. 3–4 observed segregations; |
| 4G. 5–6 observed segregations; | |
| 4H. 7 or more observed segregations. | |
| Individual case evidence, non-segregations | 4I. Variant is NOT found in another individual in the proband’s family AFFECTED with a consistent specific well-defined phenotype (no known phenocopies). |
| 4J. Variant IS found in another individual in the proband’s family UNAFFECTED with the specific well-defined phenotype observed in the proband. | |
| 4K. Variant IS found in another individual in the proband’s family UNAFFECTED with the non-specific phenotype observed in the proband. | |
| Case-control and population evidence | 4L. Statistically significant increase amongst observations in cases (with a consistent specific well-defined phenotype) compared with controls. |
| 4M. Statistically significant increase amongst observations in cases (without a consistent non-specific phenotype OR unknown phenotype) compared with controls. | |
| 4N. No statistically significant difference between observations in cases and controls. | |
| 4O. Overlap with common population variation. | |
| Section 5: Evaluation of Inheritance Pattern/Family History for Patient Being Studied | |
| Observed copy number loss is DE NOVO | 5A. Use appropriate category from de novo scoring section in Section 4. |
| Observed copy number loss is INHERITED | 5B. Patient with specific well-defined phenotype and no family history. CNV is inherited from an apparently unaffected parent. |
| 5C. Patient with non-specific phenotype and no family history. CNV is inherited from an apparently unaffected parent. | |
| 5D. CNV segregates with a consistent phenotype observed in the patient’s family. | |
| Observed copy number loss, NON- SEGREGATIONS | 5E. Use appropriate category from non-segregation section in Section 4. |
| Other | 5F. Inheritance information is unavailable or uninformative. |
| 5G. Inheritance information is unavailable or uninformative. The patient phenotype is non-specific but is consistent with what has been described in similar cases. | |
| 5H. Inheritance information is unavailable or uninformative. The patient phenotype is highly specific and consistent with what has been described in similar cases. | |



References
- Liu, Y.-C.; Wilkins, M.; Kim, T.; Malyugin, B.; Mehta, J.S. Cataracts. Lancet 2017, 390, 600–612. [Google Scholar] [CrossRef]
- Huang, B.; He, W. Molecular characteristics of inherited congenital cataracts. Eur. J. Med. Genet. 2010, 53, 347–357. [Google Scholar] [CrossRef]
- Miesfeld, J.B.; Brown, N.L. Eye organogenesis: A hierarchical view of ocular development. Curr. Top. Dev. Biol. 2019, 132, 351–393. [Google Scholar] [CrossRef]
- Khan, L.; Shaheen, N.; Hanif, Q.; Fahad, S.; Usman, M. Genetics of congenital cataract, its diagnosis and therapeutics. Egypt. J. Basic Appl. Sci. 2018, 5, 252–257. [Google Scholar] [CrossRef]
- BioRender. Available online: https://biorender.com/ (accessed on 2 January 2023).
- Hejtmancik, J.F. Congenital cataracts and their molecular genetics. Semin. Cell Dev. Biol. 2008, 19, 134–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khokhar, S.; Pillay, G.; Dhull, C.; Agarwal, E.; Mahabir, M.; Aggarwal, P. Paediatric cataract. Indian J. Ophthalmol. 2017, 65, 1340. [Google Scholar] [CrossRef] [PubMed]
- Merin, S.; Crawford, J.S. The etiology of congenital cataracts. A survey of 386 cases. Can. J. Ophthalmol. 1971, 6, 178–182. [Google Scholar] [PubMed]
- Lenhart, P.D.; Lambert, S.R. Current management of infantile cataracts. Surv. Ophthalmol. 2022, 67, 1476–1505. [Google Scholar] [CrossRef]
- Li, J.; Chen, X.; Yan, Y.; Yao, K. Molecular genetics of congenital cataracts. Exp. Eye Res. 2020, 191, 107872. [Google Scholar] [CrossRef]
- Sathish, H.A.; Koteiche, H.A.; Mchaourab, H.S. Binding of destabilized βB2-crystallin mutants to α-crystallin: The role of a folding intermediate. J. Biol. Chem. 2004, 279, 16425–16432. [Google Scholar] [CrossRef] [Green Version]
- Moreau, K.L.; King, J.A. Cataract-Causing Defect of a Mutant γ-Crystallin Proceeds through an Aggregation Pathway which Bypasses Recognition by the α-Crystallin Chaperone. PLoS ONE 2012, 7, e37256. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Yao, W.; Theendakara, V.; Chan, C.C.; Wawrousek, E.; Hejtmancik, J.F. Overexpression of human γC-crystallin 5 bp duplication disrupts lens morphology in transgenic mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5369–5375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andley, U.P.; Goldman, J.W. Autophagy and UPR in alpha-crystallin mutant knock-in mouse models of hereditary cataracts. Biochim. Biophys. Acta 2016, 1860 Pt B, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Alapure, B.V.; Stull, J.K.; Firtina, Z.; Duncan, M.K. The unfolded protein response is activated in connexin 50 mutant mouse lenses. Exp. Eye Res. 2012, 102, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Fan, D.-B.; Zhao, Y.-T.; Li, Y.; Kong, D.-Q.; Cai, F.-F.; Zheng, G.-Y. Two novel mutations identified in ADCC families impair crystallin protein distribution and induce apoptosis in human lens epithelial cells. Sci. Rep. 2017, 7, 17848. [Google Scholar] [CrossRef] [Green Version]
- Renwick, J.H.; Lawler, S.D. Probable linkage between a congenital cataract locus and the Duffy blood group locus. Ann. Hum. Genet. 1963, 27, 67–76. [Google Scholar] [CrossRef]
- Kandaswamy, D.K.; Prakash, M.V.S.; Graw, J.; Koller, S.; Magyar, I.; Tiwari, A.; Berger, W.; Santhiya, S.T. Application of WES towards molecular investigation of congenital cataracts: Identification of novel alleles and genes in a hospital-based cohort of south India. Int. J. Mol. Sci. 2020, 21, 9569. [Google Scholar] [CrossRef]
- Sun, W.; Xiao, X.; Li, S.; Guo, X.; Zhang, Q. Exome Sequencing of 18 Chinese Families with Congenital Cataracts: A New Sight of the NHS Gene. PLoS ONE 2014, 9, e100455. [Google Scholar] [CrossRef] [PubMed]
- Shiels, A.; Hejtmancik, J.F. Inherited cataracts: Genetic mechanisms and pathways new and old. Exp. Eye Res. 2021, 209, 108662. [Google Scholar] [CrossRef] [PubMed]
- Shiels, A.; Bennett, T.M.; Hejtmancik, J.F. Cat-Map: Putting cataract on the map. Mol. Vis. 2010, 16, 2007–2015. [Google Scholar]
- Weisschuh, N.; Aisenbrey, S.; Wissinger, B.; Riess, A. Identification of a novel CRYBB2 missense mutation causing congenital autosomal dominant cataract. Mol. Vis. 2012, 18, 174–180. [Google Scholar] [PubMed]
- Graw, J. Genetics of crystallins: Cataract and beyond. Exp. Eye Res. 2009, 88, 173–189. [Google Scholar] [CrossRef]
- Wistow, G. The human crystallin gene families. Hum. Genom. 2012, 6, 26. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.R.; Lubsen, N.H.; Slingsby, C. sHSP in the eye lens: Crystallin mutations, cataract and proteostasis. Am. J. Ophthalmol. 2012, 44, 1687–1697. [Google Scholar] [CrossRef]
- Khan, A.O.; Aldahmesh, M.A.; Meyer, B. Recessive Congenital Total Cataract with Microcornea and Heterozygote Carrier Signs Caused by a Novel Missense CRYAA Mutation (R54C). Am. J. Ophthalmol. 2007, 144, 949–952.e2. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Yao, W.; Chan, C.-C.; Kannabiran, C.; Wawrousek, E.; Hejtmancik, J.F. Human βA3/A1-crystallin splicing mutation causes cataracts by activating the unfolded protein response and inducing apoptosis in differentiating lens fiber cells. Biochim. Biophys. Acta 2016, 1862, 1214–1227. [Google Scholar] [CrossRef]
- Fnon, N.F.; Hassan, H.H.; Ali, H.M.; Sobh, Z.K. Sengers syndrome: A rare case of cardiomyopathy combined with congenital cataracts in an infant: Post-mortem case report. Cardiovasc. Pathol. 2021, 54, 107371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Chen, X.-J.; Zhu, J.; Xi, Y.-B.; Yang, X.; Hu, L.-D.; Ouyang, H.; Patel, S.H.; Jin, X.; Lin, D.; et al. Lanosterol reverses protein aggregation in cataracts. Nature 2015, 523, 607–611. [Google Scholar] [CrossRef]
- Castorino, J.J.; Gallagher-Colombo, S.M.; Levin, A.V.; FitzGerald, P.G.; Polishook, J.; Kloeckener-Gruissem, B.; Ostertag, E.; Philp, N.J. Juvenile Cataract-Associated Mutation of Solute Carrier SLC16A12 Impairs Trafficking of the Protein to the Plasma Membrane. Investig. Opthalmol. Vis. Sci. 2011, 52, 6774–6784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansar, M.; Chung, H.-L.; Taylor, R.L.; Nazir, A.; Imtiaz, S.; Sarwar, M.T.; Manousopoulou, A.; Makrythanasis, P.; Saeed, S.; Falconnet, E.; et al. Bi-allelic Loss-of-Function Variants in DNMBP Cause Infantile Cataracts. Am. J. Hum. Genet. 2018, 103, 568–578. [Google Scholar] [CrossRef] [Green Version]
- Quan, Y.; Du, Y.; Tong, Y.; Gu, S.; Jiang, J.X. Connexin Gap Junctions and Hemichannels in Modulating Lens Redox Homeostasis and Oxidative Stress in Cataractogenesis. Antioxidants 2021, 10, 1374. [Google Scholar] [CrossRef]
- Berthoud, V.M.; Gao, J.; Minogue, P.J.; Jara, O.; Mathias, R.T.; Beyer, E.C. Connexin Mutants Compromise the Lens Circulation and Cause Cataracts through Biomineralization. Int. J. Mol. Sci. 2020, 21, 5822. [Google Scholar] [CrossRef]
- Ramachandran, R.D.; Perumalsamy, V.; Hejtmancik, J.F. Autosomal recessive juvenile onset cataract associated with mutation in BFSP1. Hum. Genet. 2007, 121, 475–482. [Google Scholar] [CrossRef] [Green Version]
- Alizadeh, A.; Clark, J.; Seeberger, T.; Hess, J.; Blankenship, T.; FitzGerald, P.G. Targeted deletion of the lens fiber cell-specific intermediate filament protein filensin. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5252–5258. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhang, J.; Cao, Y.; You, Y.; Zhao, X. Novel mutations identified in Chinese families with autosomal dominant congenital cataracts by targeted next-generation sequencing. BMC Med. Genet. 2019, 20, 196. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.L.; McComish, B.J.; Staffieri, S.E.; Souzeau, E.; Kearns, L.S.; Elder, J.E.; Charlesworth, J.C.; Mackey, D.A.; Ruddle, J.B.; Taranath, D.; et al. Pathogenic genetic variants identified in Australian families with paediatric cataract. BMJ Open Ophthalmol. 2022, 7, e001064. [Google Scholar] [CrossRef]
- Entry—#611391—Cataract 33, Multiple Types; Ctrct33—Omim. Available online: https://www.omim.org/entry/611391 (accessed on 8 March 2023).
- Entry—#611597—Cataract 12, Multiple Types; Ctrct12—Omim. Available online: https://www.omim.org/entry/611597 (accessed on 8 March 2023).
- Nieves-Moreno, M.; Noval, S.; Peralta, J.; Palomares-Bralo, M.; del Pozo, A.; Garcia-Miñaur, S.; Santos-Simarro, F.; Vallespin, E. Expanding the Phenotypic Spectrum of PAX6 Mutations: From Congenital Cataracts to Nystagmus. Genes 2021, 12, 707. [Google Scholar] [CrossRef]
- Jarwar, P.; Sheikh, S.A.; Waryah, Y.M.; Ujjan, I.U.; Riazuddin, S.; Waryah, A.M.; Ahmed, Z.M. Biallelic Variants in EPHA2 Identified in Three Large Inbred Families with Early-Onset Cataract. Int. J. Mol. Sci. 2021, 22, 10655. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Hou, S.; Jiang, Z.; Hu, Z.; Yang, P.; Ye, J. Association of EPHA2 polymorphisms and age-related cortical cataract in a Han Chinese population. Mol. Vis. 2011, 17, 1553–1558. [Google Scholar] [PubMed]
- Iqbal, H.; Khan, S.Y.; Zhou, L.; Irum, B.; Ali, M.; Ahmed, M.R.; Shahzad, M.; Ali, M.H.; Naeem, M.A.; Riazuddin, S.; et al. Mutations in FYCO1 identified in families with congenital cataracts. Mol. Vis. 2020, 26, 334–344. [Google Scholar]
- Zhou, Y.; Bennett, T.M.; White, T.W.; Shiels, A. Charged multivesicular body protein 4b forms complexes with gap junction proteins during lens fiber cell differentiation. FASEB J. 2023, 37, e22801. [Google Scholar] [CrossRef]
- Sagona, A.P.; Nezis, I.P.; Stenmark, H. Association of CHMP4B and Autophagy with Micronuclei: Implications for Cataract Formation. BioMed Res. Int. 2014, 2014, 974393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, D.; Wang, Q.; Huang, W.; Dongye, M.; Zhang, X.; Lin, D.; Lin, Z.; Li, J.; Hu, W.; et al. Broadening the Mutation Spectrum in GJA8 and CHMP4B: Novel Missense Variants and the Associated Phenotypes in Six Chinese Han Congenital Cataracts Families. Front. Med. 2021, 8, 713284. [Google Scholar] [CrossRef] [PubMed]
- Janzen, N.; Illsinger, S.; Meyer, U.; Shin, Y.S.; Sander, J.; Lücke, T.; Das, A.M. Early Cataract Formation Due to Galactokinase Deficiency: Impact of Newborn Screening. Arch. Med. Res. 2011, 42, 608–612. [Google Scholar] [CrossRef]
- Lassen, N.; Bateman, J.B.; Estey, T.; Kuszak, J.R.; Nees, D.W.; Piatigorsky, J.; Duester, G.; Day, B.J.; Huang, J.; Hines, L.M.; et al. Multiple and Additive Functions of ALDH3A1 and ALDH1A1: Cataract phenotype and ocular oxidative damage in Aldh3a1(-/-)/Aldh1a1(-/-) knock-out mice. J. Biol. Chem. 2007, 282, 25668–25676. [Google Scholar] [CrossRef] [Green Version]
- Moreira, J.; Ribeiro, I.; Mota, Á.; Gonçalves, R.; Coelho, P.; Maio, T.; Tenedório, P. Paediatric Cataracts: A Retrospective Study of 12 Years (2004–2016). Acta Med. Port. 2017, 30, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Makrygianni, E.A.; Papadimas, G.K.; Vartzelis, G.; Georgala, M.; Tzetis, M.; Poulou, M.; Kitsiou-Tzeli, S.; Pons, R. Congenital Cataracts, Facial Dysmorphism, and Neuropathy Syndrome: Additional Clinical Features. Pediatr. Neurol. 2017, 67, e5–e6. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Z.; He, K.; Chang, P.; Zhao, Y.; Huang, X.; Li, J.; Jin, Z.; Zhao, Y.-E. Unique presentation of congenital cataract concurrent with microcornea, microphthalmia plus posterior capsule defect in monozygotic twins caused by a novel GJA8 mutation. Eye 2019, 33, 686–689. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.R.; Lynn, M.J.; Reeves, R.; Plager, D.A.; Buckley, E.G.; Wilson, M.E. Is there a latent period for the treatment of children with dense bilateral congenital cataracts? J. AAPOS 2006, 10, 30–36. [Google Scholar] [CrossRef]
- Chan, W.H.; Biswas, S.; Ashworth, J.L.; Lloyd, I.C. Congenital and infantile cataract: Aetiology and management. Eur. J. Paediatr. 2012, 171, 625–630. [Google Scholar] [CrossRef]
- Birch, E.E.; Cheng, C.; Stager, D.R., Jr.; Felius, J. Visual acuity development after the implantation of unilateral in-traocular lenses in infants and young children. J. AAPOS 2005, 9, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Autrata, R.; Řehuřek, J.; Vodičková, K. Visual Results after Primary Intraocular Lens Implantation or Contact Lens Correction for Aphakia in the First Year of Age. Int. J. Ophthalmol. 2005, 219, 72–79. [Google Scholar] [CrossRef]
- Solebo, A.L.; Russell-Eggitt, I.; Cumberland, P.M.; Rahi, J.S.; British Isles Congenital Cataract Interest Group. Risks and outcomes associated with primary intraocular lens implantation in children under 2 years of age: The IoLunder2 cohort study. Br. J. Ophthalmol. 2015, 99, 1471–1476. [Google Scholar] [CrossRef] [Green Version]
- Infant Aphakia Treatment Study Group; Lambert, S.R.; Buckley, E.G.; Drews-Botsch, C.; DuBois, L.; Hartmann, E.; Lynn, M.J.; Plager, D.A.; Wilson, M.E. The Infant Aphakia Treatment Study: Design and clinical measures at enrollment: Design and clinical measures at enrollment. Arch. Ophthalmol. 2010, 128, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, R.L.; Black, G.C.M. The Role of Molecular Genetics in the Assessment of Children with Congenital Cataract. In Congenital Cataract; Springer International Publishing: Cham, Switzerland, 2016; pp. 29–54. [Google Scholar]
- Ma, A.; Grigg, J.R.; Flaherty, M.; Smith, J.; Minoche, A.E.; Cowley, M.J.; Nash, B.M.; Ho, G.; Gayagay, T.; Lai, T.; et al. Genome sequencing in congenital cataracts improves diagnostic yield. Hum. Mutat. 2021, 42, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, S.; Gao, L.; Zhang, L.; Zheng, Y.; Cao, W.; Feng, G.; He, L.; Liu, P. A new locus in chromosome 2q37-qter is associated with posterior polar cataract. Graefe’s Arch. Clin. Exp. Ophthalmol. 2012, 250, 907–913. [Google Scholar] [CrossRef]
- VCV001174689.6—ClinVar—NCBI. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/1174689/?new_evidence=true (accessed on 25 March 2023).
- gnomAD. SNV: 14-24679928-G-A(GRCh37). Available online: https://gnomad.broadinstitute.org/variant/14-24679928-G-A?dataset=gnomad_r2_1 (accessed on 25 March 2023).
- Tang, S.; Di, G.; Hu, S.; Liu, Y.; Dai, Y.; Chen, P. AQP5 regulates vimentin expression via miR-124–3p.1 to protect lens transparency. Exp. Eye Res. 2021, 205, 108485. [Google Scholar] [CrossRef]
- Lavelle, T.A.; Feng, X.; Keisler, M.; Cohen, J.T.; Neumann, P.J.; Prichard, D.; Schroeder, B.E.; Salyakina, D.; Espinal, P.S.; Weidner, S.B.; et al. Cost-effectiveness of exome and genome sequencing for children with rare and undiagnosed conditions. Genet. Med. 2022, 24, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Bota, D.A.; Davies, K.J. Mitochondrial Lon protease in human disease and aging: Including an etiologic classification of Lon-related diseases and disorders. Free Radic. Biol. Med. 2016, 100, 188–198. [Google Scholar] [CrossRef] [Green Version]
- GeneCards Human Gene Database. LONP1 Gene—GeneCards. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=LONP1&keywords=LONP1 (accessed on 28 March 2023).
- Patel, N.; Anand, D.; Monies, D.; Maddirevula, S.; Khan, A.O.; Algoufi, T.; Alowain, M.; Faqeih, E.; Alshammari, M.; Qudair, A.; et al. Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract. Hum. Genet. 2017, 136, 205–225. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.O.; AlBakri, A. Clinical features of LONP1 -related infantile cataract. J. AAPOS 2018, 22, 229–231. [Google Scholar] [CrossRef]
- Available online: http://www.pantherdb.org/tools/csnpScore.do (accessed on 28 March 2023).
- Grid Gateway Error. Available online: http://genetics.bwh.harvard.edu/ggi/cgi-bin/ggi2.cgi (accessed on 28 March 2023).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Hansen, L.; Comyn, S.; Mang, Y.; Lind-Thomsen, A.; Myhre, L.; Jean, F.; Eiberg, H.; Tommerup, N.; Rosenberg, T.; Pilgrim, D. The myosin chaperone UNC45B is involved in lens development and autosomal dominant juvenile cataract. Eur. J. Hum. Genet. 2014, 22, 1290–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoiber, K.; Nagło, O.; Pernpeintner, C.; Zhang, S.; Koeberle, A.; Ulrich, M.; Werz, O.; Müller, R.; Zahler, S.; Lohmüller, T.; et al. Targeting de novo lipogenesis as a novel approach in anti-cancer therapy. Br. J. Cancer 2018, 118, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yu, W.; Li, S.; Guo, D.; He, J.; Wang, Y. Acetyl-CoA Carboxylases and Diseases. Front. Oncol. 2022, 12, 836058. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, E.; David, L.L.; Riviere, M.A.; Azuma, M.; Shearer, T.R. Human and Monkey Lenses Cultured with Calcium Ionophore Form αB-Crystallin Lacking the C-Terminal Lysine, a Prominent Feature of Some Human Cataracts. Investig. Opthalmol. Vis. Sci. 2009, 50, 5828–5836. [Google Scholar] [CrossRef]
- Saleh, E.; Pechhacker, M.G.; Vig, A.; Mehta, M.; Maynes, J.; Tumber, A.; Tavares, E.; Vincent, A.; Mireskandari, K.; Heon, E. Unilateral cataract and congenital stationary night blindness in a child with novel variants in TRPM1. J. AAPOS 2022, 26, 202–205. [Google Scholar] [CrossRef]
- Wirth, M.G.; Russell-Eggitt, I.M.; Craig, J.E.; Elder, J.E.; Mackey, D.A. Aetiology of congenital and paediatric cataract in an Australian population. Br. J. Ophthalmol. 2002, 86, 782–786. [Google Scholar] [CrossRef]
- Sandmeyer, L.S.; Bellone, R.R.; Archer, S.; Bauer, B.S.; Nelson, J.; Forsyth, G.; Grahn, B.H. Congenital stationary night blindness is associated with the leopard complex in the miniature horse: Miniature horse congenital stationary night blindness is associated with the leopard complex. Vet. Ophthalmol. 2012, 15, 18–22. [Google Scholar] [CrossRef]
- Zhou, Y.; Bennett, T.M.; Shiels, A. Mutation of the TRPM3 cation channel underlies progressive cataract development and lens calcification associated with pro-fibrotic and immune cell responses. FASEB J. 2021, 35, e21288. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Giannone, A.A.; Li, L.; Sellitto, C.; White, T.W. Physiological Mechanisms Regulating Lens Transport. Front. Physiol. 2021, 12, 818649. [Google Scholar] [CrossRef]
- Kumari, S.S.; Gupta, N.; Shiels, A.; FitzGerald, P.G.; Menon, A.G.; Mathias, R.T.; Varadaraj, K. Role of Aquaporin 0 in lens biomechanics. Biochem. Biophys. Res. Commun. 2015, 462, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ederra, J.; Verkman, A.S. Accelerated Cataract Formation and Reduced Lens Epithelial Water Permeability in Aquaporin-1-Deficient Mice. Investig. Opthalmol. Vis. Sci. 2006, 47, 3960–3967. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Lerner, J.; Wyatt, M.K.; Cai, P.; Peterson, K.; Dong, L.; Wistow, G. The klotho-related protein KLPH (lctl) has preferred expression in lens and is essential for expression of clic5 and normal lens suture formation. Exp. Eye Res. 2018, 169, 111–121. [Google Scholar] [CrossRef]
- SIFT—Predict Effects of Nonsynonmous/Missense Variants. Available online: https://sift.bii.a-star.edu.sg/ (accessed on 9 April 2023).
- Use—Align GVGD. Available online: http://agvgd.hci.utah.edu/agvgd_input.php (accessed on 9 April 2023).
- GeneCards Human Gene Database. HSPE1 Gene—GeneCards. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=HSPE1&keywords=hspe1 (accessed on 9 April 2023).
- Kappé, G.; Franck, E.; Verschuure, P.; Boelens, W.C.; Leunissen, J.A.M.; de Jong, W.W. The human genome encodes 10 α-crystallin–related small heat shock proteins: HspB1–10. Cell Stress Chaperones 2003, 8, 53. [Google Scholar] [CrossRef]
- Laskowska, E. Small heat shock proteins—Role in apoptosis, cancerogenesis and diseases associated with protein aggregation. Postep. Biochem. 2007, 53, 19–26. [Google Scholar]
- Sooraj, K.; Shukla, S.; Kaur, R.; Titiyal, J.S.; Kaur, J. The protective role of HSP27 in ocular diseases. Mol. Biol. Rep. 2022, 49, 5107–5115. [Google Scholar] [CrossRef]
- Fernández-Alcalde, C.; Nieves-Moreno, M.; Noval, S.; Peralta, J.M.; Montaño, V.E.F.; del Pozo, Á.; Santos-Simarro, F.; Vallespín, E. Molecular and Genetic Mechanism of Non-Syndromic Congenital Cataracts. Mutation Screening in Spanish Families. Genes 2021, 12, 580. [Google Scholar] [CrossRef]
- Rivero-Ríos, P.; Weisman, L.S. Roles of PIKfyve in multiple cellular pathways. Curr. Opin. Cell Biol. 2022, 76, 102086. [Google Scholar] [CrossRef] [PubMed]
- Ikonomov, O.C.; Sbrissa, D.; Shisheva, A. Mammalian Cell Morphology and Endocytic Membrane Homeostasis Require Enzymatically Active Phosphoinositide 5-Kinase PIKfyve. J. Biol. Chem. 2001, 276, 26141–26147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, S.A.; Uderhardt, S.; Saric, A.; Collins, R.F.; Buckley, C.M.; Mylvaganam, S.; Boroumand, P.; Plumb, J.; Germain, R.N.; Ren, D.; et al. Lipid-gated monovalent ion fluxes regulate endocytic traffic and support immune surveillance. Science 2020, 367, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Wu, Y.; Wang, Y.; Cui, Y.; Zhang, M.; Zhang, T.; Huang, X.; Yu, S.; Yu, T.; Zhao, J. Disruption of PIKFYVE causes congenital cataract in human and zebrafish. eLife 2022, 11, e71256. [Google Scholar] [CrossRef] [PubMed]
- GeneCards Human Gene Database. CHMP4A Gene—GeneCards. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=CHMP4A&keywords=chmp4a (accessed on 12 April 2023).
- GeneCards Human Gene Database. GeneCards—Human Genes. Available online: https://www.genecards.org/ (accessed on 2 February 2023).
- Cataract 32, Multiple Types Disease: Malacards—Research Articles, Drugs, Genes, Clinical Trials. Available online: https://www.malacards.org/card/cataract_32_multiple_types (accessed on 12 April 2023).
- Zhou, Y.; Bennett, T.M.; Shiels, A. A charged multivesicular body protein (CHMP4B) is required for lens growth and differentiation. Differentiation 2019, 109, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Shiels, A.; Bennett, T.M.; Knopf, H.L.; Yamada, K.; Yoshiura, K.-I.; Niikawa, N.; Shim, S.; Hanson, P.I. CHMP4B, a Novel Gene for Autosomal Dominant Cataracts Linked to Chromosome 20q. Am. J. Hum. Genet. 2007, 81, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Kemény, K.K.; Ducza, E. Physiological Cooperation between Aquaporin 5 and TRPV4. Int. J. Mol. Sci. 2022, 23, 11634. [Google Scholar] [CrossRef]
- Bassnett, S.; Wilmarth, P.A.; David, L.L. The membrane proteome of the mouse lens fiber cell. Mol. Vis. 2009, 15, 2448–2463. [Google Scholar]
- Grey, A.C.; Walker, K.L.; Petrova, R.S.; Han, J.; Wilmarth, P.A.; David, L.L.; Donaldson, P.J.; Schey, K.L. Verification and spatial localization of aquaporin-5 in the ocular lens. Exp. Eye Res. 2013, 108, 94–102. [Google Scholar] [CrossRef] [Green Version]
- NCBI. Available online: https://pubmed.ncbi.nlm.nih.gov/ (accessed on 24 May 2022).
- DECIPHER v11.12: Mapping the Clinical Genome. Available online: https://www.deciphergenomics.org/ (accessed on 2 February 2023).
- gnomAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 3 February 2023).
- UCSC Genome Browser Home. Available online: https://genome.ucsc.edu/ (accessed on 3 February 2023).
- Cat-Map. Available online: https://cat-map.wustl.edu/ (accessed on 3 February 2023).
- VarSome the Human Genomics Community. Available online: https://varsome.com/ (accessed on 24 March 2023).
- Franklin. Available online: https://franklin.genoox.com/clinical-db/hom (accessed on 3 February 2023).
- GeneMatcher. Available online: https://genematcher.org/ (accessed on 24 March 2023).




| Family ID | Relationship | Eye | Cataract Type | Microphthalmia | Nystagmus | Age at Surgery | Glaucoma |
|---|---|---|---|---|---|---|---|
| OFT-00172 | Proband | RE | Nuclear | Yes | No | <1 year | Yes |
| LE | Nuclear | Yes | No | <1 year | No | ||
| OFT-00247 | Proband | RE | Posterior polar | Yes | No | N/A | N/A |
| LE | PFV | Yes | No | <1 year | Yes | ||
| OFT-00289 | Proband | RE | Posterior subcapsular | No | No | >1 year | No |
| LE | Posterior subcapsular | No | No | >1 year | No | ||
| Sister | RE | Lamellar | No | No | >1 year | No | |
| LE | Lamellar | No | No | >1 year | No | ||
| OFT-00222 | Proband | RE | Nuclear | No | Yes | <1 year | Yes |
| LE | Cuneiform | No | Yes | N/A | N/A | ||
| OFT-00334 | Proband | RE | Posterior subcapsular | No | No | N/A | N/A |
| LE | Posterior polar | No | No | N/A | N/A | ||
| OFT-00350 | Proband | RE | Posterior polar | No | No | >1 year | No |
| LE | Posterior polar | No | No | N/A | N/A | ||
| OFT-00214 | Proband | RE | Nuclear | Yes | Yes | <1 year | Yes |
| LE | Nuclear | Yes | Yes | <1 year | No | ||
| OFT-00322 | Proband | RE | Posterior polar | No | No | N/A | N/A |
| LE | Posterior subcapsular | No | No | N/A | N/A | ||
| OFT-00338 | Proband | RE | Posterior subcapsular | No | No | N/A | N/A |
| LE | Posterior subcapsular | No | No | N/A | N/A | ||
| Sister | RE | Transparent lens | No | No | N/A | N/A | |
| LE | Posterior lenticonus | No | No | >1 year | No | ||
| OFT-00346 | Proband | RE | Lamellar | No | No | >1 year | No |
| LE | Lamellar | No | No | >1 year | No | ||
| OFT-00302 | Proband | RE | Lamellar | Yes | No | <1 year | No |
| LE | Lamellar | Yes | No | <1 year | No | ||
| OFT-00215 | Proband | RE | Lamellar | No | No | >1 year | No |
| LE | Lamellar | No | No | >1 year | No | ||
| OFT-00235 | Proband | RE | Lamellar | No | No | N/A | N/A |
| LE | Lamellar | No | No | N/A | N/A | ||
| OFT-00377 | Proband | RE | Pulverulent | No | No | N/A | N/A |
| LE | Pulverulent | No | No | N/A | N/A | ||
| Proband | RE | Pulverulent | No | No | N/A | N/A | |
| LE | Pulverulent | No | No | N/A | N/A | ||
| OFT-00040 | Proband | RE | Nuclear | No | Yes | <1 year | No |
| LE | Nuclear | No | Yes | <1 year | No | ||
| OFT-00446 | Proband | RE | Nuclear | Yes | Yes | <1 year | Yes |
| LE | Nuclear | Yes | Yes | <1 year | Yes | ||
| OFT-00345 | Proband | RE | Lamellar | No | No | >1 year | No |
| LE | Lamellar | No | No | >1 year | No | ||
| OFT-00456 | Proband | RE | Nuclear | No | No | <1 year | No |
| LE | Nuclear | No | No | <1 year | No | ||
| OFT-00487 | Proband | RE | Posterior subcapsular | No | No | N/A | N/A |
| LE | Posterior subcapsular | No | No | N/A | N/A | ||
| OFT-00522 | Proband | RE | Nuclear | No | No | <1 year | No |
| LE | Posterior polar | No | No | <1 year | No |
| Family ID | Sex | Relationship | Gene | Mutation | Location | ACMG Criteria * | ACMG Result | Coding Impact | Zygosity | Inheritance | De Novo/ Inherited | Reported By |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OFT-00172 | M | Proband | NC | NC | - | - | - | - | - | - | - | - |
| OFT-00247 | F | Proband | LONP1 | NM_004793.3:c.1939G>A p.(Glu647Lys) | Exon 13 | PM2, PP3, BP6 | VUS | Missense | Hetero | AD/AR | Paternal | (Ma, A., 2021) [59] |
| OFT-00289 | M | Proband | ACACA | NM_198839.2:c.1126C>T p.(Arg376Cys) | Exon 15 | PM2, PP2, PP3 | VUS | Missense | Hetero | AD/AR | Maternal Germline Mosaic | Novel ** |
| F | Sister | |||||||||||
| OFT-00222 | M | Proband | NC | NC | - | - | - | - | - | - | - | - |
| OFT-00334 | F | Proband | TRPM1 | NM_001252024.2:c.4720dup p.(Ser1574LysfsTer7) | Exon 27 | PVS1, PM2 | LP | Frameshift | Hetero | AR | Maternal | Novel ** |
| OFT-00350 | M | Proband | Locus 2q37.3 | Deletion of 661,2 Kb (chr2:241526318–242187541) | Chr 2 | 1A, 2A-2E, 2H, 3A, 5A | P | - | Hetero | AD/AR | De novo | (Ouyang, 2012) [60] |
| OFT-00214 | F | Proband | NC | NC | - | - | - | - | - | - | - | - |
| OFT-00322 | F | Proband | NC | NC | - | - | - | - | - | - | - | - |
| OFT-00338 | M | Proband | CLIC5 | NM_016929.4:c.514C>T p.(Arg172Trp) | Exon 5 | PM2, BS2 | VUS | Missense | Hetero | AR | Maternal | ClinVar [61] |
| F | Sister | |||||||||||
| OFT-00346 | F | Proband | HSPE1 | NM_002157.2:c.61_62insACCA: p.(Ser21AsnfsTer5) | Exon 2 | PM2 | VUS | Frameshift | Hetero | AD/AR | De Novo | Novel ** |
| ODF1 | NM_024410.4:c.678_686del p.(Cys227_Pro229del) | Exon 2 | PM2, PM4 | VUS | Inframe Deletion | Hetero | AD/AR | De Novo | Novel ** | |||
| OFT-00302 | F | Proband | NC | NC | - | - | - | - | - | - | - | - |
| OFT-00215 | M | Proband | PIKFYVE | NM_015040.3:c.5844+3A>G | Exon 39 | PM2, PS3, BS4 | VUS | Splicing | Hetero | AD | Maternal | Novel ** |
| OFT-00235 | M | Proband | CHMP4A | NM_014169.5:c.406C>T p.(Gln136Ter) | Exon 4 | PM2, BS2 | VUS | Nonsense | Hetero | AD/AR | Paternal | gnomAD [62] |
| NM_014169.5:c.65del p.(Ala22GlufsTer5) | Exon 2 | PM2 | VUS | Frameshift | Hetero | Maternal | Novel ** | |||||
| OFT-00377 | F | Proband | NC | NC | - | - | - | - | - | - | - | - |
| F | Sister | NC | NC | - | - | - | - | - | - | - | - | |
| OFT-00040 | F | Proband | AQP5 | NM_001651.3:c.152T>C p.(Leu51Pro) | Exon 1 | PM2, PP3, PS2, PS3 | P | Missense | Hetero | AD | De Novo | (Tang, 2021) [63] |
| OFT-00446 | M | Proband | NC | NC | - | - | - | - | - | - | - | - |
| OFT-00345 | F | Proband | NC | NC | - | - | - | - | - | - | - | - |
| OFT-00456 | M | Proband | NC | NC | - | - | - | - | - | - | - | - |
| OFT-00487 | F | Proband | NC | NC | - | - | - | - | - | - | - | - |
| OFT-00522 | M | Proband | NC | NC | - | - | - | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Solana, P.; Arruti, N.; Nieves-Moreno, M.; Mena, R.; Rodríguez-Jiménez, C.; Guerrero-Carretero, M.; Acal, J.C.; Blasco, J.; Peralta, J.M.; Del Pozo, Á.; et al. Whole Exome Sequencing of 20 Spanish Families: Candidate Genes for Non-Syndromic Pediatric Cataracts. Int. J. Mol. Sci. 2023, 24, 11429. https://doi.org/10.3390/ijms241411429
Rodríguez-Solana P, Arruti N, Nieves-Moreno M, Mena R, Rodríguez-Jiménez C, Guerrero-Carretero M, Acal JC, Blasco J, Peralta JM, Del Pozo Á, et al. Whole Exome Sequencing of 20 Spanish Families: Candidate Genes for Non-Syndromic Pediatric Cataracts. International Journal of Molecular Sciences. 2023; 24(14):11429. https://doi.org/10.3390/ijms241411429
Chicago/Turabian StyleRodríguez-Solana, Patricia, Natalia Arruti, María Nieves-Moreno, Rocío Mena, Carmen Rodríguez-Jiménez, Marta Guerrero-Carretero, Juan Carlos Acal, Joana Blasco, Jesús M. Peralta, Ángela Del Pozo, and et al. 2023. "Whole Exome Sequencing of 20 Spanish Families: Candidate Genes for Non-Syndromic Pediatric Cataracts" International Journal of Molecular Sciences 24, no. 14: 11429. https://doi.org/10.3390/ijms241411429