
Exploring the Pathophysiologic Cascade Leading to Osteoclastogenic Activation in Gaucher Disease Monocytes Generated via CRISPR/Cas9 Technology
 , , and
, , and 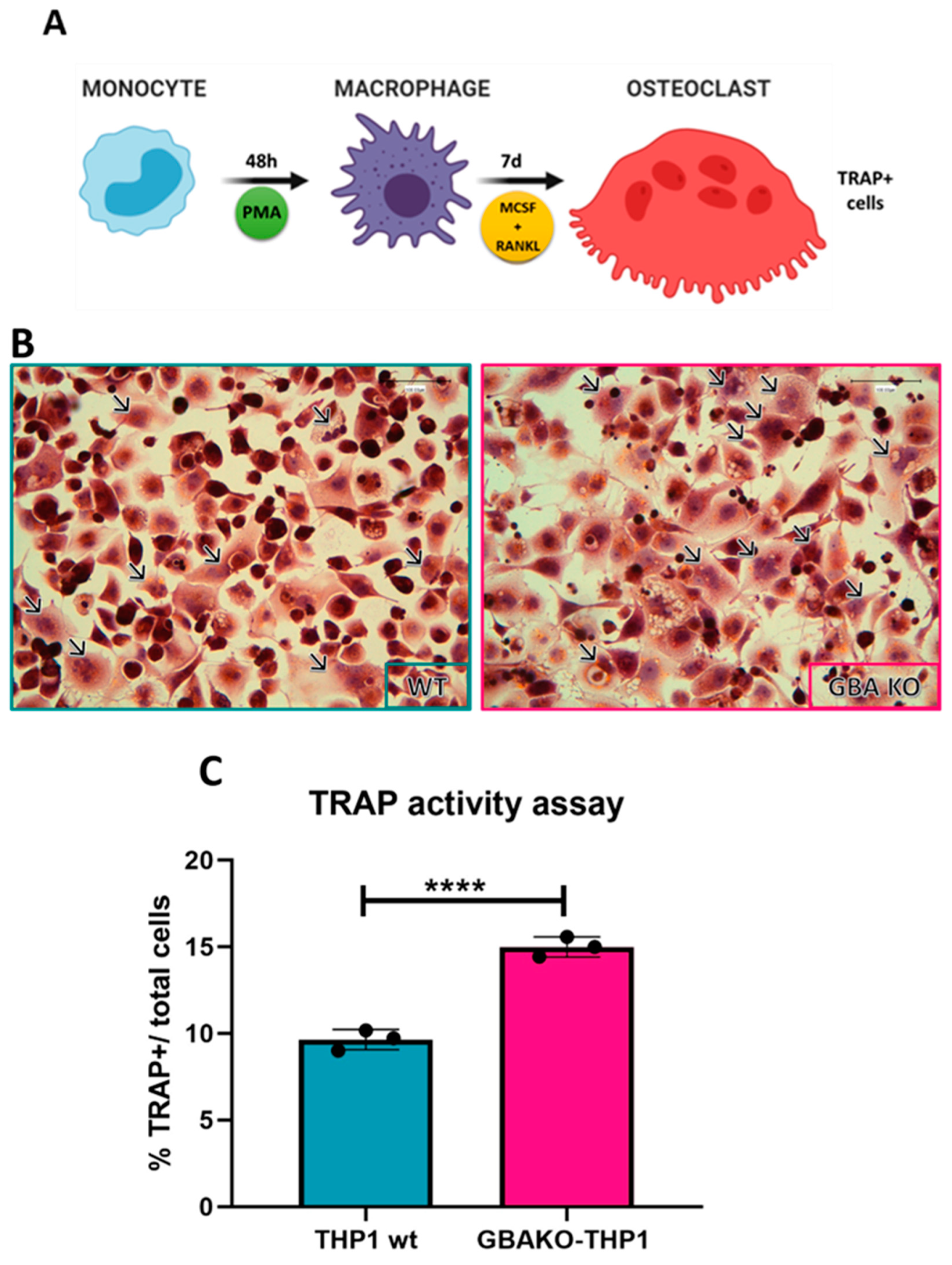{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Characterization of the Monocyte GBA1 Knockout Cells
2.1.1. Increased Osteoclastogenesis in GBAKO-THP1 Cells
2.1.2. Proinflammatory Cytokines Released by GBAKO-THP1 Cells
2.1.3. Role of Increased IL-1β Levels in Osteoclastogenesis
2.2. Evaluation of the Effect of Treatments on the Phenotype of the GBAKO-THP1
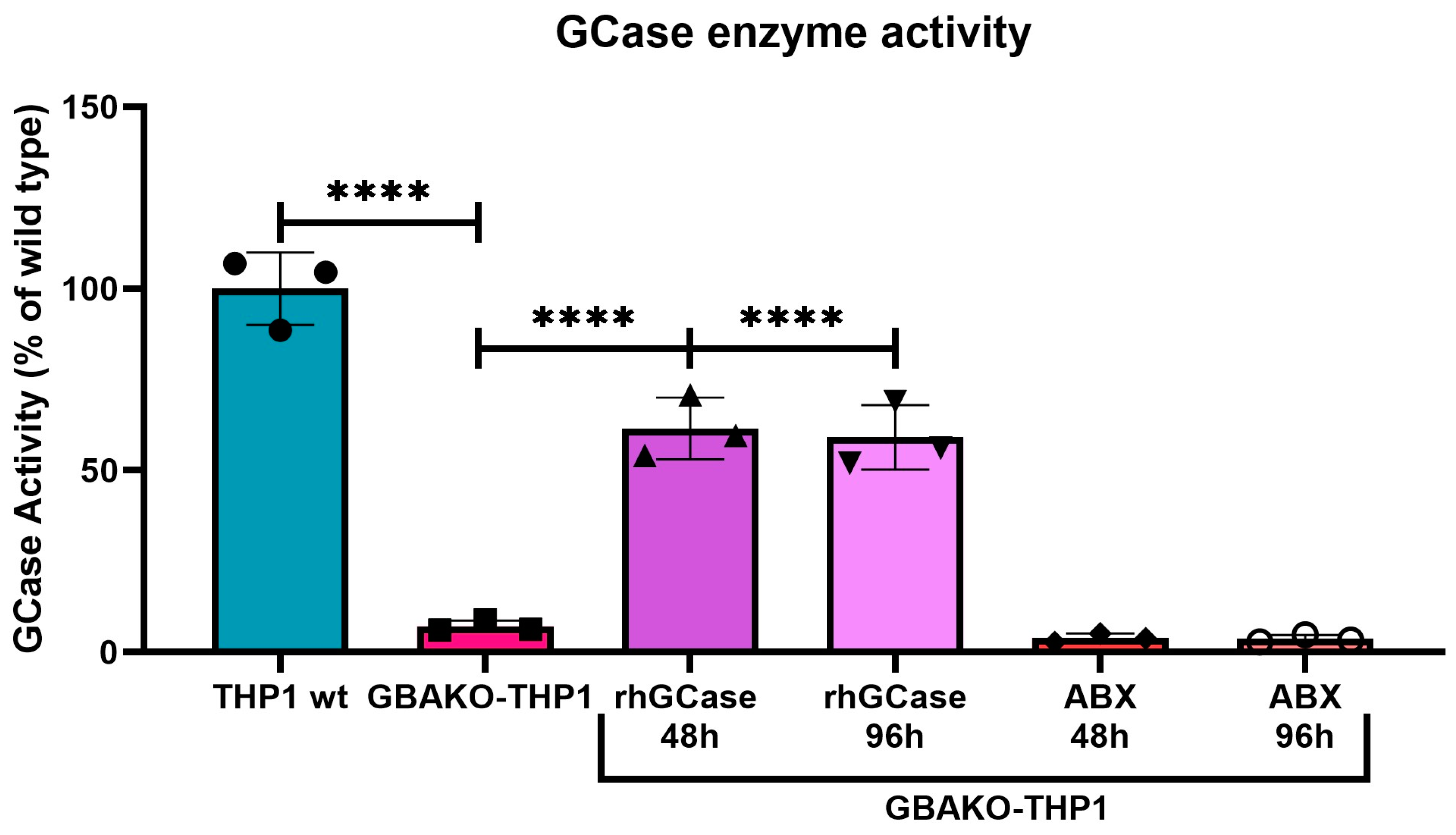
2.2.1. Effect on GCase Activity
2.2.2. Effect on Substrate Accumulation
2.2.3. Effect of Ambroxol on Substrate Reduction
2.2.4. Effect on Osteoclastogenesis
2.2.5. Effect on Proinflammatory Cytokines Release
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Cell Culture
4.3. Osteoclast Differentiation Assay
4.4. Enzymatic Activity
4.5. Cytokine Measurement
4.6. Glucosylsphingosine Measurement (GlcSph)
4.7. Cells Treatments
4.8. Statistical Analysis
5. Limitations and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stirnemann, J.Ô.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mistrya, P.K.; Liua, J.; Yanga, M.; Nottolic, T.; McGratha, J.; Jaine, D.; Zhangf, K.; Keutzerf, J.; Chuangf, W.L.; Mehalb, W.Z.; et al. Glucocerebrosidase Gene-Deficient Mouse Recapitulates Gaucher Disease Displaying Cellular and Molecular Dysregulation beyond the Macrophage. Proc. Natl. Acad. Sci. USA 2010, 107, 19473–19478. [Google Scholar] [CrossRef]
- Aflaki, E.; Moaven, N.; Borger, D.K.; Lopez, G.; Westbroek, W.; Chae, J.J.; Marugan, J.; Patnaik, S.; Maniwang, E.; Gonzalez, A.N.; et al. Lysosomal Storage and Impaired Autophagy Lead to Inflammasome Activation in Gaucher Macrophages. Aging Cell 2016, 15, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Bembi, B.; Marsala, S.Z.; Sidransky, E.; Ciana, G.; Carrozzi, M.; Zorzon, M.; Martini, C.; Gioulis, M.; Pittis, M.G.; Capus, L. Gaucher’s Disease with Parkinson’s Disease: Clinical and Pathological Aspects. Neurology 2003, 61, 99–101. [Google Scholar] [CrossRef]
- Itzchaki, M.; Lebel, E.; Dweck, A.; Patlas, M.; Hadas-Halpern, I.; Zimran, A.; Elstein, D. Orthopedic Considerations in Gaucher Disease since the Advent of Enzyme Replacement Therapy. Acta Orthop. Scand. 2004, 75, 641–653. [Google Scholar] [CrossRef]
- Hughes, D.; Mikosch, P.; Belmatoug, N.; Carubbi, F.; Cox, T.M.; Goker-Alpan, O.; Kindmark, A.; Mistry, P.K.; Poll, L.; Weinreb, N.; et al. Gaucher Disease in Bone: From Pathophysiology to Practice; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2019; Volume 34, pp. 996–1013. [Google Scholar]
- Mela, A.; Poniatowski, Ł.A.; Drop, B.; Furtak-Niczyporuk, M.; Jaroszyński, J.; Wrona, W.; Staniszewska, A.; Dąbrowski, J.; Czajka, A.; Jagielska, B.; et al. Overview and Analysis of the Cost of Drug Programs in Poland: Public Payer Expenditures and Coverage of Cancer and Non-Neoplastic Diseases Related Drug Therapies from 2015–2018 Years. Front. Pharmacol. 2020, 11, 1123. [Google Scholar] [CrossRef]
- Manea, S.; Visonà Dalla Pozza, L.; Minichiello, C.; Altieri, L.; Mazzucato, M.; Bonin, M.; De Ambrosis, P.; Borgonovi, E.; Facchin, P. High-Cost Drugs for Rare Diseases: Their Expenditure and Value Based on a Regional Area-Based Study. Health Serv. Manag. Res. 2023, 09514848231151814. [Google Scholar] [CrossRef] [PubMed]
- Sirrs, S.; Nakagawa, B.; Anderson, H.; Regier, D.; Jiwani, B.; Rizzardo, S.; Lun, E.; McFarlane, A. Expensive Drugs for Rare Diseases in Canada: Time for Action Everywhere and by Everyone. Healthc. Pap. 2023, 21, 74–80. [Google Scholar] [CrossRef]
- Raggatt, L.J.; Partridge, N.C. Cellular and Molecular Mechanisms of Bone Remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef] [Green Version]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef]
- Bolamperti, S.; Villa, I.; Rubinacci, A. Bone Remodeling: An Operational Process Ensuring Survival and Bone Mechanical Competence. Bone Res. 2022, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Delaisse, J.M.; Andersen, T.L.; Kristensen, H.B.; Jensen, P.R.; Andreasen, C.M.; Søe, K. Re-Thinking the Bone Remodeling Cycle Mechanism and the Origin of Bone Loss. Bone 2020, 141, 115628. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.; Baker, R.J.; Mehta, A.B.; Hughes, D.A. Enhanced Differentiation of Osteoclasts from Mononuclear Precursors in Patients with Gaucher Disease. Blood Cells Mol. Dis. 2013, 51, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Reed, M.C.; Bauernfreund, Y.; Cunningham, N.; Beaton, B.; Mehta, A.B.; Hughes, D.A. Generation of Osteoclasts from Type 1 Gaucher Patients and Correlation with Clinical and Genetic Features of Disease. Gene 2018, 678, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Bondar, C.; Ormazabal, M.; Crivaro, A.; Ferreyra-Compagnucci, M.; Delpino, M.; Rozenfeld, P.A.P.A.; Mucci, J.M.J.M.J.M. Osteocyte Alterations Induce Osteoclastogenesis in an in Vitro Model of Gaucher Disease. Int. J. Mol. Sci. 2017, 18, 112. [Google Scholar] [CrossRef] [Green Version]
- Rigante, D.; Cipolla, C.; Basile, U.; Gulli, F.; Savastano, M.C. Overview of Immune Abnormalities in Lysosomal Storage Disorders. Immunol. Lett. 2017, 188, 79–85. [Google Scholar] [CrossRef]
- Rozenfeld, P.A.; Crivaro, A.N.; Ormazabal, M.; Mucci, J.M.; Bondar, C.; Delpino, M.V. Unraveling the Mystery of Gaucher Bone Density Pathophysiology. Mol. Genet. Metab. 2021, 132, 76–85. [Google Scholar] [CrossRef]
- Lampiasi, N.; Russo, R.; Zito, F. The Alternative Faces of Macrophage Generate Osteoclasts. BioMed Res. Int. 2016. [Google Scholar] [CrossRef] [Green Version]
- Lecourt, S.; Vanneaux, V.; Cras, A.; Freida, D.; Heraoui, D.; Herbi, L.; Caillaud, C.; Chomienne, C.; Marolleau, J.P.; Belmatoug, N.; et al. Bone Marrow Microenvironment in an in Vitro Model of Gaucher Disease: Consequences of Glucocerebrosidase Deficiency. Stem Cells Dev. 2012, 21, 239–248. [Google Scholar] [CrossRef]
- Crivaro, A.; Bondar, C.; Mucci, J.M.; Ormazabal, M.; Feldman, R.A.; Delpino, M.V.; Rozenfeld, P.A. Gaucher Disease-Associated Alterations in Mesenchymal Stem Cells Reduce Osteogenesis and Favour Adipogenesis Processes with Concomitant Increased Osteoclastogenesis. Mol. Genet. Metab. 2020, 130, 274–282. [Google Scholar] [CrossRef]
- Lorenz, F.; Pawłowicz, E.; Klimkowska, M.; Beshara, S.; Bulanda Brustad, A.; Skotnicki, A.B.; Wahlin, A.; Machaczka, M. Ferritinemia and Serum Inflammatory Cytokines in Swedish Adults with Gaucher Disease Type 1. Blood Cells Mol. Dis. 2018, 68, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Matta, M.C.; Vairo, F.; Torres, L.C.; Schwartz, I. Could Enzyme Replacement Therapy Promote Immune Tolerance in Gaucher Disease Type 1? Blood Cells Mol. Dis. 2018, 68, 200–202. [Google Scholar] [CrossRef] [PubMed]
- Enquist, I.B.; Nilsson, E.; Månsson, J.-E.; Ehinger, M.; Richter, J.; Karlsson, S.; Sun, Y.; Florer, J.; Mayhew, C.N.; Jia, Z.; et al. Properties of Neurons Derived from Induced Pluripotent Stem Cells of Gaucher Disease Type 2 Patient Fibroblasts: Potential Role in Neuropathology. PLoS ONE 2015, 10, e0118771. [Google Scholar] [CrossRef] [Green Version]
- Rybová, J.; Ledvinová, J.; Sikora, J.; Kuchař, L.; Dobrovolný, R. Neural Cells Generated from Human Induced Pluripotent Stem Cells as a Model of CNS Involvement in Mucopolysaccharidosis Type II. J. Inherit. Metab. Dis. 2018, 41, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Zunke, F.; Mazzulli, J.R. Modeling Neuronopathic Storage Diseases with Patient-Derived Culture Systems. Neurobiol. Dis. 2019, 127, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Srikanth, M.P.; Deleidi, M.; Hallett, P.J.; Isacson, O.; Feldman, R.A. Acid Ceramidase Involved in Pathogenic Cascade Leading to Accumulation of α-Synuclein in IPSC Model of GBA1 -Associated Parkinson’s Disease. Hum. Mol. Genet. 2023, 32, 1888–1900. [Google Scholar] [CrossRef]
- Peng, Y.; Liou, B.; Lin, Y.; Mayhew, C.N.; Fleming, S.M.; Sun, Y. IPSC-Derived Neural Precursor Cells Engineering GBA1 Recovers Acid β-Glucosidase Deficiency and Diminishes α-Synuclein and Neuropathology. Mol. Ther. Methods Clin. Dev. 2023, 29, 185–201. [Google Scholar] [CrossRef]
- Aflaki, E.; Stubblefield, B.K.; Maniwang, E.; Lopez, G.; Moaven, N.; Goldin, E.; Marugan, J.; Patnaik, S.; Dutra, A.; Southall, N.; et al. Macrophage Models of Gaucher Disease for Evaluating Disease Pathogenesis and Candidate Drugs. Sci. Transl. Med. 2014, 6, 240ra73. [Google Scholar] [CrossRef] [Green Version]
- Panicker, L.M.; Miller, D.; Awad, O.; Bose, V.; Lun, Y.; Park, T.S.; Zambidis, E.T.; Sgambato, J.A.; Feldman, R.A. Gaucher IPSC-Derived Macrophages Produce Elevated Levels of Inflammatory Mediators and Serve as a New Platform for Therapeutic Development. Stem Cells 2014, 32, 2338–2349. [Google Scholar] [CrossRef] [Green Version]
- Messelodi, D.; Bertuccio, S.N.; Indio, V.; Strocchi, S.; Taddia, A.; Serravalle, S.; Bandini, J.; Astolfi, A.; Pession, A. IPSC-Derived Gaucher Macrophages Display Growth Impairment and Activation of Inflammation-Related Cell Death. Cells 2021, 10, 2822. [Google Scholar] [CrossRef]
- Borger, D.K.; Sidransky, E.; Aflaki, E. New Macrophage Models of Gaucher Disease Offer New Tools for Drug Development. Macrophage 2015, 2, e712. [Google Scholar]
- Pavan, E.; Ormazabal, M.; Peruzzo, P.; Vaena, E.; Rozenfeld, P.; Dardis, A. Crispr/Cas9 Editing for Gaucher Disease Modelling. Int. J. Mol. Sci. 2020, 21, 3268. [Google Scholar] [CrossRef]
- Matafora, V.; Cuccurullo, M.; Beneduci, A.; Petrazzuolo, O.; Simeone, A.; Anastasio, P.; Mignani, R.; Feriozzi, S.; Pisani, A.; Comotti, C.; et al. Early Markers of Fabry Disease Revealed by Proteomics. Mol. Biosyst. 2015, 11, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucci, J.M.; Cuello, M.F.; Kisinovsky, I.; Larroude, M.; Delpino, M.V.; Rozenfeld, P.A. Proinflammatory and Proosteoclastogenic Potential of Peripheral Blood Mononuclear Cells from Gaucher Patients: Implication for Bone Pathology. Blood Cells Mol. Dis. 2015, 55, 134–143. [Google Scholar] [CrossRef]
- Michelakakis, H.; Spanou, C.; Kondyli, A.; Dimitriou, E.; Van Weely, S.; Hollak, C.E.M.; Van Oers, M.H.J.; Aerts, J.M.F.G. Plasma Tumor Necrosis Factor-a (TNF-a) Levels in the Gaucher Disease. Biochim. Biophys. Acta Mol. Basis Dis. 1996, 1317, 219–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barak, V.; Acker, M.; Nisman, B.; Kalickman, I.; Abrahamov, A.; Zimran, A.; Yatziv, S. Cytokines in Gaucher’s Disease. Eur. Cytokine Netw. 1999, 10, 205–210. [Google Scholar]
- Vitner, E.B.; Farfel-Becker, T.; Eilam, R.; Biton, I.; Futerman, A.H. Contribution of Brain Inflammation to Neuronal Cell Death in Neuronopathic Forms of Gaucher’s Disease. Brain 2012, 135, 1724–1735. [Google Scholar] [CrossRef]
- Rolph, D.; Das, H. Transcriptional Regulation of Osteoclastogenesis: The Emerging Role of KLF2. Front. Immunol. 2020, 11, 937. [Google Scholar] [CrossRef]
- Kim, J.H.; Jin, H.M.; Kim, K.; Song, I.; Youn, B.U.; Matsuo, K.; Kim, N. The Mechanism of Osteoclast Differentiation Induced by IL-1. J. Immunol. 2009, 183, 1862–1870. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Schapira, A.H. Effects of Ambroxol on the Autophagy-Lysosome Pathway and Mitochondria in Primary Cortical Neurons. Sci. Rep. 2018, 8, 1385. [Google Scholar] [CrossRef] [Green Version]
- Panicker, L.M.; Srikanth, M.P.; Castro-Gomes, T.; Miller, D.; Andrews, N.W.; Feldman, R.A. Gaucher Disease IPSC-Derived Osteoblasts Have Developmental and Lysosomal Defects That Impair Bone Matrix Deposition. Hum. Mol. Genet. 2018, 27, 811–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, G.; D’Amelio, P.; Faccio, R.; Brunetti, G. The Interplay between the Bone and the Immune System. Clin. Dev. Immunol. 2013, 2013, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crivaro, A.N.; Mucci, J.M.; Bondar, C.M.; Ormazabal, M.E.; Ceci, R.; Simonaro, C.; Rozenfeld, P.A. Efficacy of Pentosan Polysulfate in Vitro Models of Lysosomal Storage Disorders: Fabry and Gaucher Disease. PLoS ONE 2019, 14, e0217780. [Google Scholar] [CrossRef] [PubMed]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-Ray Structure of Human Acid-Beta-Glucosidase, the Defective Enzyme in Gaucher Disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef] [Green Version]
- Fois, G.; Hobi, N.; Felder, E.; Ziegler, A.; Miklavc, P.; Walther, P.; Radermacher, P.; Haller, T.; Dietl, P. A New Role for an Old Drug: Ambroxol Triggers Lysosomal Exocytosis via PH-Dependent Ca2+ Release from Acidic Ca2+ Stores. Cell Calcium 2015, 58, 628–637. [Google Scholar] [CrossRef]
- Ciana, G.; Dardis, A.; Pavan, E.; Da Riol, R.M.; Biasizzo, J.; Ferino, D.; Zanatta, M.; Boni, A.; Antonini, L.; Crichiutti, G.; et al. In Vitro and in Vivo Effects of Ambroxol Chaperone Therapy in Two Italian Patients Affected by Neuronopathic Gaucher Disease and Epilepsy. Mol. Genet. Metab. Rep. 2020, 25, 100678. [Google Scholar] [CrossRef]
- Polo, G.; Burlina, A.P.; Kolamunnage, T.B.; Zampieri, M.; Dionisi-Vici, C.; Strisciuglio, P.; Zaninotto, M.; Plebani, M.; Burlina, A.B. Diagnosis of Sphingolipidoses: A New Simultaneous Measurement of Lysosphingolipids by LC-MS/MS. Clin. Chem. Lab. Med. 2017, 55, 403–414. [Google Scholar] [CrossRef]
- Sillence, D.J.; Puri, V.; Marks, D.L.; Butters, T.D.; Dwek, R.A.; Pagano, R.E.; Platt, F.M. Glucosylceramide Modulates Membrane Traffic along the Endocytic Pathway Supplementary Key Words Glycosphingolipid Gaucher Disease Golgi Endocytic Trafficking Sphingolipid Sorting BODIPY Micro-Domain Cholesterol. J. Lipid Res. 2002, 43, 1837–1845. [Google Scholar] [CrossRef] [Green Version]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ormazabal, M.E.; Pavan, E.; Vaena, E.; Ferino, D.; Biasizzo, J.; Mucci, J.M.; Serra, F.; Cifù, A.; Scarpa, M.; Rozenfeld, P.A.; et al. Exploring the Pathophysiologic Cascade Leading to Osteoclastogenic Activation in Gaucher Disease Monocytes Generated via CRISPR/Cas9 Technology. Int. J. Mol. Sci. 2023, 24, 11204. https://doi.org/10.3390/ijms241311204
Ormazabal ME, Pavan E, Vaena E, Ferino D, Biasizzo J, Mucci JM, Serra F, Cifù A, Scarpa M, Rozenfeld PA, et al. Exploring the Pathophysiologic Cascade Leading to Osteoclastogenic Activation in Gaucher Disease Monocytes Generated via CRISPR/Cas9 Technology. International Journal of Molecular Sciences. 2023; 24(13):11204. https://doi.org/10.3390/ijms241311204
Chicago/Turabian StyleOrmazabal, Maximiliano Emanuel, Eleonora Pavan, Emilio Vaena, Dania Ferino, Jessica Biasizzo, Juan Marcos Mucci, Fabrizio Serra, Adriana Cifù, Maurizio Scarpa, Paula Adriana Rozenfeld, and et al. 2023. "Exploring the Pathophysiologic Cascade Leading to Osteoclastogenic Activation in Gaucher Disease Monocytes Generated via CRISPR/Cas9 Technology" International Journal of Molecular Sciences 24, no. 13: 11204. https://doi.org/10.3390/ijms241311204