
Structural Modifications Introduced by NS2B Cofactor Binding to the NS3 Protease of the Kyasanur Forest Disease Virus

Abstract
:1. Introduction
2. Results
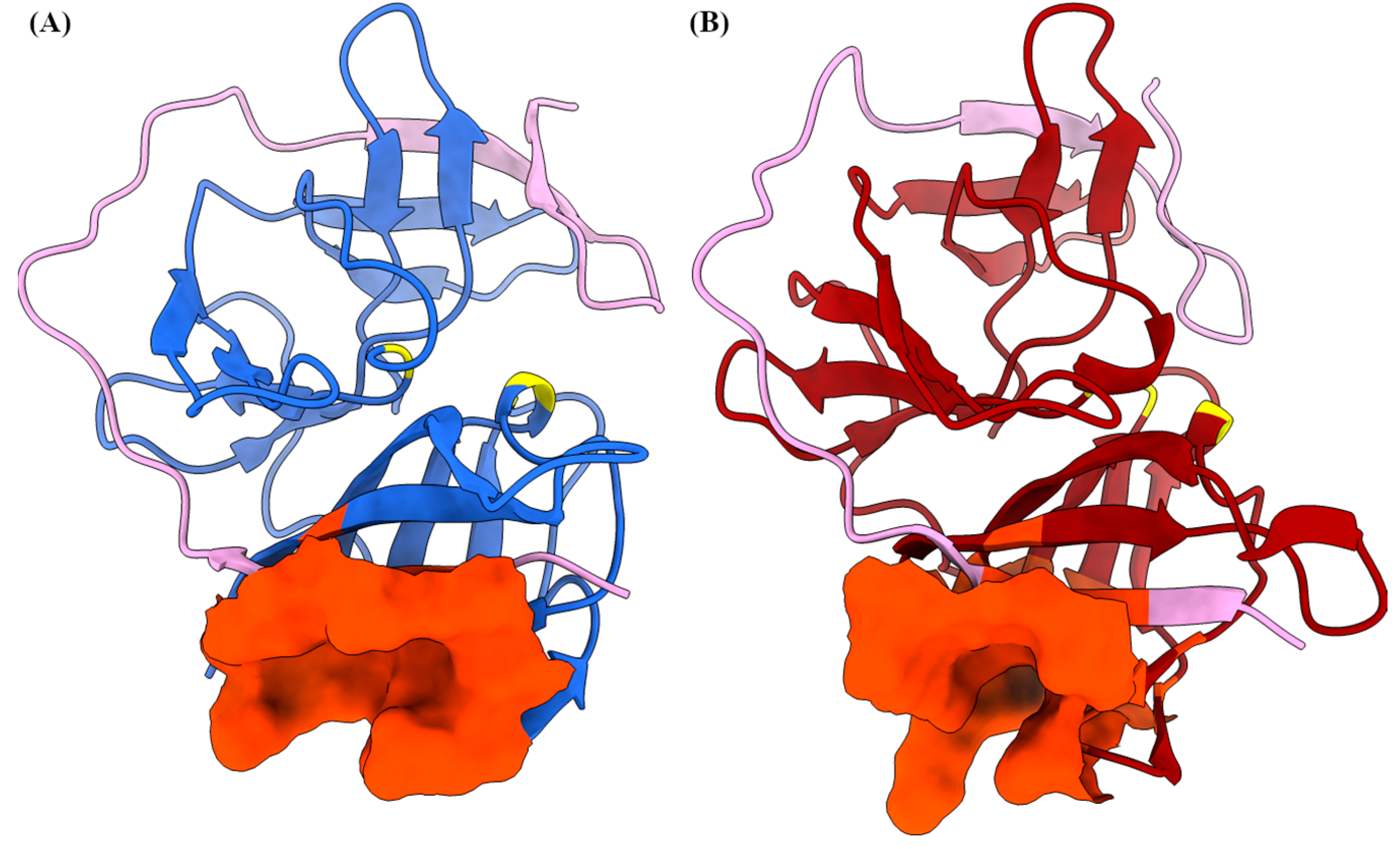
2.1. AlphaFold Predicts a Reliable Model of the NS2B/NS3
2.2. NS2B Cofactor Binding Reduces NS3 Flexibility
2.3. Clustering Identifies the Conformations of NS2B/NS3 and NS3
2.4. PCA Shows Large-Amplitude Motion Changes after Cofactor Binding
2.5. Potential Druggable Pockets
2.6. Allosteric Binding Sites—Hit Molecules Identification
3. Discussion
4. Materials and Methods
4.1. Prediction of the Structure of the NS2B/NS3 Protease with AlphaFold
4.2. Molecular Dynamics (MD) Simulation
4.3. Principal Component Analysis (PCA)
4.4. Clustering Analysis
4.5. Identification of Potential Druggable Pockets
4.6. Allosteric Binding Sites—Molecular Docking and MD Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Work, T.H.; Trapido, H. Summary of Preliminary Report of Investigations of the Virus Research Centre on an Epidemic Disease Affecting Forest Villagers and Wild Monkeys of Shimoga District, Mysore. Indian J. Med. Sci. 1957, 11, 341–342. [Google Scholar] [PubMed]
- Bhatt, P.N.; Work, T.H.; Varma, M.G.; Trapido, H.; Murthy, D.P.; Rodrigues, F.M. Kyasanur Forest Diseases. IV. Isolation of Kyasanur Forest Disease Virus from Infected Humans and Monkeys of Shimogadistrict, Mysore State. Indian J. Med. Sci. 1996, 20, 316–320. [Google Scholar]
- Holbrook, M.R. Kyasanur Forest Disease. Antiviral Res. 2012, 96, 353–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhaka, R.; Verma, R.; Kumar, R.; Dhankar, M.; Bhalla, K.; Agrawal, G. Kyasanur Forest Disease: A Rare Viral Hemorrhagic Disease in India. Int. J. Community Med. Public Health 2018, 5, 3149–3151. [Google Scholar] [CrossRef] [Green Version]
- Trapido, H.; Rajagopalan, P.K.; Work, T.H.; Varma M G Kyasanur Forest Disease. VIII. Isolation of Kyasanur Forest Disease Virus from Naturally Infected Ticks of the Genus Haemaphysalis. Indian J. Med. Res. 1959, 47, 133–138. [Google Scholar]
- Sreenivasan, M.A.; Bhat, H.R.; Naik, S.V. Experimental Transmission of Kyasanur Forest Disease Virus by Dermacentor Auratus Supino. Indian J. Med. Res. 1979, 69, 701–707. [Google Scholar]
- Mourya, D.; Yadav, P.; Patil, D. Expediency of Dengue Illness Classification: The Sri Lankan Perspective Highly Infectious Tick-Borne Viral Diseases: Kyasanur Forest Disease and Crimean-Congo Haemorrhagic Fever in India. WHO South East Asia J. Public Health 2014, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Rajaiah, P. Kyasanur Forest Disease in India: Innovative Options for Intervention. Hum. Vaccin. Immunother. 2019, 15, 2243. [Google Scholar] [CrossRef]
- Stone, R. Monkey Fever Unbound. Science 2014, 345, 130–133. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, H.; Fu, S.; Wang, H.; Ni, D.; Nasci, R.; Tang, Q.; Liang, G. Isolation of Kyasanur Forest Disease Virus from Febrile Patient, Yunnan, China. Emerg. Infect. Dis. 2009, 15, 326–328. [Google Scholar] [CrossRef]
- Dodd, K.A.; Bird, B.H.; Khristova, M.L.; Albariño, C.G.; Carroll, S.A.; Comer, J.A.; Erickson, B.R.; Rollin, P.E.; Nichol, S.T. Ancient Ancestry of KFDV and AHFV Revealed by Complete Genome Analyses of Viruses Isolated from Ticks and Mammalian Hosts. PLoS Negl. Trop. Dis. 2011, 5, e1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazan, J.F.; Fletterick, R.J. Detection of a Trypsin-like Serine Protease Domain in Flaviviruses and Pestiviruses. Virology 1989, 171, 637–639. [Google Scholar] [CrossRef]
- Li, H.; Clum, S.; You, S.; Ebner, K.E.; Padmanabhan, R. The Serine Protease and RNA-Stimulated Nucleoside Triphosphatase and RNA Helicase Functional Domains of Dengue Virus Type 2 NS3 Converge within a Region of 20 Amino Acids. J. Virol. 1999, 73, 3108–3116. [Google Scholar] [CrossRef] [Green Version]
- Warrener, P.; Tamura, J.K.; Collett, M.S. RNA-Stimulated NTPase Activity Associated with Yellow Fever Virus NS3 Protein Expressed in Bacteria. J. Virol. 1993, 67, 989–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wengler, G.; Wengler, G. The Carboxy-Terminal Part of the NS 3 Protein of the West Nile Flavivirus Can Be Isolated as a Soluble Protein after Proteolytic Cleavage and Represents an RNA-Stimulated NTPase. Virology 1991, 184, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.F.; Preugschat, F.; Strass, J.H. Dengue 2 Virus Ns2b and Ns3 Form a Stable Complex That Can Cleave Ns3 within the Helicase Domain. Virology 1993, 193, 888–899. [Google Scholar] [CrossRef]
- Chambers, T.J.; Nestorowicz, A.; Amberg, S.M.; Rice, C.M. Mutagenesis of the Yellow Fever Virus NS2B Protein: Effects on Proteolytic Processing, NS2B-NS3 Complex Formation, and Viral Replication. J. Virol. 1993, 67, 6797–6807. [Google Scholar] [CrossRef] [Green Version]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus Genome Organization, Expression, and Replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef]
- Gouvea, I.E.; Izidoro, M.A.; Judice, W.A.S.; Cezari, M.H.S.; Caliendo, G.; Santagada, V.; dos Santos, C.N.D.; Queiroz, M.H.; Juliano, M.A.; Young, P.R.; et al. Substrate Specificity of Recombinant Dengue 2 Virus NS2B-NS3 Protease: Influence of Natural and Unnatural Basic Amino Acids on Hydrolysis of Synthetic Fluorescent Substrates. Arch. Biochem. Biophys. 2007, 457, 187–196. [Google Scholar] [CrossRef]
- Aleshin, A.E.; Shiryaev, S.A.; Strongin, A.Y.; Liddington, R.C. Structural Evidence for Regulation and Specificity of Flaviviral Proteases and Evolution of the Flaviviridae Fold. Protein Sci. 2007, 16, 795. [Google Scholar] [CrossRef] [Green Version]
- Assenberg, R.; Mastrangelo, E.; Walter, T.S.; Verma, A.; Milani, M.; Owens, R.J.; Stuart, D.I.; Grimes, J.M.; Mancini, E.J. Crystal Structure of a Novel Conformational State of the Flavivirus NS3 Protein: Implications for Polyprotein Processing and Viral Replication. J. Virol. 2009, 83, 12895–12906. [Google Scholar] [CrossRef] [Green Version]
- Chandramouli, S.; Joseph, J.S.; Daudenarde, S.; Gatchalian, J.; Cornillez-Ty, C.; Kuhn, P. Serotype-Specific Structural Differences in the Protease-Cofactor Complexes of the Dengue Virus Family. J. Virol. 2010, 84, 3059. [Google Scholar] [CrossRef] [Green Version]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural Basis for the Activation of Flaviviral NS3 Proteases from Dengue and West Nile Virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L. Molecular Basis of Human Immunodeficiency Virus Drug Resistance: An Update. Antivir. Res. 2010, 85, 210–231. [Google Scholar] [CrossRef]
- Novak, J.; Pathak, P.; Grishina, M.A.; Potemkin, V.A. The Design of Compounds with Desirable Properties—The AntiHIV Case Study. J. Comput. Chem. 2023, 44, 1016–1030. [Google Scholar] [CrossRef]
- Brecher, M.; Zhang, J.; Li, H. The Flavivirus Protease as a Target for Drug Discovery. Virol. Sin. 2013, 28, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, C.G.; Shi, P.Y. Structural Biology of Dengue Virus Enzymes: Towards Rational Design of Therapeutics. Antiviral Res. 2012, 96, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Sampath, A.; Padmanabhan, R. Molecular Targets for Flavivirus Drug Discovery. Antivir. Res. 2009, 81, 6. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.P.; Wang, Q.Y.; Noble, C.G.; Chen, Y.L.; Dong, H.; Zou, B.; Yokokawa, F.; Nilar, S.; Smith, P.; Beer, D.; et al. Ten Years of Dengue Drug Discovery: Progress and Prospects. Antivir. Res. 2013, 100, 500–519. [Google Scholar] [CrossRef]
- Luo, D.; Vasudevan, S.G.; Lescar, J. The Flavivirus NS2B-NS3 Protease-Helicase as a Target for Antiviral Drug Development. Antivir. Res. 2015, 118, 148–158. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, J.; Li, H. Flavivirus NS2B/NS3 Protease: Structure, Function, and Inhibition. In Viral Proteases and Their Inhibitors; Elsevier: Amsterdam, The Netherlands, 2017; pp. 163–188. ISBN 9780128097120. [Google Scholar]
- Falgout, B.; Pethel, M.; Zhang, Y.M.; Lai, C.J. Both Nonstructural Proteins NS2B and NS3 Are Required for the Proteolytic Processing of Dengue Virus Nonstructural Proteins. J. Virol. 1991, 65, 2467. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Mohan, P.M.; Padmanabhan, R. Processing and Localization of Dengue Virus Type 2 Polyprotein Precursor NS3-NS4A-NS4B-NS5. J. Virol. 1992, 66, 7549. [Google Scholar] [CrossRef] [Green Version]
- Lim, L.; Gupta, G.; Roy, A.; Kang, J.; Srivastava, S.; Shi, J.; Song, J. Structurally- and Dynamically-Driven Allostery of the Chymotrypsin-like Proteases of SARS, Dengue and Zika Viruses. Prog. Biophys. Mol. Biol. 2019, 143, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Clum, S.; Ebner, K.E.; Padmanabhan, R. Cotranslational Membrane Insertion of the Serine Proteinase Precursor NS2B-NS3(Pro) of Dengue Virus Type 2 Is Required for Efficient in Vitro Processing and Is Mediated through the Hydrophobic Regions of NS2B. J. Biol. Chem. 1997, 272, 30715–30723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robin, G.; Chappell, K.; Stoermer, M.J.; Hu, S.H.; Young, P.R.; Fairlie, D.P.; Martin, J.L. Structure of West Nile Virus NS3 Protease: Ligand Stabilization of the Catalytic Conformation. J. Mol. Biol. 2009, 385, 1568–1577. [Google Scholar] [CrossRef]
- Gupta, G.; Lim, L.; Song, J. NMR and MD Studies Reveal That the Isolated Dengue NS3 Protease Is an Intrinsically Disordered Chymotrypsin Fold Which Absolutely Requests NS2B for Correct Folding and Functional Dynamics. PLoS ONE 2015, 10, e0134823. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Roe, D.R.; Swails, J.; Case, D.A. PYTRAJ v1.0.0.Dev1: Interactive Data Analysis for Molecular Dynamics Simulations. 2016. Available online: https://zenodo.org/record/44612 (accessed on 20 August 2022). [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Kronenberger, T.; Sá Magalhães Serafim, M.; Kumar Tonduru, A.; Gonçalves Maltarollo, V.; Poso, A. Ligand Accessibility Insights to the Dengue Virus NS3-NS2B Protease Assessed by Long-Timescale Molecular Dynamics Simulations. Chem. Med. Chem. 2021, 16, 2524–2534. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [Green Version]
- Tomar, S.; Mudgal, R.; Fatma, B. Flavivirus Protease: An Antiviral Target. In Viral Proteases and Their Inhibitors; Elsevier: Amsterdam, The Netherlands, 2017; pp. 137–161. ISBN 9780128097120. [Google Scholar]
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining Global and Local Measures for Structure-Based Druggability Predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Brecher, M.; Deng, Y.Q.; Zhang, J.; Sakamuru, S.; Liu, B.; Huang, R.; Koetzner, C.A.; Allen, C.A.; Jones, S.A.; et al. Existing Drugs as Broad-Spectrum and Potent Inhibitors for Zika Virus by Targeting NS2B-NS3 Interaction. Cell Res. 2017, 27, 1046–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Sakamuru, S.; Huang, R.; Brecher, M.; Koetzner, C.A.; Zhang, J.; Chen, H.; Qin, C.F.; Zhang, Q.Y.; Zhou, J.; et al. Erythrosin B Is a Potent and Broad-Spectrum Orthosteric Inhibitor of the Flavivirus NS2B-NS3 Protease. Antivir. Res. 2018, 150, 217. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, J.; Lang, Y.; Fan, X.; Kuo, L.; D’Brant, L.; Hu, S.; Samrat, S.K.; Trudeau, N.; Tharappel, A.M.; et al. JMX0207, a niclosamide derivative with improved pharmacokinetics, suppresses Zika virus infection both in vitro and in vivo. ACS Infect. Dis. 2020, 6, 2616–2628. [Google Scholar] [CrossRef] [PubMed]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug Repurposing: Progress, Challenges and Recommendations. Nat. Rev. Drug Discov. 2018, 18, 41–58. [Google Scholar] [CrossRef]
- Achenbach, J.; Tiikkainen, P.; Franke, L.; Proschak, E. Computational Tools for Polypharmacology and Repurposing. Future Med. Chem. 2011, 3, 961–968. [Google Scholar] [CrossRef]
- Novak, J.; Potemkin, V.A. A New Glimpse on the Active Site of SARS-CoV-2 3CLpro, Coupled with Drug Repurposing Study. Mol. Divers. 2022, 26, 2631–2645. [Google Scholar] [CrossRef]
- Novak, J.; Rimac, H.; Kandagalla, S.; Pathak, P.; Naumovich, V.; Grishina, M.; Potemkin, V. Proposition of a New Allosteric Binding Site for Potential SARS-CoV-2 3CL Protease Inhibitors by Utilizing Molecular Dynamics Simulations and Ensemble Docking. J. Biomol. Struct. Dyn. 2022, 40, 9347–9360. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C. Beware of Docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Macip, G.; Garcia-Segura, P.; Mestres-Truyol, J.; Saldivar-Espinoza, B.; Ojeda-Montes, M.J.; Gimeno, A.; Cereto-Massagué, A.; Garcia-Vallvé, S.; Pujadas, G. Haste Makes Waste: A Critical Review of Docking-Based Virtual Screening in Drug Repurposing for SARS-CoV-2 Main Protease (M-pro) Inhibition. Med. Res. Rev. 2022, 42, 744–769. [Google Scholar] [CrossRef]
- Roy, A.; Lim, L.; Srivastava, S.; Lu, Y.; Song, J. Solution Conformations of Zika NS2B-NS3pro and Its Inhibition by Natural Products from Edible Plants. PLoS ONE 2017, 12, e0180632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Sousa, L.R.F.; Wu, H.; Nebo, L.; Fernandes, J.B.; Da Silva, M.F.D.G.F.; Kiefer, W.; Kanitz, M.; Bodem, J.; Diederich, W.E.; Schirmeister, T.; et al. Flavonoids as Noncompetitive Inhibitors of Dengue Virus NS2B-NS3 Protease: Inhibition Kinetics and Docking Studies. Bioorg. Med. Chem. 2015, 23, 466–470. [Google Scholar] [CrossRef]
- Othman, R.; Kiat, T.S.; Khalid, N.; Yusof, R.; Newhouse, E.I.; Newhouse, J.S.; Alam, M.; Rahman, N.A. Docking of Noncompetitive Inhibitors into Dengue Virus Type 2 Protease: Understanding the Interactions with Allosteric Binding Sites. J. Chem. Inf. Model. 2008, 48, 1582–1591. [Google Scholar] [CrossRef]
- Brecher, M.; Li, Z.; Liu, B.; Zhang, J.; Koetzner, C.A.; Alifarag, A.; Jones, S.A.; Lin, Q.; Kramer, L.D.; Li, H. A Conformational Switch High-Throughput Screening Assay and Allosteric Inhibition of the Flavivirus NS2B-NS3 Protease. PLoS Pathog. 2017, 13, e1006411. [Google Scholar] [CrossRef] [Green Version]
- Millies, B.; Von Hammerstein, F.; Gellert, A.; Hammerschmidt, S.; Barthels, F.; Göppel, U.; Immerheiser, M.; Elgner, F.; Jung, N.; Basic, M.; et al. Proline-Based Allosteric Inhibitors of Zika and Dengue Virus NS2B/NS3 Proteases. J. Med. Chem. 2019, 62, 11359–11382. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold-Making Protein Folding Accessible to All. Nat. Methods 2021, 19, 679–682. [Google Scholar] [CrossRef]
- Morris, A.L.; MacArthur, M.W.; Hutchinson, E.G.; Thornton, J.M. Stereochemical Quality of Protein Structure Coordinates. Proteins Struct. Funct. Bioinform. 1992, 12, 345–364. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Te, C.I.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. Amber 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Kandagalla, S.; Novak, J.; Shekarappa, S.B.; Grishina, M.A.; Potemkin, V.A.; Kumbar, B. Exploring Potential Inhibitors against Kyasanur Forest Disease by Utilizing Molecular Dynamics Simulations and Ensemble Docking. J. Biomol. Struct. Dyn. 2022, 40, 13547–13563. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS Biomolecular Solvation Software Suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, J.; Tanner, S.W.; Thompson, N.; Cheatham, T.E. Clustering Molecular Dynamics Trajectories: 1. Characterizing the Performance of Different Clustering Algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software News and Updates AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Li, Z.; Lang, Y.; Sakamuru, S.; Samrat, S.; Trudeau, N.; Kuo, L.; Rugenstein, N.; Tharappel, A.; D’Brant, L.; Koetzner, C.A.; et al. Methylene blue is a potent and broad-spectrum inhibitor against Zika virus in vitro and in vivo. Emerg. Microbes Infect. 2020, 9, 2404. [Google Scholar] [CrossRef]
- Li, Z.; Xu, J.; Lang, Y.; Wu, X.; Hu, S.; Samrat, S.K.; Tharappel, A.M.; Kuo, L.; Butler, D.; Song, Y.; et al. In vitro and in vivo characterization of erythrosin B and derivatives against Zika virus. Acta Pharm. Sin. B 2022, 12, 1662–1670. [Google Scholar] [CrossRef]
- Yao, Y.; Huo, T.; Lin, Y.L.; Nie, S.; Wu, F.; Hua, Y.; Wu, J.; Kneubehl, A.R.; Vogt, M.B.; Rico-Hesse, R.; et al. Discovery, X-ray Crystallography and Antiviral Activity of Allosteric Inhibitors of Flavivirus NS2B-NS3 Protease. J. Am. Chem. Soc. 2019, 141, 6832–6836. [Google Scholar] [CrossRef]
- Nie, S.; Yao, Y.; Wu, F.; Wu, X.; Zhao, J.; Hua, Y.; Wu, J.; Huo, T.; Lin, Y.L.; Kneubehl, A.R.; et al. Synthesis, Structure-Activity Relationships, and Antiviral Activity of Allosteric Inhibitors of Flavivirus NS2B-NS3 Protease. J. Med. Chem. 2021, 64, 2777–2800. [Google Scholar] [CrossRef]
- Batool, F.; Saeed, M.; Saleem, H.N.; Kirschner, L.; Bodem, J. Facile Synthesis and in Vitro Activity of N-Substituted 1,2-Benzisothiazol-3(2H)-Ones against Dengue Virus NS2BNS3 Protease. Pathogens 2021, 10, 464. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Important Structural Properties | NS2B/NS3 a | NS3 a | ||
|---|---|---|---|---|
| MolProbity | atom contacts | clash score | 4.98 (94th percentile) | 4.47 (95th percentile) |
| protein geometry | poor rotamers | 0 (0.00%) | 0 (0.00%) | |
| favored rotamers | 144 (97.96%) | 112 (97.39%) | ||
| Ramachandran outliers | 0 (0.00%) | 0 (0.00%) | ||
| Ramachandran favored | 182 (99.45%) | 146 (99.32%) | ||
| MolProbity score | 1.26 (99th percentile) | 1.22 (99th percentile) | ||
| PROCHECK | Ramachandran plot | most favored | 138 (93.2%) | 107 (91.5%) |
| additional allowed | 10 (6.8%) | 10 (8.5%) | ||
| generously allowed | 0 (0.0%) | 0 (0.0%) | ||
| disallowed | 0 (0.0%) | 0 (0.0%) | ||
| NS2B Residues | ΔGbind/kcal mol−1 | NS3 Residues | ΔGbind/kcal mol−1 |
|---|---|---|---|
| Trp12 | −8.6 | Arg44 | −7.8 |
| Leu2 | −8.4 | Tyr43 | −4.6 |
| Trp6 | −7.3 | Val80 | −4.5 |
| Val10 | −5.5 | Gln116 | −4.4 |
| Ala4 | −4.9 | Ile137 | −4.3 |
| System | Cluster | Conformation | Population | d a | csd b | RMSD against A |
|---|---|---|---|---|---|---|
| NS2B/NS3 | 1 | A | 0.636 | 2.394 | 0.285 | 0 |
| 2 | B | 0.364 | 2.622 | 0.369 | 1.859 | |
| NS3 | 1 | A | 0.637 | 3.130 | 0.530 | 0 |
| 2 | B | 0.363 | 3.226 | 0.670 | 3.537 |
| Conformation | Binding Pocket | Volume (Å3) | Surfaces (Å2) | Depth (Å) | Drug Score | Pocket Amino Acids |
|---|---|---|---|---|---|---|
| A | BP1 | 216.38 | 455.93 | 11.13 | 0.417 | Thr3, Ala4 *, Glu5, Trp6 *, Tyr43 *, Lys64, Gly65, Val80*, Val81, Asp82, Glu83 |
| BP2 | 136 | 324.37 | 7.47 | 0.238 | Gly20, Gly21, Glu22, Val23, Leu25, Pro148, Thr177, Glu179, Ala180, Val182 | |
| BP3 | 134.34 | 310.16 | 7.87 | 0.235 | Gly65, Val66, Leu79, Val81, Asp82, Ala84, Ile85, Ser86, Gly87, Tyr89, Tyr100, Gly101 | |
| BP4 | 103.62 | 315.94 | 6.84 | 0.156 | Glu22, Trp110, Gly112, Glu113, Thr114, Gln132, Pro133, Gly134, Glu135 | |
| B | BP1 | 320.19 | 533.63 | 14.34 | 0.620 | Ala4 *, Glu5, Trp6 *, Lyn39, Tyr43 *, Gly62, Ala63, Lyn64, Gly65, Val66, Leu67, Leu79, Val81, Asp82, Ser86, Tyr100, Gly101 |
| BP2 | 270.14 | 530.71 | 14.72 | 0.595 | Val66, Leu67, Hie68, Cys99, Tyr100, Gly102, Ala103, Trp104, Ser105, Leu106, Glu107, Ser108, Arg109, Gln187 | |
| BP3 | 259.14 | 484.09 | 11.22 | 0.450 | Glu9, Val10, Glu11, Asp40, Gly41, Tyr61, Gly62, Ala63, Trp104, Ser105, Leu106, Glu107, Gly165, Glu166, Val167 | |
| BP4 | 179.71 | 392.03 | 9.69 | 0.309 | Val27, Arg28, Gln29, Asp30, Ala31, Gly33, Arg94, Glu95, Leu138, Glu139, Asn140, Arg142 | |
| BP5 | 122.3 | 254.17 | 6.34 | 0.183 | Gly20, Gly21, Glu22, Val23, Pro148, Thr177, Glu179, Ala180, Val182 |
| Compound Name | Structural Formula | Drug Bank ID | A | B | Score |
|---|---|---|---|---|---|
| zosuquidar |  | DB06191 | −8.4 | −6.7 | −7.8 |
| pramiconazole |  | DB06295 | −8.3 | −7.8 | −8.1 |
| 2-(4-fluorophenyl)-N-{[3-fluoro-4-(1H-pyrrolo [2,3-b]pyridin-4-yloxy)phenyl]carbamoyl}acetamide |  | DB06997 | −8.4 | −8.1 | −8.3 |
| MK-3207 |  | DB12424 | −8.8 | −8.4 | −8.7 |
| DB06191 | DB06295 | DB06997 | DB12424 | ||||
|---|---|---|---|---|---|---|---|
| Residue | ΔGbind | Residue | ΔGbind | Residue | ΔGbind | Residue | ΔGbind |
| Leu50 | −1.87 ± 0.55 | Trp6 | −2.56 ± 0.22 | Trp6 | −2.32 ± 0.09 | Trp6 | −2.89 ± 0.10 |
| His54 | −1.32 ± 0.42 | Val81 | −2.56 ± 0.22 | Thr3 | −1.42 ± 0.10 | Glu5 | −1.64 ± 0.08 |
| Pro48 | −1.28 ± 0.14 | Asp82 | −1.83 ± 0.22 | Val81 | −1.35 ± 0.22 | ||
| Lyn64 | −1.55 ± 0.22 | Thr3 | −1.20 ± 0.02 | ||||
| Val80 | −1.49 ± 0.46 | ||||||
| Glu83 | −1.40 ± 0.13 | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kandagalla, S.; Kumbar, B.; Novak, J. Structural Modifications Introduced by NS2B Cofactor Binding to the NS3 Protease of the Kyasanur Forest Disease Virus. Int. J. Mol. Sci. 2023, 24, 10907. https://doi.org/10.3390/ijms241310907
Kandagalla S, Kumbar B, Novak J. Structural Modifications Introduced by NS2B Cofactor Binding to the NS3 Protease of the Kyasanur Forest Disease Virus. International Journal of Molecular Sciences. 2023; 24(13):10907. https://doi.org/10.3390/ijms241310907
Chicago/Turabian StyleKandagalla, Shivananda, Bhimanagoud Kumbar, and Jurica Novak. 2023. "Structural Modifications Introduced by NS2B Cofactor Binding to the NS3 Protease of the Kyasanur Forest Disease Virus" International Journal of Molecular Sciences 24, no. 13: 10907. https://doi.org/10.3390/ijms241310907