
Identification of Potential Lead Compounds Targeting Novel Druggable Cavity of SARS-CoV-2 Spike Trimer by Molecular Dynamics Simulations

 , and
, and
Abstract
:
1. Introduction
2. Results and Disussion
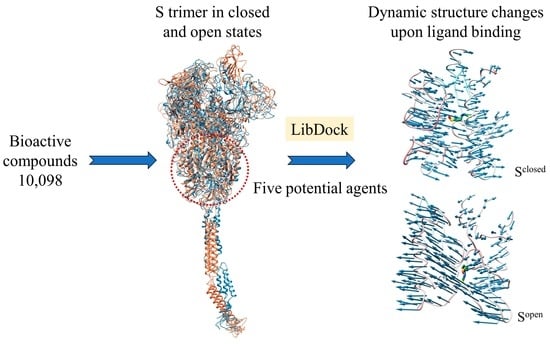
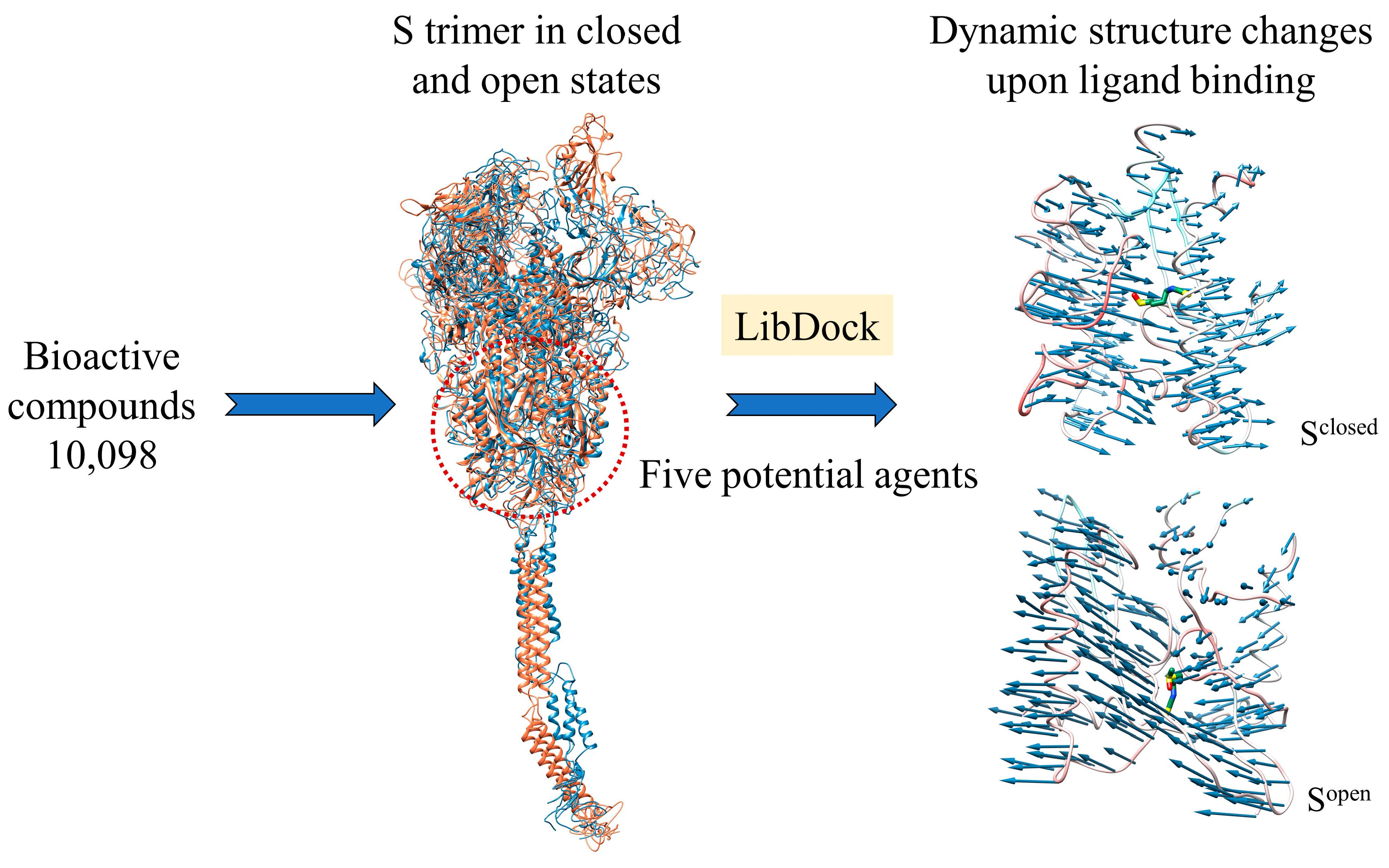
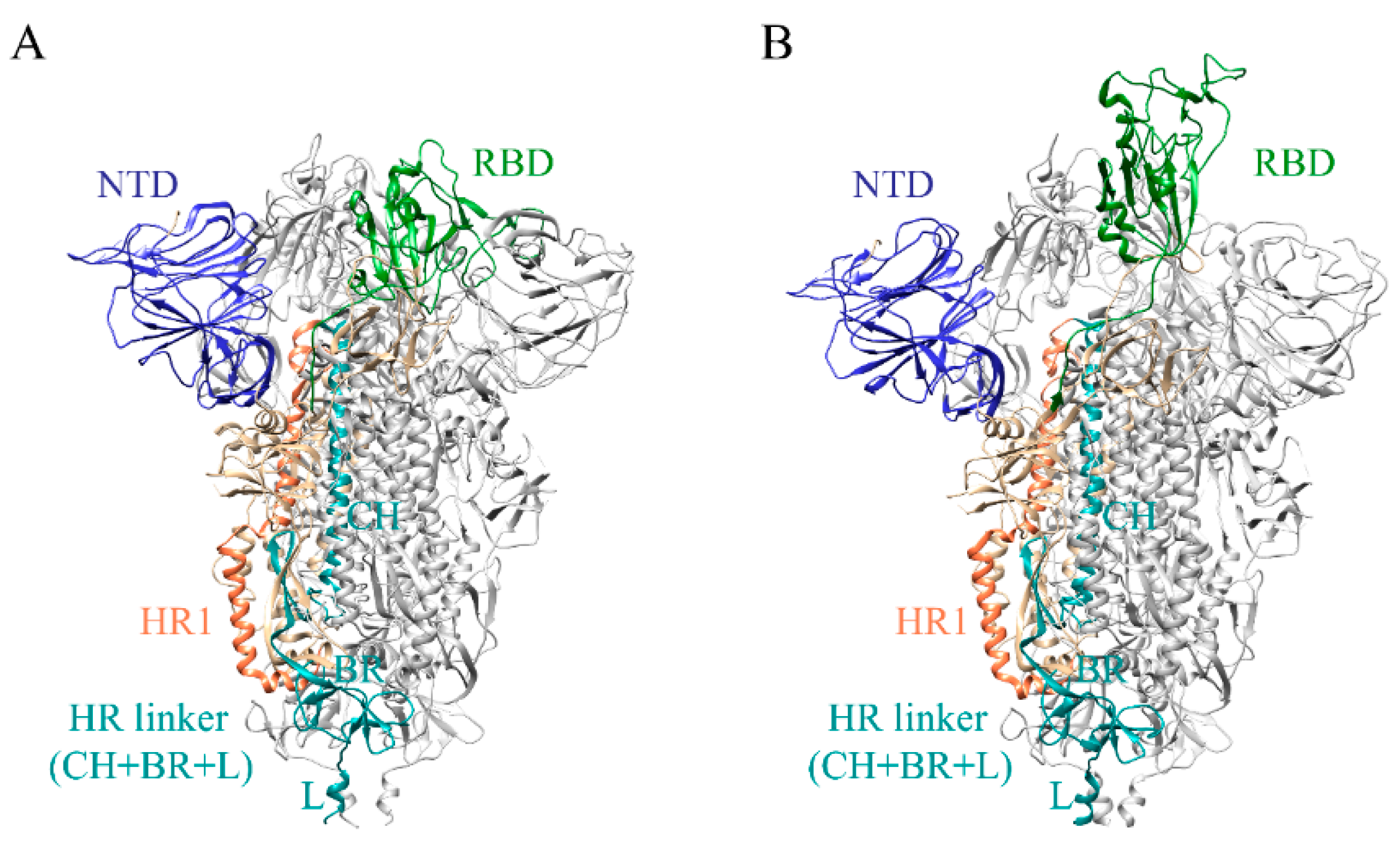
2.1. Full-Length S Structure Model
2.2. Docking Hits
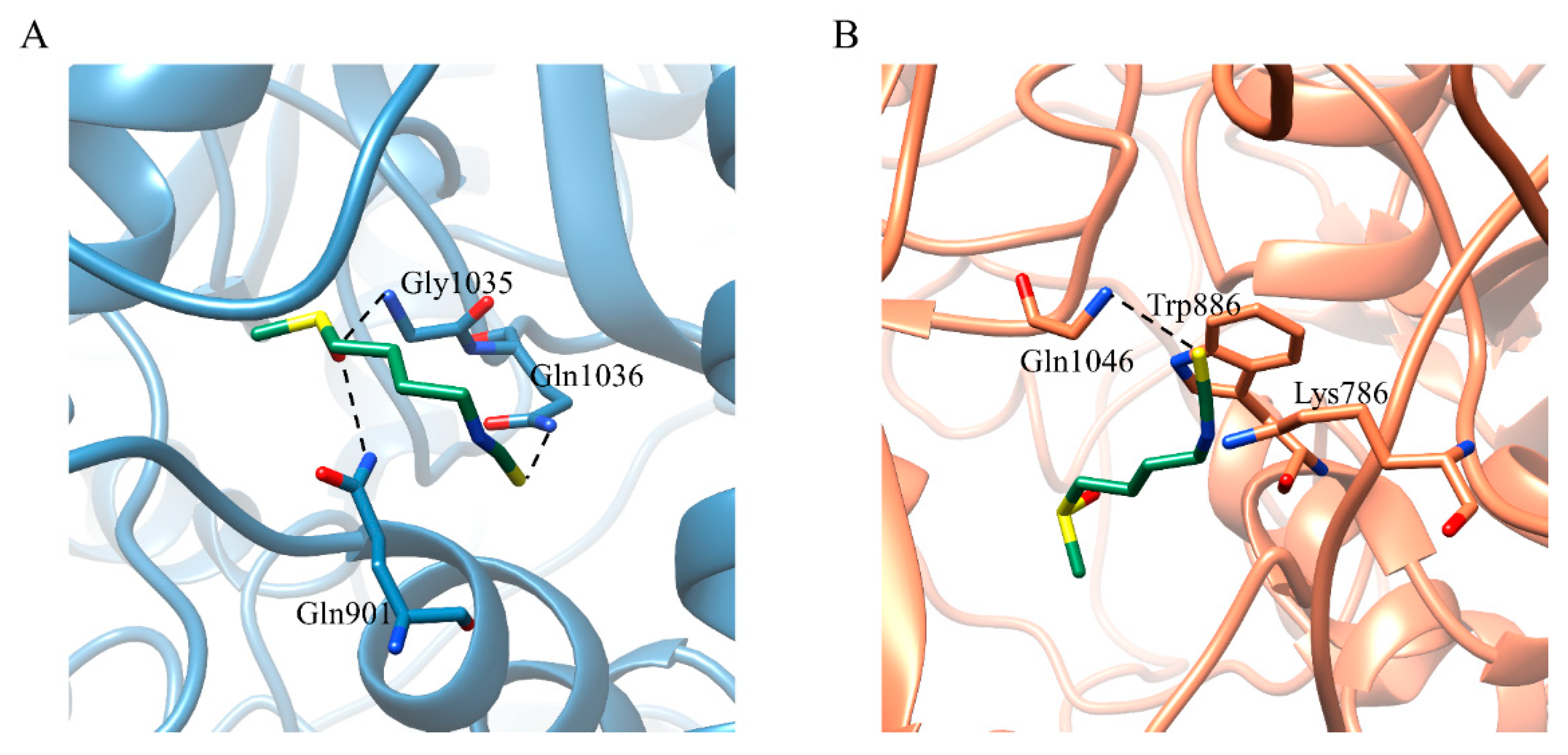
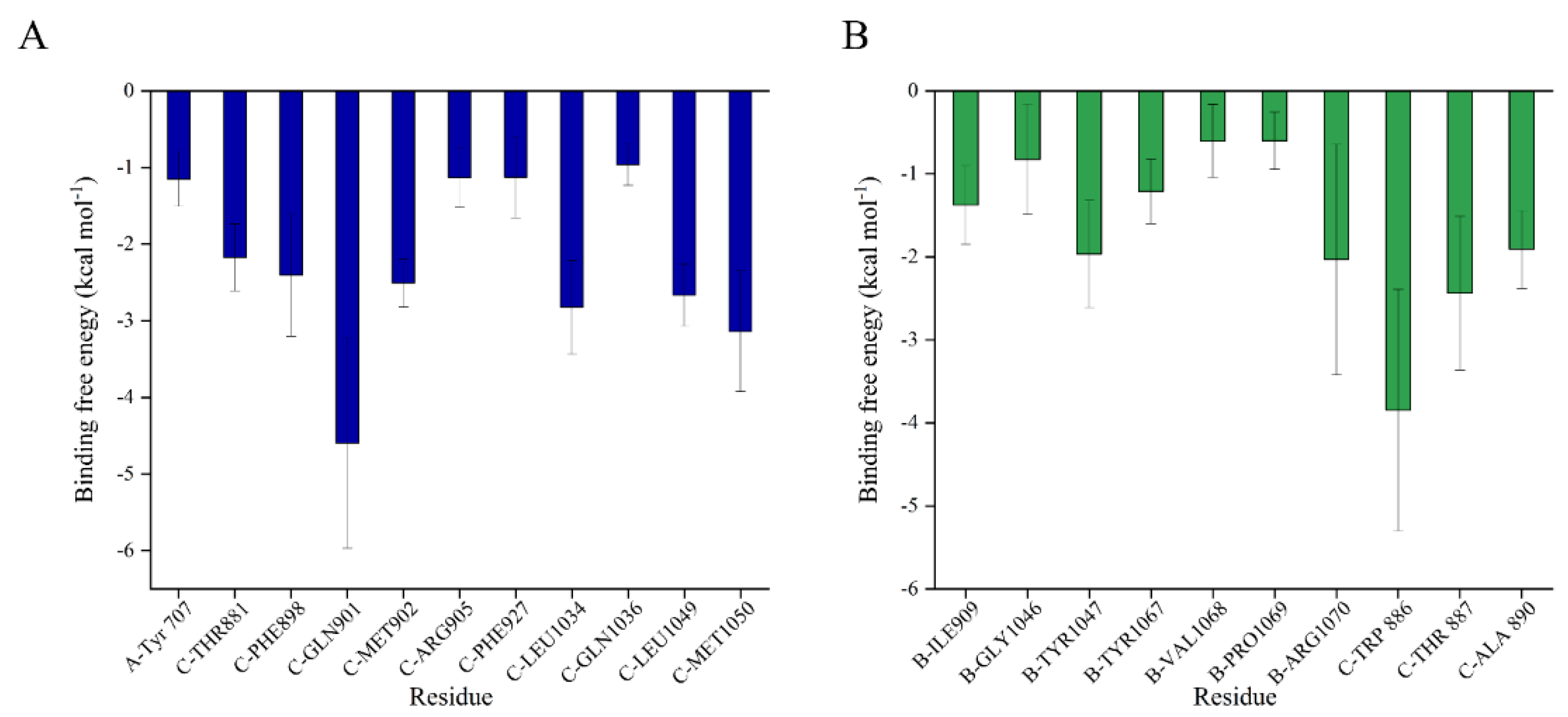
2.3. Binding Free Energy between SFN and S Trimer

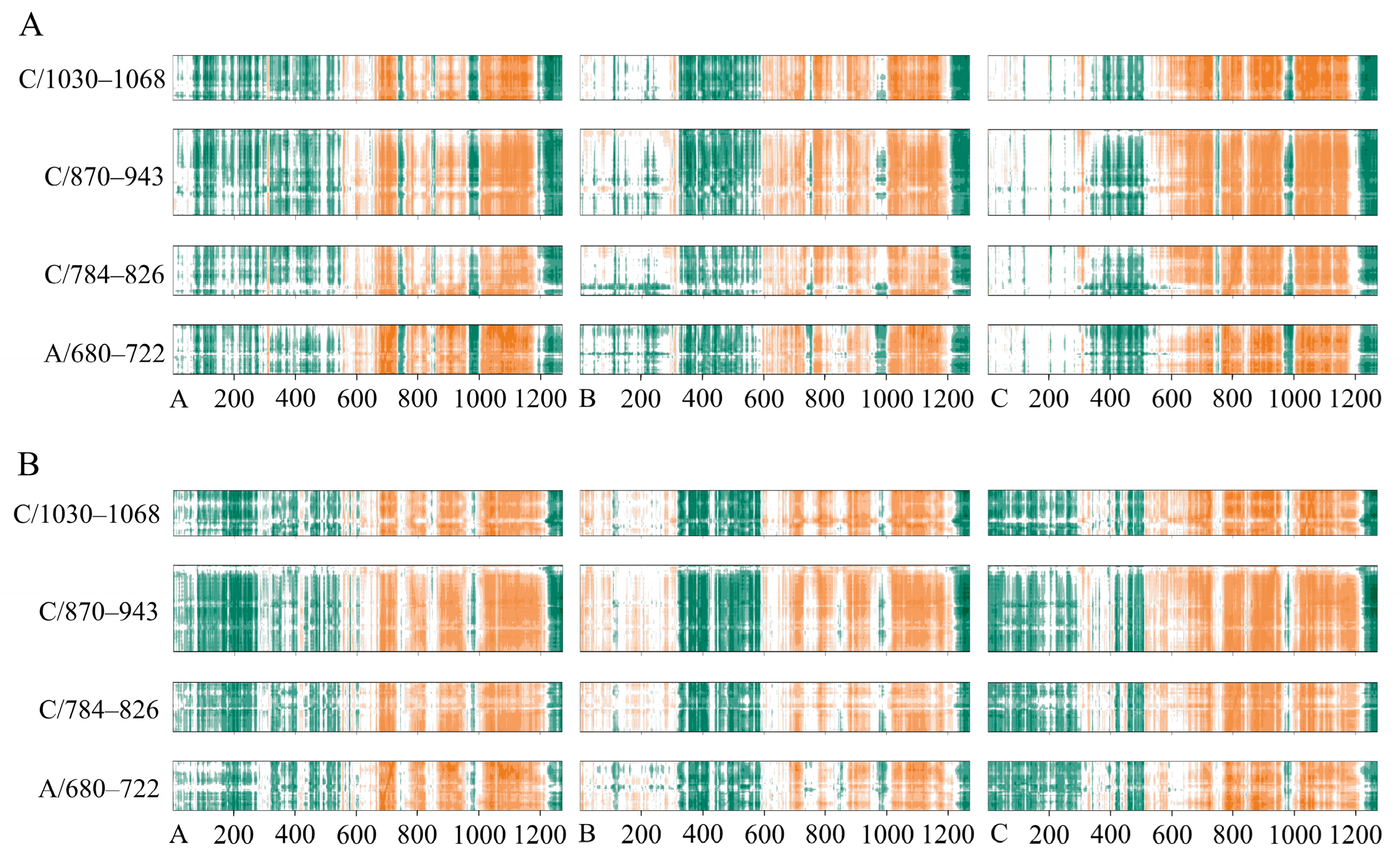
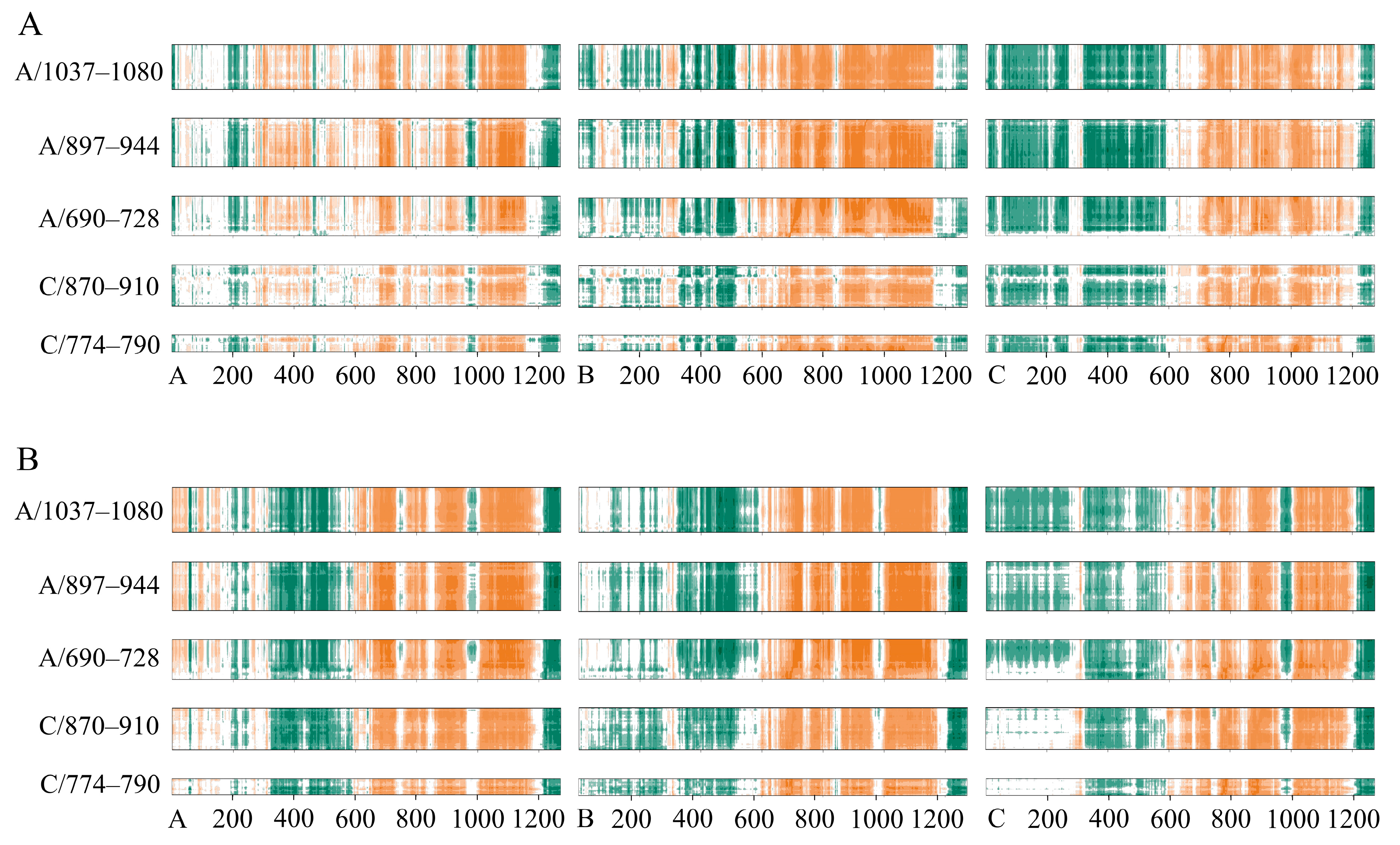
2.4. Conformational Changes upon Ligand Binding
3. Materials and Methods
3.1. In Situ Full-Length S Trimer Structure Modeling
3.2. Molecular Dynamics Flexible Fitting (MDFF)
3.3. Docking with Multiple Conformations
3.4. Molecular Dynamics (MD) Simulations
3.5. Binding Free Energy Calculation
3.6. Structural Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cano-Munoz, M.; Polo-Megias, D.; Camara-Artigas, A.; Gavira, J.A.; Lopez-Rodriguez, M.J.; Laumond, G.; Schmidt, S.; Demiselle, J.; Bahram, S.; Moog, C.; et al. Novel chimeric proteins mimicking SARS-CoV-2 spike epitopes with broad inhibitory activity. Int. J. Biol. Macromol. 2022, 222, 2467–2478. [Google Scholar] [CrossRef] [PubMed]
- Li, D.P.; Sempowski, G.D.; Saunders, K.O.; Acharya, P.; Haynes, B.F. SARS-CoV-2 Neutralizing Antibodies for COVID-19 Prevention and Treatment. Ann. Rev. Med. 2022, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.H.; Wang, P.F.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.W.; Sahi, V.; Figueroa, A.; et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature 2020, 584, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Rees-Spear, C.; Muir, L.; Griffith, S.A.; Heaney, J.; Aldon, Y.; Snitselaar, J.L.; Thomas, P.; Graham, C.; Seow, J.; Lee, N.; et al. The effect of spike mutations on SARS-CoV-2 neutralization. Cell Rep. 2021, 34, 108890. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.J.; Wu, N.C.; Yuan, M.; Bangaru, S.; Torres, J.L.; Caniels, T.G.; van Schooten, J.; Zhu, X.Y.; Lee, C.C.D.; Brouwer, P.J.M.; et al. Cross-Neutralization of a SARS-CoV-2 Antibody to a Functionally Conserved Site Is Mediated by Avidity. Immunity 2020, 53, 1272–1280. [Google Scholar] [CrossRef]
- Barnes, C.O.; West, A.P.; Huey-Tubman, K.E.; Hoffmann, M.A.G.; Sharaf, N.G.; Hoffman, P.R.; Koranda, N.; Gristick, H.B.; Gaebler, C.; Muecksch, F.; et al. Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies. Cell 2020, 182, 828–842. [Google Scholar] [CrossRef]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Ag. 2020, 56, 105998. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, L.; Zhang, Y.; Zhang, X.; Zhang, L.; Shang, W.; Bai, F. Probing the Allosteric Inhibition Mechanism of a Spike Protein Using Molecular Dynamics Simulations and Active Compound Identifications. J. Med. Chem. 2022, 65, 2827–2835. [Google Scholar] [CrossRef]
- Wang, Q.; Meng, F.; Xie, Y.; Wang, W.; Meng, Y.; Li, L.; Liu, T.; Qi, J.; Ni, X.; Zheng, S.; et al. In Silico Discovery of Small Molecule Modulators Targeting the Achilles’ Heel of SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2023, 9, 252–265. [Google Scholar] [CrossRef]
- Wang, Y.C.; Zhang, L.; Li, Q.Q.; Liang, Z.T.; Li, T.; Liu, S.; Cui, Q.Q.; Nie, J.H.; Wu, Q.; Qu, X.W.; et al. The significant immune escape of pseudotyped SARS-CoV-2 variant Omicron. Emerg. Microbes. Infec. 2022, 11, 1–5. [Google Scholar] [CrossRef]
- Nishima, W.; Kulik, M. Full-Length Computational Model of the SARS-CoV-2 Spike Protein and Its Implications for a Viral Membrane Fusion Mechanism. Viruses 2021, 13, 1126. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, Y.X.; Liu, C.X.; Zhang, C.; Han, W.Y.; Hong, X.Y.; Wang, Y.F.; Hong, Q.; Wang, S.T.; Zhao, Q.Y.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Pan, X.Y.; Huang, Y.; Cheng, C.; Xu, X.F.; Wu, Y.; Xu, Y.X.; Shang, W.J.; Niu, X.G.; Wan, Y.H.; et al. Drug Repurposing of Itraconazole and Estradiol Benzoate against COVID-19 by Blocking SARS-CoV-2 Spike Protein-Mediated Membrane Fusion. Adv. Ther. 2021, 4, 2000224. [Google Scholar] [CrossRef]
- Marchetti, C.; Vaglietti, S.; Rizzo, F.; Di Nardo, G.; Colnaghi, L.; Ghirardi, M.; Fiumara, F. Heptad stereotypy, S/Q layering, and remote origin of the SARS-CoV-2 fusion core. Virus Evol. 2021, 7, veab097. [Google Scholar] [CrossRef]
- Ita, K. Coronavirus Disease (COVID-19): Current Status and Prospects for Drug and Vaccine Development. Arch. Med. Res. 2021, 52, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Coco, A.; Patamia, V.; Zagni, C.; Floresta, G.; Rescifina, A. Targeting the SARS-CoV-2 HR1 with Small Molecules as Inhibitors of the Fusion Process. Int. J. Mol. Sci. 2022, 23, 10067. [Google Scholar] [CrossRef]
- Canales, F.J.R.; Mayoral, L.P.C.; Hernandez-Huerta, M.T.; Navarro, L.M.S.; Matias-Cervantes, C.A.; Cruz, M.M.; Parada, E.C.; Zenteno, E.; Ramos-Martinez, E.G.; Mayoral, E.P.C.; et al. Interaction of Spike protein and lipid membrane of SARS-CoV-2 with Ursodeoxycholic acid, an in-silico analysis. Sci. Rep. 2021, 11, 22288. [Google Scholar] [CrossRef]
- Zhong, H.J.; Ma, V.P.Y.; Cheng, Z.; Chan, D.S.H.; He, H.Z.; Leung, K.H.; Ma, D.L.; Leung, C.H. Discovery of a natural product inhibitor targeting protein neddylation by structure-based virtual screening. Biochimie 2012, 94, 2457–2460. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.F.; Zhu, Y.F.; Liu, C.X.; Gu, C.J.; Xu, S.Q.; Wang, Y.L.; Zhou, Y.; Wang, Y.X.; Han, W.Y.; et al. Development and structural basis of a two-MAb cocktail for treating SARS-CoV-2 infections. Nat. Commun. 2021, 12, 264. [Google Scholar] [CrossRef]
- Accelrys. Discovery Studio 3.1. Available online: http://accelrys.com (accessed on 8 November 2022).
- Woo, H.; Park, S.J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.W.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef]
- Turonova, B.; Sikora, M.; Schurmann, C.; Hagen, W.J.H.; Welsch, S.; Blanc, F.E.C.; von Bulow, S.; Gecht, M.; Bagola, K.; Horner, C.; et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science 2020, 370, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Dolan, M.A.; Noah, J.W.; Hurt, D. Comparison of common homology modeling algorithms: Application of user-defined alignments. Methods Mol. Biol. 2012, 857, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Tupina, D.; Krah, A.; Marzinek, J.K.; Zuzic, L.; Moverley, A.A.; Constantinidou, C.; Bond, P.J. Bridging the N-terminal and middle domains in FliG of the flagellar rotor. Curr. Res. Struct. Biol. 2022, 4, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Hakansson-McReynolds, S.; Jiang, S.K.; Rong, L.J.; Caffrey, M. Solution structure of the severe acute respiratory syndrome-coronavirus heptad repeat 2 domain in the prefusion state. J. Biol. Chem. 2006, 281, 11965–11971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itskanov, S.; Kuo, K.M.; Gumbart, J.C.; Park, E. Stepwise gating of the Sec61 protein-conducting channel by Sec63 and Sec62. Nat. Struct. Mol. Biol. 2021, 28, 162–172. [Google Scholar] [CrossRef]
- Schultze, P.; Worgotter, E.; Braun, W.; Wagner, G.; Vasak, M.; Kagi, J.H.R.; Wuthrich, K. Conformation of [Cd7]-Metallothionein-2 from Rat-Liver in Aqueous-Solution Determined by Nuclear Magnetic-Resonance Spectroscopy. J. Mol. Biol. 1988, 203, 251–268. [Google Scholar] [CrossRef]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef]
- Ke, Z.L.; Oton, J.Q.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 65. [Google Scholar] [CrossRef]
- He, Y.; Yan, C.L.; Fang, J.; Inouye, C.; Tjian, R.; Ivanov, I.; Nogales, E. Near-atomic resolution visualization of human transcription promoter opening. Acta Crystallogr. A 2017, 73, A256. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Kalathiya, U.; Padariya, M.; Mayordomo, M.; Lisowska, M.; Nicholson, J.; Singh, A.; Baginski, M.; Fahraeus, R.; Carragher, N.; Ball, K.; et al. Highly Conserved Homotrimer Cavity Formed by the SARS-CoV-2 Spike Glycoprotein: A Novel Binding Site. J. Clin. Med. 2020, 9, 1473. [Google Scholar] [CrossRef] [PubMed]
- Romeo, A.; Iacovelli, F.; Falconi, M. Targeting the SARS-CoV-2 spike glycoprotein prefusion conformation: Virtual screening and molecular dynamics simulations applied to the identification of potential fusion inhibitors. Virus Res. 2020, 286, 198068. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.N.; Head, M.S.; Kulkarni, A.; LaLonde, J.M. Validation studies of the site-directed docking program LibDock. J. Chem. Inf. Model 2007, 47, 2159–2171. [Google Scholar] [CrossRef] [Green Version]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and done?. Targeting the NRF2 pathway with sulforaphane. Trends Food Sci. Tech. 2017, 69, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.S.; Al Mamun, A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2020, 707, 135624. [Google Scholar] [CrossRef] [PubMed]
- Gasparello, J.; D’Aversa, E.; Papi, C.; Gambari, L.; Grigolo, B.; Borgatti, M.; Finotti, A.; Gambari, R. Sulforaphane inhibits the expression of interleukin-6 and interleukin-8 induced in bronchial epithelial IB3-1 cells by exposure to the SARS-CoV-2 Spike protein. Phytomedicine 2021, 87, 153583. [Google Scholar] [CrossRef]
- Labastida-Ramirez, A.; Rubio-Beltran, E.; Hernandez-Abreu, O.; Daugherty, B.L.; MaassenVanDenBrink, A.; Villalon, C.M. Pharmacological analysis of the increases in heart rate and diastolic blood pressure produced by (S)-isometheptene and (R)-isometheptene in pithed rats. J. Headache Pain 2017, 18, 52. [Google Scholar] [CrossRef] [Green Version]
- Lima, F.V.; Martins, T.E.A.; Morocho-Jacome, A.L.; Almeida, I.F.; Rosado, C.F.; Velasco, M.V.R.; Baby, A.R. Analytical tools for urocanic acid determination in human samples: A review. J. Sep. Sci. 2021, 44, 438–447. [Google Scholar] [CrossRef]
- Nahab, F.B.; Handforth, A.; Brown, T.; Shin, C.; Quesada, A.; Dong, C.H.; Haubenberger, D.; Hallett, M. Octanoic Acid Suppresses Harmaline-Induced Tremor in Mouse Model of Essential Tremor. Neurotherapeutics 2012, 9, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Borlinghaus, J.; Albrecht, F.; Gruhlke, M.C.H.; Nwachukwu, I.D.; Slusarenko, A.J. Allicin: Chemistry and Biological Properties. Molecules 2014, 19, 12591–12618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, B.; Marasini, B.P.; Rayamajhee, B.; Bhattarai, B.R.; Lamichhane, G.; Khadayat, K.; Adhikari, A.; Khanal, S.; Parajuli, N. Potential roles of medicinal plants for the treatment of viral diseases focusing on COVID-19: A review. Phytother Res. 2021, 35, 1298–1312. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.J.C.; Mou, Z.; Lei, R.; Ouyang, W.O.; Yuan, M.; Song, G.; Andrabi, R.; Wilson, I.A.; Kieffer, C.; Dai, X.; et al. High-throughput identification of prefusion-stabilizing mutations in SARS-CoV-2 spike. bioRxiv 2022. [Google Scholar] [CrossRef]
- Somadi, G.; Sivan, S.K. Identification of therapeutic target in S2 domain of SARS nCov-2 Spike glycoprotein: Key to design and discover drug candidates for inhibition of viral entry into host cell. J. Theor. Comput. Chem. 2020, 19, 2050028. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, I.T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Basso, L.G.M.; Vicente, E.F.; Crusca, E.; Cilli, E.M.; Costa, A.J. SARS-CoV fusion peptides induce membrane surface ordering and curvature. Sci. Rep. 2016, 6, 37131. [Google Scholar] [CrossRef] [Green Version]
- Zannella, C.; Chianese, A.; Greco, G.; Santella, B.; Squillaci, G.; Monti, A.; Doti, N.; Sanna, G.; Manzin, A.; Morana, A.; et al. Design of Three Residues Peptides against SARS-CoV-2 Infection. Viruses 2022, 14, v14102103. [Google Scholar] [CrossRef]
- Xia, J.W.; Yang, L.; Dong, L.; Niu, M.J.; Zhang, S.L.; Yang, Z.W.; Wumaier, G.; Li, Y.; Wei, X.M.; Gong, Y.; et al. Cefminox, a Dual Agonist of Prostacyclin Receptor and Peroxisome Proliferator-Activated Receptor-Gamma Identified by Virtual Screening, Has Therapeutic Efficacy against Hypoxia-Induced Pulmonary Hypertension in Rats. Front. Pharmacol. 2018, 9, 134. [Google Scholar] [CrossRef] [Green Version]
- Sergelius, C.; Yamaguchi, S.; Yamamoto, T.; Engberg, O.; Katsumura, S.; Slotte, J.P. Cholesterol’s interactions with serine phospholipids—A comparison of N-palmitoyl ceramide phosphoserine with dipalmitoyl phosphatidylserine. Biochim. Biophys. Acta-Biomembr. 2013, 1828, 785–791. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 104–110. [Google Scholar] [CrossRef]
- Balakumar, C.; Ramesh, M.; Tham, C.L.; Khathi, S.P.; Kozielski, F.; Srinivasulu, C.; Hampannavar, G.A.; Sayyad, N.; Soliman, M.E.; Karpoormath, R. Ligand- and structure-based in silico studies to identify kinesin spindle protein (KSP) inhibitors as potential anticancer agents. J. Biomol. Struct. Dyn. 2018, 36, 3687–3704. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Cheatham, T.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Grand, S.L.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, C.J.; Madej, B.D.; Skjevik, A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar]
- Kale, L.; Skeel, R.; Bhandarkar, M.; Brunner, R.; Gursoy, A.; Krawetz, N.; Phillips, J.; Shinozaki, A.; Varadarajan, K.; Schulten, K. NAMD2: Greater scalability for parallel molecular dynamics. J. Comput. Phys. 1999, 151, 283–312. [Google Scholar] [CrossRef]
- Zhong, X.W.; Liu, Y.; Zhu, L.; Meng, X.; Wang, R.W.; Van Petegem, F.; Wagenknecht, T.; Chen, S.R.W.; Liu, Z. Conformational Dynamics inside Amino-Terminal Disease Hotspot of Ryanodine Receptor. Structure 2013, 21, 2051–2060. [Google Scholar] [CrossRef] [Green Version]
- Trabuco, L.G.; Villa, E.; Mitra, K.; Frank, J.; Schulten, K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 2008, 16, 673–683. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.S.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, Y.; Hao, D.; Wang, H.; Li, S.; Jia, L.; Yuan, X.; Zhang, L.; Meng, L.; Zhang, S. Computational identification of potential chemoprophylactic agents according to dynamic behavior of peroxisome proliferator-activated receptor gamma. RSC Adv. 2020, 11, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.W.; Zang, Y.J.; Wang, H.; Kang, Y.; Zhang, J.W.; Li, X.H.; Zhang, L.; Zhang, S.L. Recognition between CD147 and cyclophilin A deciphered by accelerated molecular dynamics simulations. Phys. Chem. Chem. Phys. 2022, 24, 18905–18914. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, D.A.; Case, D.A.; Caldwell, J.W.; Ross, W.S.; Cheatham, T.E.; Debolt, S.; Ferguson, D.; Seibel, G.; Kollman, P. Amber, a Package of Computer-Programs for Applying Molecular Mechanics, Normal-Mode Analysis, Molecular-Dynamics and Free-Energy Calculations to Simulate the Structural and Energetic Properties of Molecules. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar] [CrossRef]
- Yang, Z.W.; Zhao, Y.Z.; Zang, Y.J.; Wang, H.; Zhu, X.; Meng, L.J.; Yuan, X.H.; Zhang, L.; Zhang, S.L. Rapid Structure-Based Screening Informs Potential Agents for Coronavirus Disease (COVID-19) Outbreak. Chinese Phys. Lett. 2020, 37, 058701. [Google Scholar] [CrossRef]
- Hou, T.J.; Wang, J.M.; Li, Y.Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Sun, H.Y.; Li, Y.Y.; Tian, S.; Xu, L.; Hou, T.J. Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys. Chem. Chem. Phys. 2014, 16, 16719–16729. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.W.; Li, G.Y.; Zhao, Y.Z.; Zhang, L.; Yuan, X.H.; Meng, L.J.; Liu, H.D.; Han, Y.; Jia, L.T.; Zhang, S.L. Molecular Insights into the Recruiting Between UCP2 and DDX5/UBAP2L in the Metabolic Plasticity of Non-Small-Cell Lung Cancer. J. Chem. Inf. Model 2021, 61, 3978–3987. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wires Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. Software News and Updates MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Fataftah, H.; Karain, W. Detecting protein atom correlations using correlation of probability of recurrence. Proteins 2014, 82, 2180–2189. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zannella, C.; Rinaldi, L.; Boccia, G.; Chianese, A.; Sasso, F.C.; De Caro, F.; Franci, G.; Galdiero, M. Regulation of m6A Methylation as a New Therapeutic Option against COVID-19. Pharmaceuticals 2021, 14, ph14111135. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZINC ID | Compound | Structure | Original Purpose | LibDockScore | |
|---|---|---|---|---|---|
| Sclosed | Sopen | ||||
| ZINC000002557133 | Sulforaphane |  | anticarcinogenic | 70.62 | 78.16 |
| ZINC000001683250 | Isometheptene |  | antihyperglycemic | 69.26 | 58.46 |
| ZINC000034633903 | Urocanic acid |  | antihyperglycemic | 68.20 | 68.90 |
| ZINC000001532735 | 1-Octanol |  | anti-cancer, antimicrobial | 66.89 | 65.42 |
| ZINC000004097409 | Allicin |  | anti-inflammatory, anticancer | 64.54 | 70.52 |
| Complex | ΔEele | ΔEvdw | ΔGSA | ΔGGB | ΔGbind |
|---|---|---|---|---|---|
| Sulforaphane-Sclosed | −10.90 ± 3.24 | −30.92 ± 1.17 | −4.51 ± 0.14 | 24.06 ± 3.28 | −22.27 ± 1.12 |
| Sulforaphane-Sopen | −8.70 ± 3.52 | −24.34 ± 1.81 | −3.62 ± 0.27 | 22.44 ± 1.98 | −14.21 ± 2.53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Zhao, Y.; Xie, L.; Li, Q.; Zhang, Y.; Zang, Y.; Li, X.; Zhang, L.; Yang, Z. Identification of Potential Lead Compounds Targeting Novel Druggable Cavity of SARS-CoV-2 Spike Trimer by Molecular Dynamics Simulations. Int. J. Mol. Sci. 2023, 24, 6281. https://doi.org/10.3390/ijms24076281
Zhao Y, Zhao Y, Xie L, Li Q, Zhang Y, Zang Y, Li X, Zhang L, Yang Z. Identification of Potential Lead Compounds Targeting Novel Druggable Cavity of SARS-CoV-2 Spike Trimer by Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2023; 24(7):6281. https://doi.org/10.3390/ijms24076281
Chicago/Turabian StyleZhao, Yizhen, Yifan Zhao, Linke Xie, Qian Li, Yuze Zhang, Yongjian Zang, Xuhua Li, Lei Zhang, and Zhiwei Yang. 2023. "Identification of Potential Lead Compounds Targeting Novel Druggable Cavity of SARS-CoV-2 Spike Trimer by Molecular Dynamics Simulations" International Journal of Molecular Sciences 24, no. 7: 6281. https://doi.org/10.3390/ijms24076281