
microRNA Expression of Renal Proximal Tubular Epithelial Cells and Their Extracellular Vesicles in an Inflammatory Microenvironment In Vitro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
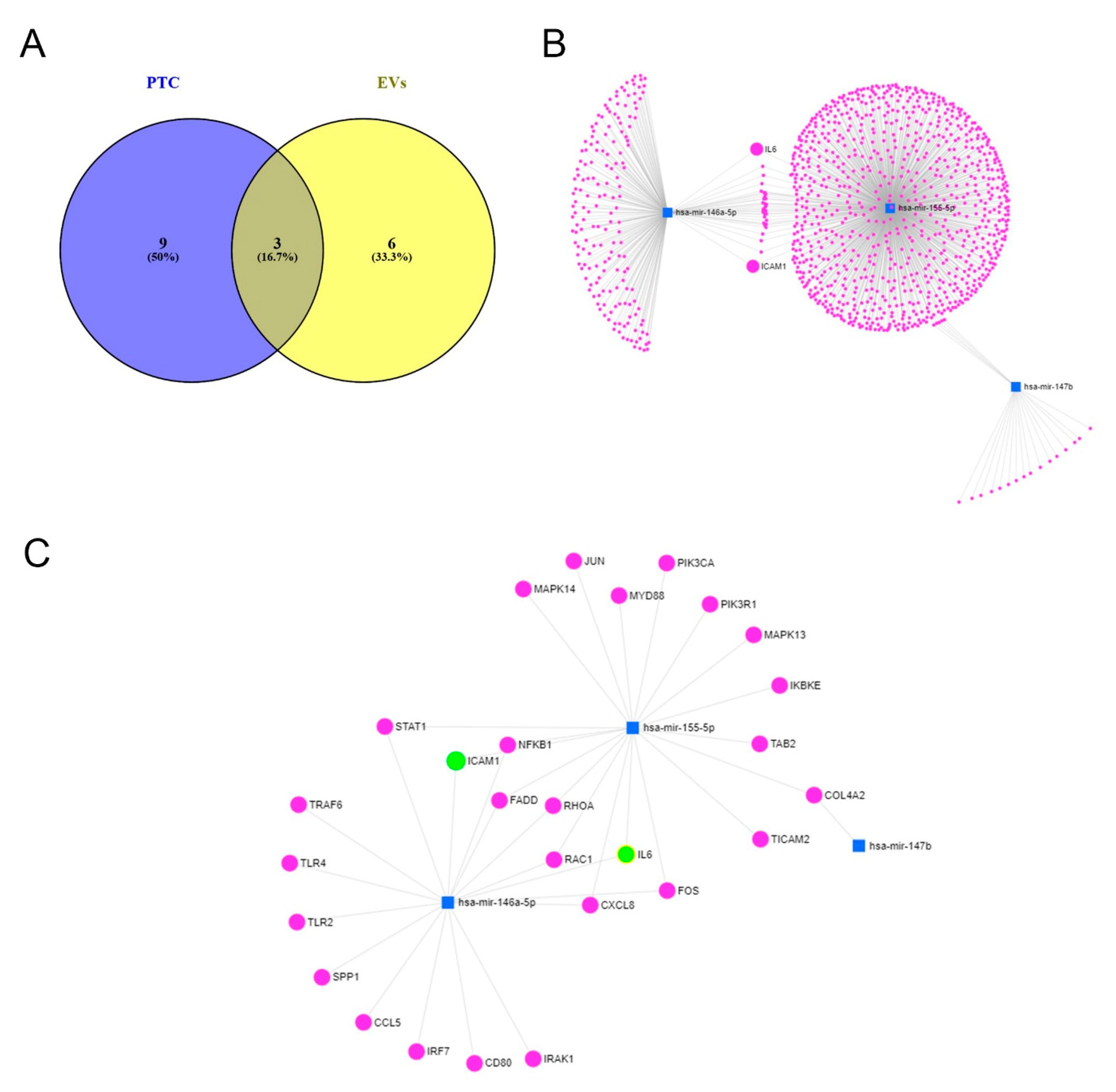
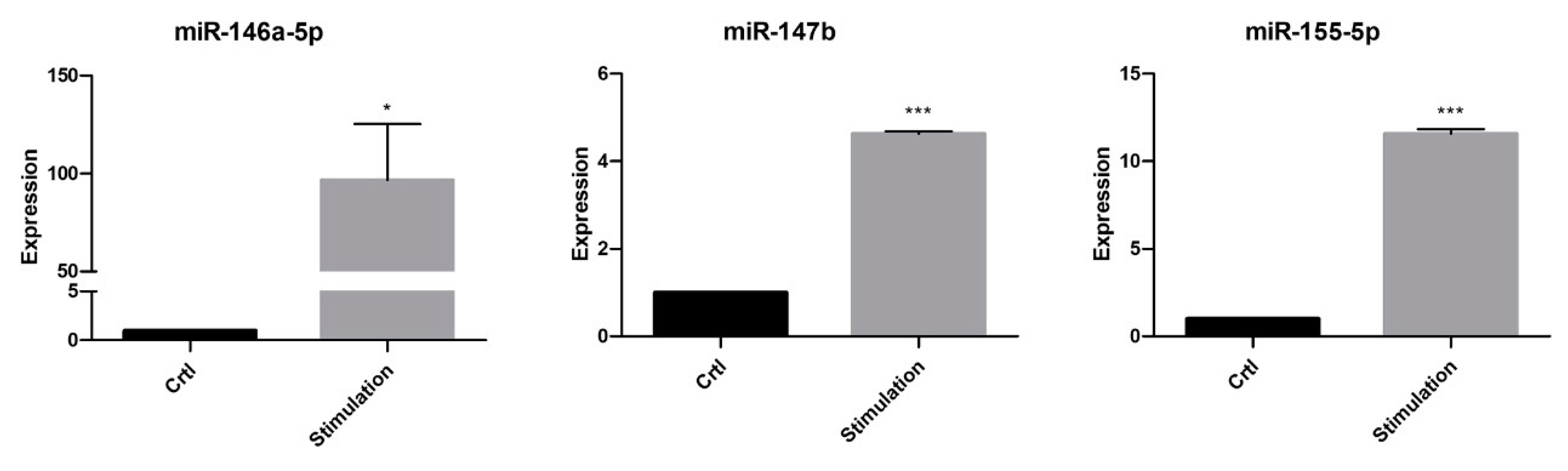
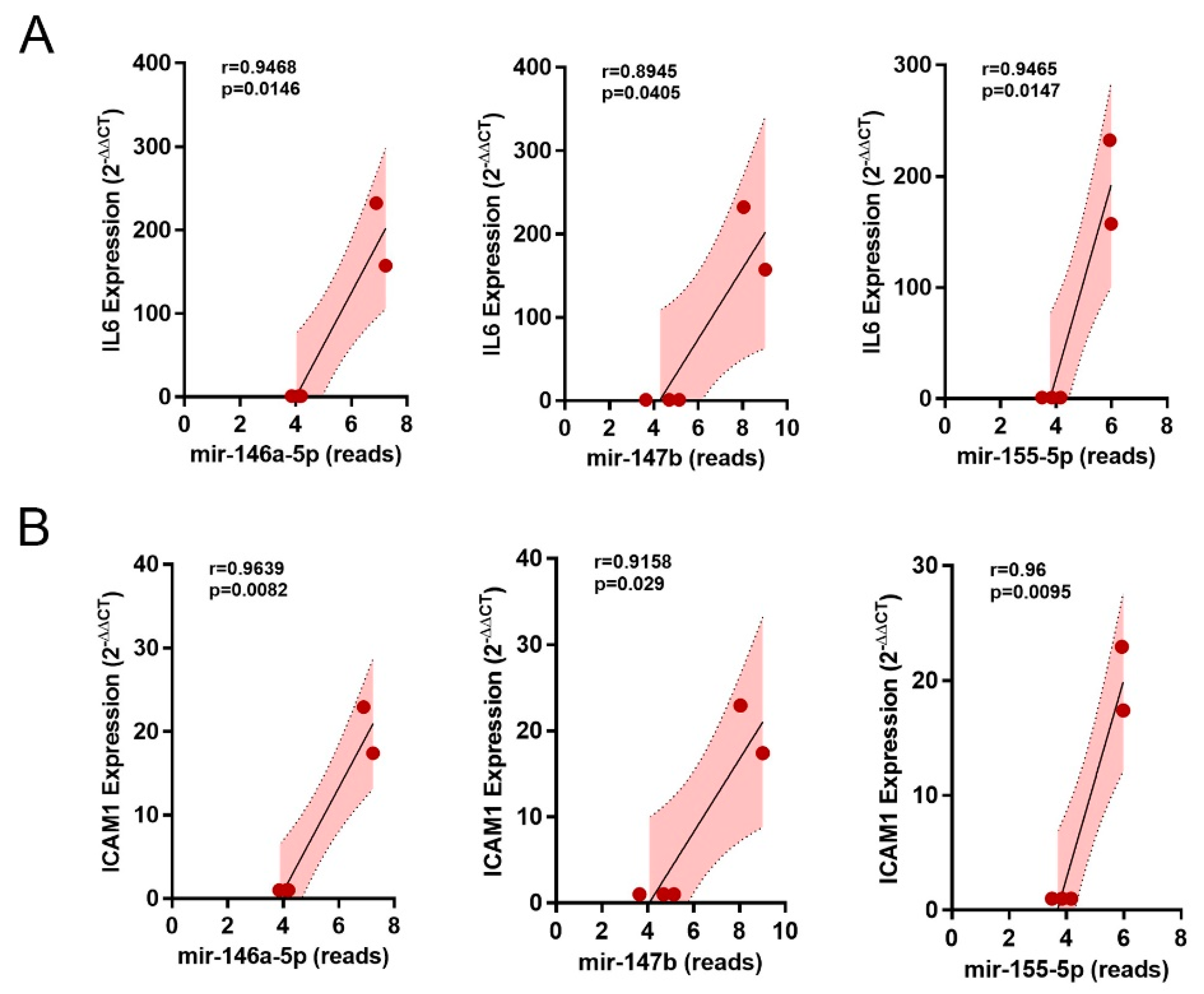
2. Results
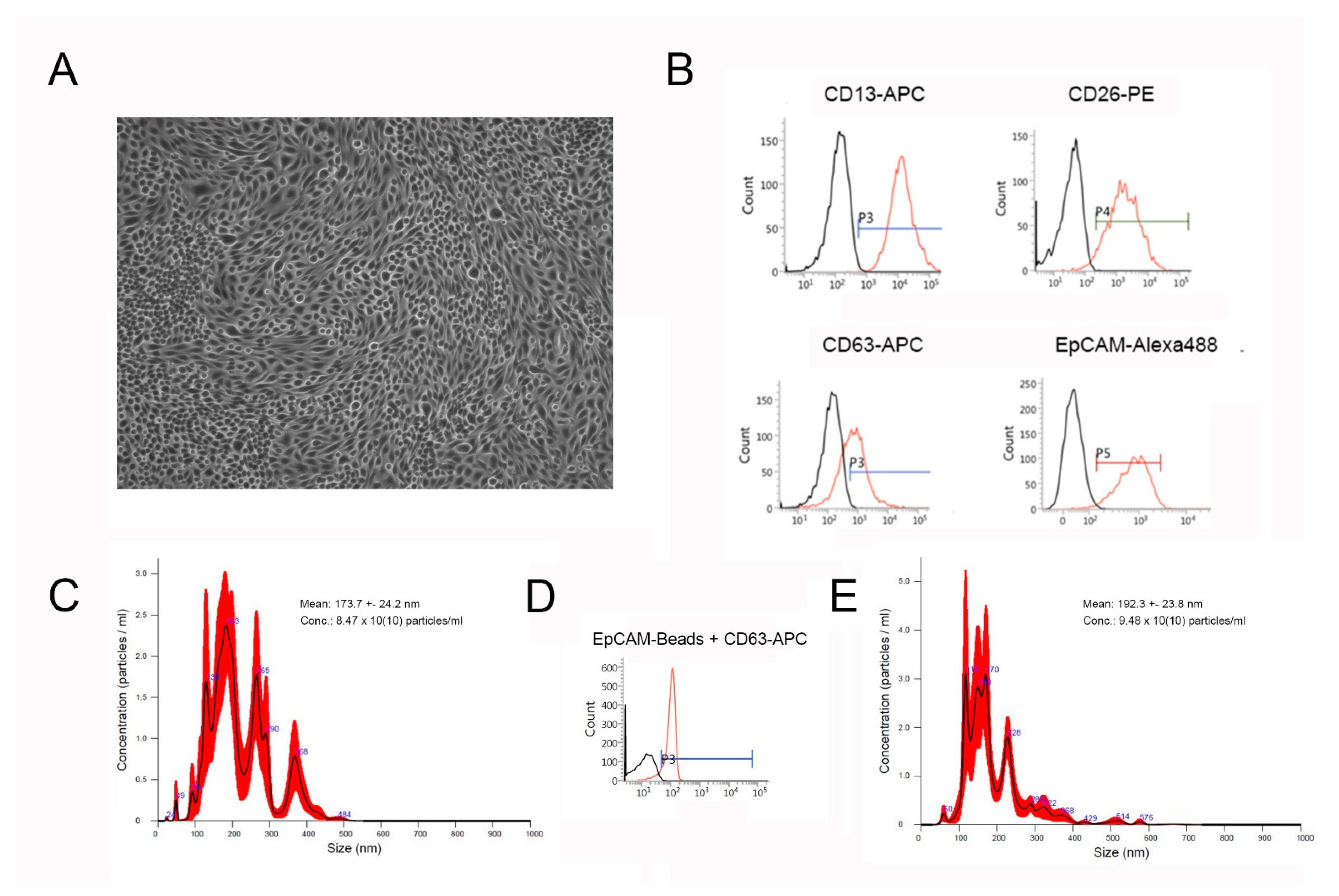
2.1. Characterization of PTCs and Their EVs
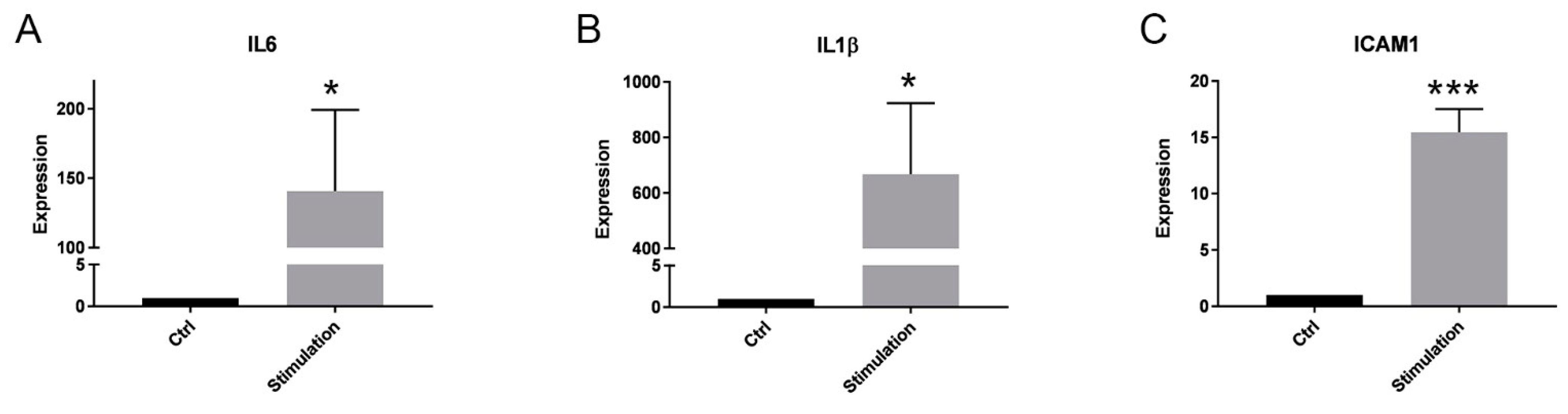
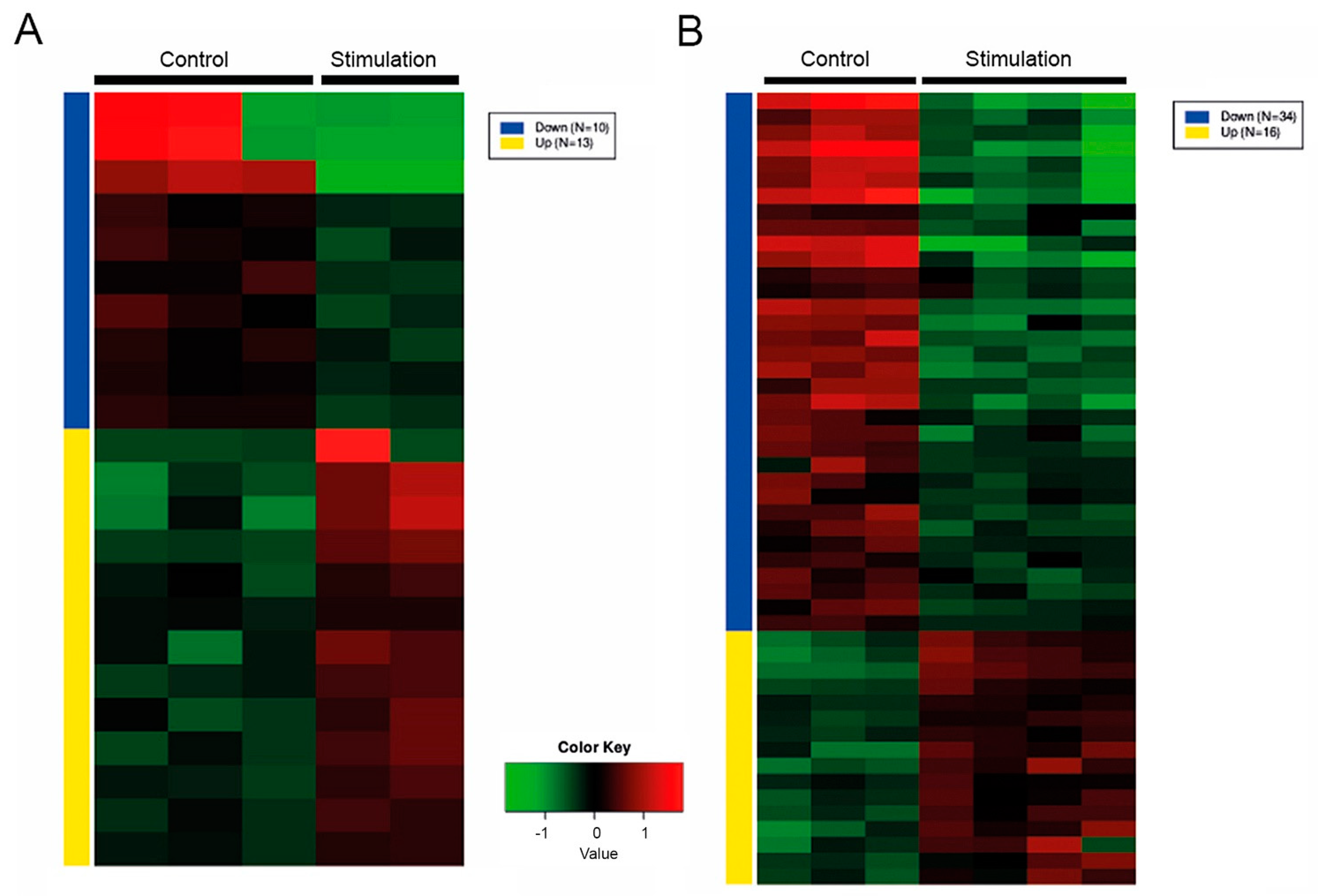
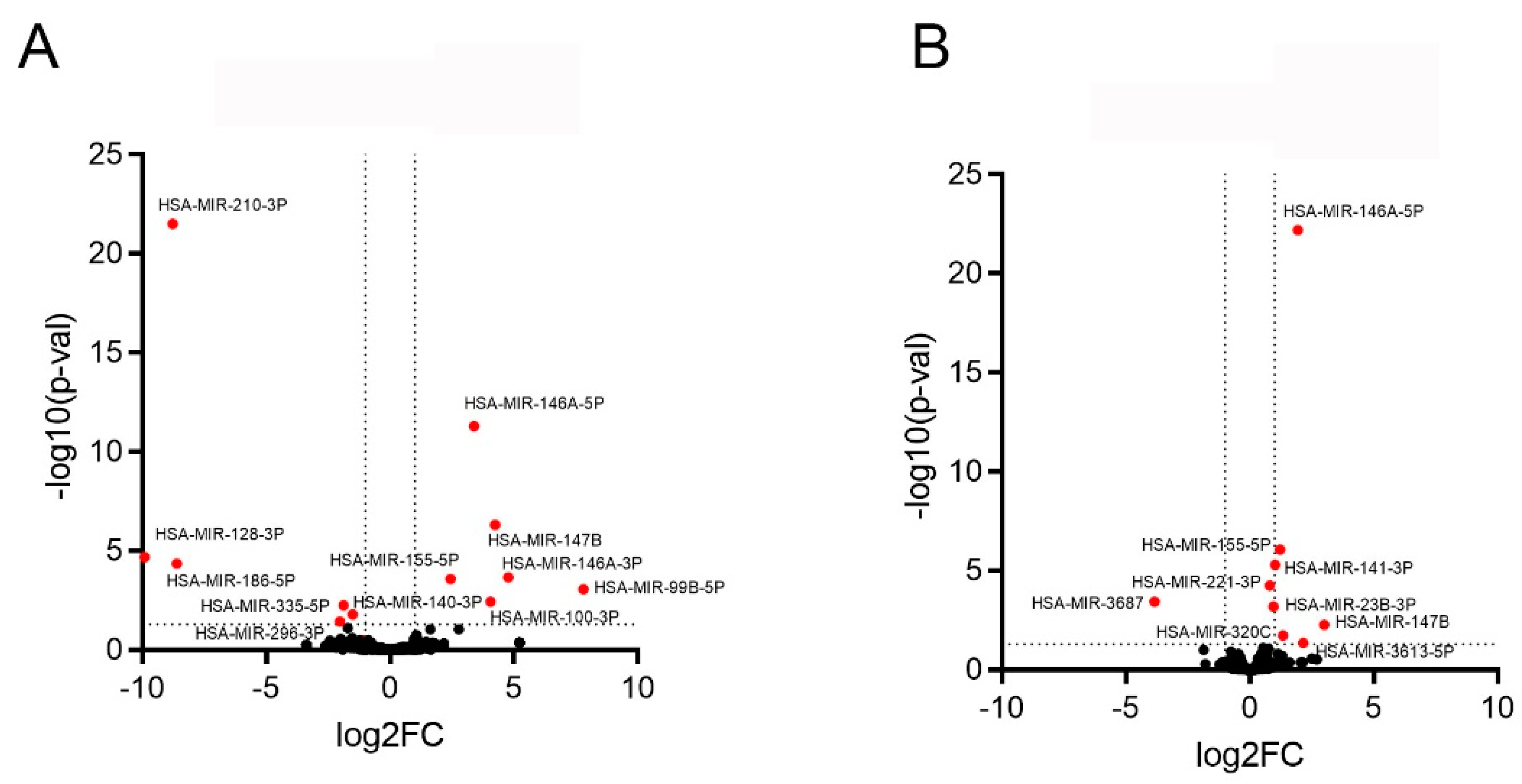
2.2. Effect of Inflammation on the miR Expression in PTC and Their EVs
3. Discussion
4. Materials and Methods
4.1. Isolation and Culture of Human Renal Proximal Tubular Epithelial Cells
4.2. Preconditioning in an Inflammatory Microenvironment
4.3. Isolation of Extracellular Vesicles
4.4. Characterization of Extracellular Vesicles
4.5. Isolation of miR from PTC
4.6. Isolation of miR from EVs
4.7. miR Sequencing
4.8. Validation of miR Expression
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schlondorff, D.O. Overview of factors contributing to the pathophysiology of progressive renal disease. Kidney Int. 2008, 74, 860–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliam, S.J.; Wright, R.D.; Welsh, G.I.; Tuffin, J.; Budge, K.L.; Swan, L.; Wilm, T.; Martinas, I.-R.; Littlewood, J.; Oni, L. The complex interplay between kidney injury and inflammation. Clin. Kidney J. 2021, 14, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V. The proximal tubule in the pathophysiology of the diabetic kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1009–R1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade-Oliveira, V.; Foresto-Neto, O.; Watanabe, I.K.M.; Zatz, R.; Câmara, N.O.S. Inflammation in Renal Diseases: New and Old Players. Front. Pharmacol. 2019, 10, 1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wilkinson, R.; Kildey, K.; Ungerer, J.P.J.; Hill, M.M.; Shah, A.K.; Mohamed, A.; Dutt, M.; Molendijk, J.; Healy, H.; et al. Molecular and functional profiling of apical versus basolateral small extracellular vesicles derived from primary human proximal tubular epithelial cells under inflammatory conditions. J. Extracell. Vesicles 2021, 10, e12064. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Battistelli, M.; Falcieri, E. Apoptotic Bodies: Particular Extracellular Vesicles Involved in Intercellular Communication. Biology 2020, 9, 21. [Google Scholar] [CrossRef] [Green Version]
- Tkach, M.; Théry, C. Communication by Extracellular Vesicles: Where We Are and Where We Need to Go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.S.; Kim, E.; Bae, Y.-U.; Yang, W.M.; Lee, H.; Kim, H.; Noh, H.; Han, D.C.; Ryu, S.; Kwon, S.H. microRNA in Extracellular Vesicles Released by Damaged Podocytes Promote Apoptosis of Renal Tubular Epithelial Cells. Cells 2020, 9, 1409. [Google Scholar] [CrossRef]
- Chung, A.C.-K.; Lan, H.Y. MicroRNAs in renal fibrosis. Front. Physiol. 2015, 6, 50. [Google Scholar] [CrossRef] [Green Version]
- Khoury, S.; Tran, N. Circulating microRNAs: Potential biomarkers for common malignancies. Biomark. Med. 2015, 9, 131–151. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Wei, S.; Zhou, C.; Yu, J.; Wang, G.; Wang, W.; Zhao, L. Extracellular vesicles isolated by size-exclusion chromatography present suitability for RNomics analysis in plasma. J. Transl. Med. 2021, 19, 104. [Google Scholar] [CrossRef] [PubMed]
- Daha, M.R.; van Kooten, C. Is the proximal tubular cell a proinflammatory cell? Nephrol. Dial. Transpl. 2000, 15 (Suppl. S6), 41–43. [Google Scholar] [CrossRef]
- Wang, X.; Wilkinson, R.; Kildey, K.; Potriquet, J.; Mulvenna, J.; Lobb, R.J.; Möller, A.; Cloonan, N.; Mukhopadhyay, P.; Kassianos, A.J.; et al. Unique molecular profile of exosomes derived from primary human proximal tubular epithelial cells under diseased conditions. J. Extracell. Vesicles 2017, 6, 1314073. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.; Hu, R.; Runtsch, M.C.; Kagele, D.A.; Mosbruger, T.L.; Tolmachova, T.; Seabra, M.C.; Round, J.L.; Ward, D.M.; O’Connell, R.M. Exosome-delivered microRNAs modulate the inflammatory response to endotoxin. Nat. Commun. 2015, 6, 7321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Wang, Y.; Shu, S.; Cai, J.; Tang, C.; Dong, Z. Non-coding RNAs in kidney injury and repair. Am. J. Physiol. Cell Physiol. 2019, 317, C177–C188. [Google Scholar] [CrossRef]
- Ranganathan, P.; Jayakumar, C.; Tang, Y.; Park, K.; Teoh, J.; Su, H.; Li, J.; Kim, I.; Ramesh, G. MicroRNA-150 deletion in mice protects kidney from myocardial infarction-induced acute kidney injury. Am. J. Physiol. Renal Physiol. 2015, 309, F551–F558. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, K.; Wei, Q.; Pabla, N.; Dong, G.; Mi, Q.-S.; Liang, M.; Mei, C.; Dong, Z. MicroRNA-687 Induced by Hypoxia-Inducible Factor-1 Targets Phosphatase and Tensin Homolog in Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. 2015, 26, 1588–1596. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Shu, L. Upregulation of miR-21 by Ghrelin Ameliorates Ischemia/Reperfusion-Induced Acute Kidney Injury by Inhibiting Inflammation and Cell Apoptosis. DNA Cell Biol. 2016, 35, 417–425. [Google Scholar] [CrossRef]
- Joo, M.S.; Lee, C.G.; Koo, J.H.; Kim, S.G. miR-125b transcriptionally increased by Nrf2 inhibits AhR repressor, which protects kidney from cisplatin-induced injury. Cell Death Dis. 2013, 4, e899. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, K.L.; Han, T.; Bijol, V.; Saikumar, J.; Craciun, F.L.; Chen, W.W.; Fuscoe, J.C.; Vaidya, V.S. MicroRNA-155 deficient mice experience heightened kidney toxicity when dosed with cisplatin. Toxicol. Sci. 2014, 141, 484–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattai, P.P.; Maquigussa, E.; Da Silva Novaes, A.; Da Silva Ribeiro, R.; Varela, V.A.; Ormanji, M.S.; Boim, M.A. miR-26a modulates HGF and STAT3 effects on the kidney repair process in a glycerol-induced AKI model in rats. J. Cell. Biochem. 2018, 119, 7757–7766. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.-L.; Feng, Y.; Tang, T.-T.; Liu, B.-C. New insight into the role of extracellular vesicles in kidney disease. J. Cell. Mol. Med. 2019, 23, 731–739. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Hu, K. MiR-147: Functions and Implications in Inflammation and Diseases. Microrna 2021, 10, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Nahid, M.A.; Satoh, M.; Chan, E.K. MicroRNA in TLR signaling and endotoxin tolerance. Cell. Mol. Immunol. 2011, 8, 388–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trung, N.T.; Lien, T.T.; Sang, V.V.; Hoan, N.X.; Manh, N.D.; Thau, N.S.; Quyen, D.T.; Hien, T.T.T.; Hoan, P.Q.; Bang, M.H.; et al. Circulating miR-147b as a diagnostic marker for patients with bacterial sepsis and septic shock. PLoS ONE 2021, 16, e0261228. [Google Scholar] [CrossRef]
- Xu, Q.; Huang, G.-D.; Duan, G.-C.; Qin, H.-J. MicroRNA-147b alleviates inflammation and apoptosis in acute lung injury via inhibition of p38 MAPK signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1974–1981. [Google Scholar] [CrossRef]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-kappaB signaling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef]
- Feng, L.-L.; Xin, W.-N.; Tian, X.-L. MALAT1 modulates miR-146’s protection of microvascular endothelial cells against LPS-induced NF-κB activation and inflammatory injury. Innate Immun. 2019, 25, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Zhang, J.; Liu, J.; Guo, J.; Wei, Y.; Li, J.; He, P.; Lan, T.; Peng, L.; Li, H. MicroRNA miR-155-5p knockdown attenuates Angiostrongylus cantonensis-induced eosinophilic meningitis by downregulating MMP9 and TSLP proteins. Int. J. Parasitol. 2021, 51, 13–22. [Google Scholar] [CrossRef]
- Mahesh, G.; Biswas, R. MicroRNA-155: A Master Regulator of Inflammation. J. Interferon Cytokine Res. 2019, 39, 321–330. [Google Scholar] [CrossRef]
- Mortazavi-Jahromi, S.S.; Aslani, M.; Mirshafiey, A. A comprehensive review on miR-146a molecular mechanisms in a wide spectrum of immune and non-immune inflammatory diseases. Immunol. Lett. 2020, 227, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Huang, S.; Liu, X.; Zhang, Y.; Wei, S.; Hu, X. miR-155: An Important Role in Inflammation Response. J. Immunol. Res. 2022, 2022, 7437281. [Google Scholar] [CrossRef]
- Mann, M.; Mehta, A.; Zhao, J.L.; Lee, K.; Marinov, G.K.; Garcia-Flores, Y.; Lu, L.-F.; Rudensky, A.Y.; Baltimore, D. An NF-κB-microRNA regulatory network tunes macrophage inflammatory responses. Nat. Commun. 2017, 8, 851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saikumar, J.; Hoffmann, D.; Kim, T.-M.; Gonzalez, V.R.; Zhang, Q.; Goering, P.L.; Brown, R.P.; Bijol, V.; Park, P.J.; Waikar, S.S.; et al. Expression, circulation, and excretion profile of microRNA-21, -155, and -18a following acute kidney injury. Toxicol. Sci. 2012, 129, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.-B.; Chen, X.; Li, D.-J.; Qi, G.-N.; Dai, Y.-Q.; Gu, J.; Chen, M.-Q.; Hu, S.; Liu, Z.-Y.; Yang, Z.-M. Inhibition of miR-155 Ameliorates Acute Kidney Injury by Apoptosis Involving the Regulation on TCF4/Wnt/β-Catenin Pathway. Nephron 2019, 143, 135–147. [Google Scholar] [CrossRef]
- Fu, H.-X.; Fan, X.-P.; Li, M.; Liu, M.-J.; Sun, Q.-L. MiR-146a relieves kidney injury in mice with systemic lupus erythematosus through regulating NF-κB pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7024–7032. [Google Scholar] [CrossRef]
- Yu, H.-Y.; Meng, L.-F.; Lu, X.-H.; Liu, L.-H.; Ci, X.; Zhuo, Z. Protective effect of miR-146 against kidney injury in diabetic nephropathy rats through mediating the NF-κB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 6445. [Google Scholar] [CrossRef]
- Zhu, J.; Xiang, X.; Hu, X.; Li, C.; Song, Z.; Dong, Z. miR-147 Represses NDUFA4, Inducing Mitochondrial Dysfunction and Tubular Damage in Cold-storage Kidney Transplantation. J. Am. Soc. Nephrol. 2023; Online ahead of print. [Google Scholar] [CrossRef]
- Baer, P.C.; Nockher, W.A.; Haase, W.; Scherberich, J.E. Isolation of proximal and distal tubule cells from human kidney by immunomagnetic separation. Technical note. Kidney Int. 1997, 52, 1321–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, P.C.; Bereiter-Hahn, J.; Schubert, R.; Geiger, H. Differentiation status of human renal proximal and distal tubular epithelial cells in vitro: Differential expression of characteristic markers. Cells Tissues Organs (Print) 2006, 184, 16–22. [Google Scholar] [CrossRef]
- Koch, B.; Geßner, A.; Farmand, S.; Fuhrmann, D.C.; Chiocchetti, A.G.; Schubert, R.; Baer, P.C. Effects of Hypoxia on RNA Cargo in Extracellular Vesicles from Human Adipose-Derived Stromal/Stem Cells. Int. J. Mol. Sci. 2022, 23, 7384. [Google Scholar] [CrossRef] [PubMed]
- Mol, E.A.; Goumans, M.-J.; Doevendans, P.A.; Sluijter, J.P.G.; Vader, P. Higher functionality of extracellular vesicles isolated using size-exclusion chromatography compared to ultracentrifugation. Nanomedicine 2017, 13, 2061–2065. [Google Scholar] [CrossRef]
- Böing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.W.; Sturk, A.; Nieuwland, R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles 2014, 3, 23430. [Google Scholar] [CrossRef] [PubMed]
- Ge, X. iDEP Web Application for RNA-Seq Data Analysis. Methods Mol. Biol. 2021, 2284, 417–443. [Google Scholar] [CrossRef]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 22 May 2023).
- Faraldi, M.; Gomarasca, M.; Sansoni, V.; Perego, S.; Banfi, G.; Lombardi, G. Normalization strategies differently affect circulating miRNA profile associated with the training status. Sci. Rep. 2019, 9, 1584. [Google Scholar] [CrossRef] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baer, P.C.; Neuhoff, A.-K.; Schubert, R. microRNA Expression of Renal Proximal Tubular Epithelial Cells and Their Extracellular Vesicles in an Inflammatory Microenvironment In Vitro. Int. J. Mol. Sci. 2023, 24, 11069. https://doi.org/10.3390/ijms241311069
Baer PC, Neuhoff A-K, Schubert R. microRNA Expression of Renal Proximal Tubular Epithelial Cells and Their Extracellular Vesicles in an Inflammatory Microenvironment In Vitro. International Journal of Molecular Sciences. 2023; 24(13):11069. https://doi.org/10.3390/ijms241311069
Chicago/Turabian StyleBaer, Patrick C., Ann-Kathrin Neuhoff, and Ralf Schubert. 2023. "microRNA Expression of Renal Proximal Tubular Epithelial Cells and Their Extracellular Vesicles in an Inflammatory Microenvironment In Vitro" International Journal of Molecular Sciences 24, no. 13: 11069. https://doi.org/10.3390/ijms241311069