

Effects of miR-33 Deficiency on Metabolic and Cardiovascular Diseases: Implications for Therapeutic Intervention

{kind=link}
{kind=link}
Abstract
:1. Introduction
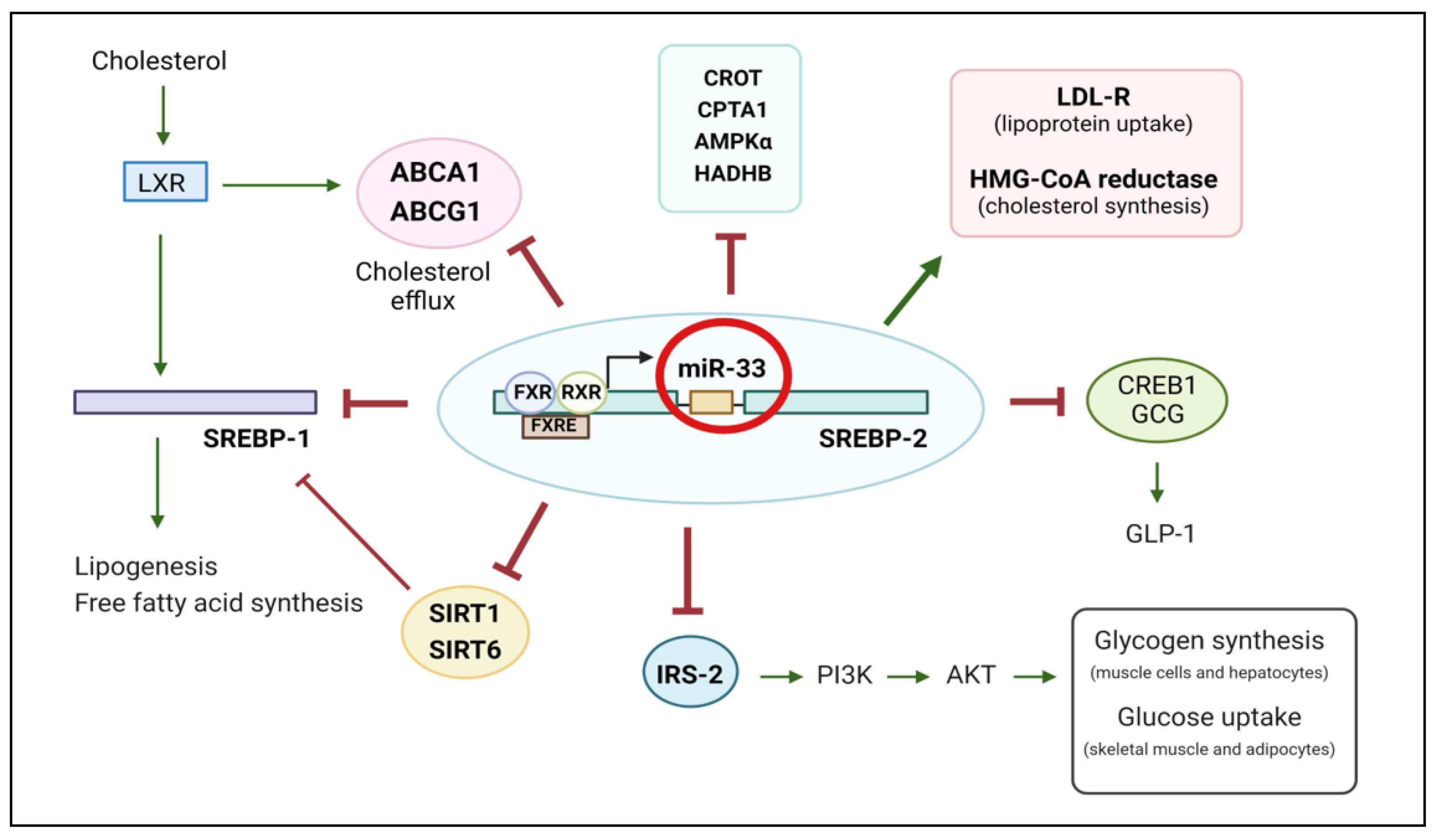
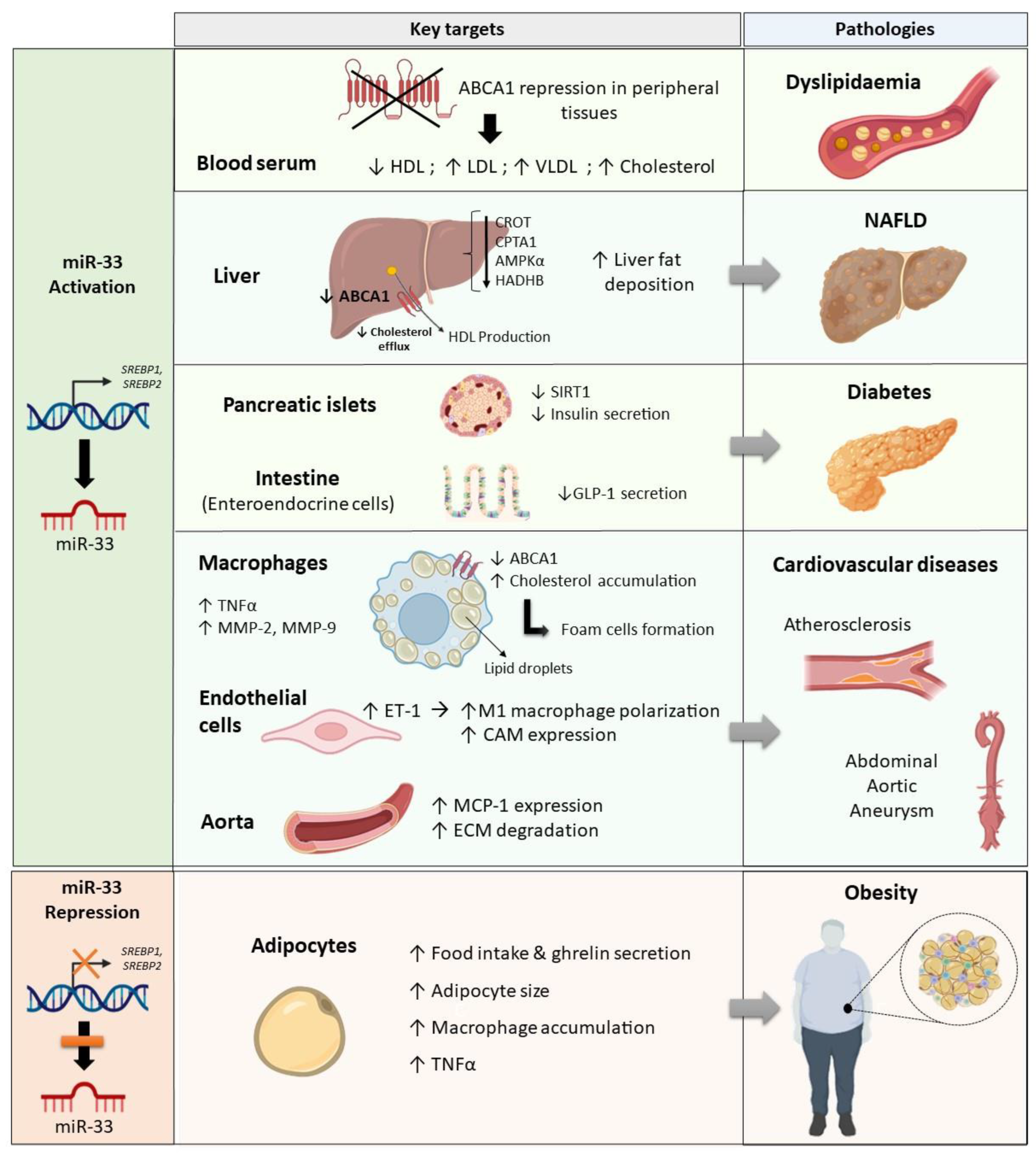
2. The Role of miR-33 in Dyslipidaemia
3. MiR-33 and the Development of Hepatic Steatosis
4. The Role of miR-33 in Adipose Tissue Dysfunction and Obesity
5. MiR-33 Impairments in Insulin Resistance and Diabetes
6. MiR-33 Is Involved in Endothelial Dysfunction and Contributes to the Development of Cardiovascular Diseases
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAA | abdominal aortic aneurysm |
| ABC | ATP-binding cassette transporter |
| AKT | protein kinase B |
| AMPKα | AMP-activated protein kinase alpha |
| AT | adipose tissue |
| CAMs | cell adhesion molecules |
| CPT1A | carnitine palmitoyltransferase 1A |
| CREB1 | cAMP response element binding protein 1 |
| CROT | carnitine O-octanoyltransferase |
| ECM | extracellular matrix |
| ET-1 | endothelin-1 |
| FXR | farnesoid X receptor |
| FXRE | FXR response element |
| GLP-1 | glucagon-like peptide-1 |
| GLUT4 | glucose transporter type 4 |
| HADHB | hydroxyacyl-CoA-dehydrogenase |
| HDL | high density lipoprotein |
| HFD | high fat diet |
| HMG-CoA | 3-hydroxy-3-methylglutaryl-CoA |
| IRS-2 | insulin receptor substrate-2 |
| KO | knockout |
| LDL | low density lipoprotein |
| LDL-R | low density lipoprotein receptor |
| LXR | liver X receptor |
| MCP-1 | monocyte chemoattractant protein-1 |
| MiRNA | microRNA |
| MMP | metalloproteinase |
| NAFLD | nonalcoholic fatty liver disease |
| NFκB | nuclear factor κB |
| PDK-1 | phosphoinositide-dependent kinase-1 |
| PI3K | phosphoinositide 3-kinase |
| RCT | reverse cholesterol transport |
| SIRT | sirtuin |
| SREBP | sterol regulatory element-binding protein |
| RXR | retinoid X receptor |
| TNFα | tumour necrosis factor α |
| UTR | untranslated region |
| VLDL | very low density lipoprotein |
References
- Pritchard, C.C.; Cheng, H.H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 2012, 13, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.B.; Wang, M.Y.; Zhu, H.Y.; Tang, S.Q.; You, Y.D.; Xie, Y.Q. Overexpression of microRNA-21 and microRNA-126 in the patients of bronchial asthma. Int. J. Clin. Exp. Med. 2014, 7, 1307–1312. [Google Scholar] [PubMed]
- Sun, P.; Zhang, S.; Wu, D.; Qian, Y.; Xiao, X.; Zhang, Q. MiR-21 modulates proliferation and apoptosis of human airway smooth muscle cells by regulating autophagy via PARP-1/AMPK/mTOR signalling pathway. Respir. Physiol. Neurobiol. 2022, 301, 103891. [Google Scholar] [CrossRef] [PubMed]
- Najm, A.; Masson, F.M.; Preuss, P.; Georges, S.; Ory, B.; Quillard, T.; Sood, S.; Goodyear, C.S.; Veale, D.J.; Fearon, U.; et al. MicroRNA-17-5p Reduces Inflammation and Bone Erosions in Mice With Collagen-Induced Arthritis and Directly Targets the JAK/STAT Pathway in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol. 2020, 72, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Lovat, F.; Nigita, G.; Distefano, R.; Nakamura, T.; Gasparini, P.; Tomasello, L.; Fadda, P.; Ibrahimova, N.; Catricalà, S.; Palamarchuk, A.; et al. Combined loss of function of two different loci of miR-15/16 drives the pathogenesis of acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2020, 117, 12332–12340. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.B.; Li, Q.H.; Zhang, N.; Li, M.; Li, K. MiR-142 inhibits lung cancer cell proliferation and promotes apoptosis by targeting XIAP. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7430–7437. [Google Scholar]
- Zhang, W.; Chen, C.J.; Guo, G.L. MiR-155 promotes the proliferation and migration of breast cancer cells via targeting SOCS1 and MMP16. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7323–7332. [Google Scholar]
- Wang, Y.; Xu, H.; Si, L.; Li, Q.; Zhu, X.; Yu, T.; Gang, X. MiR-206 inhibits proliferation and migration of prostate cancer cells by targeting CXCL11. Prostate 2018, 78, 479–490. [Google Scholar] [CrossRef]
- Mishra, S.; Yadav, T.; Rani, V. Exploring miRNA based approaches in cancer diagnostics and therapeutics. Crit. Rev. Oncol. Hematol. 2016, 98, 12–23. [Google Scholar] [CrossRef]
- Li, W.; Yuan, X.; He, X.; Yang, L.; Wu, Y.; Deng, X.; Zeng, Y.; Hu, K.; Tang, B. The downregulation of miR-22 and miR-372 may contribute to gestational diabetes mellitus through regulating glucose metabolism via the PI3K/AKT/GLUT4 pathway. J. Clin. Lab. Anal. 2022, 36, e24557. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Sobolewski, C.; Ay, A.S.; Abegg, D.; Correia de Sousa, M.; Portius, D.; Berthou, F.; Fournier, M.; Maeder, C.; Rantakari, P.; et al. Genetic Ablation of MiR-22 Fosters Diet-Induced Obesity and NAFLD Development. J. Pers. Med. 2020, 10, 170. [Google Scholar] [CrossRef]
- Barajas, J.M.; Reyes, R.; Guerrero, M.J.; Jacob, S.T.; Motiwala, T.; Ghoshal, K. The role of miR-122 in the dysregulation of glucose-6-phosphate dehydrogenase (G6PD) expression in hepatocellular cancer. Sci. Rep. 2018, 8, 9105. [Google Scholar] [CrossRef] [PubMed]
- Agbu, P.; Carthew, R.W. MicroRNA-mediated regulation of glucose and lipid metabolism. Nat. Rev. Mol. Cell Biol. 2021, 22, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Nishiga, M.; Horie, T.; Kuwabara, Y.; Nagao, K.; Baba, O.; Nakao, T.; Nishino, T.; Hakuno, D.; Nakashima, Y.; Nishi, H.; et al. MicroRNA-33 Controls Adaptive Fibrotic Response in the Remodeling Heart by Preserving Lipid Raft Cholesterol. Circ. Res. 2017, 120, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Ediriweera, H.N.; Gundra, U.M.; Sheedy, F.J.; Ramkhelawon, B.; Hutchison, S.B.; Rinehold, K.; van Solingen, C.; Fullerton, M.D.; Cecchini, K.; et al. MicroRNA-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J. Clin. Investig. 2015, 125, 4334–4348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, P.C.; Chang, K.C.; Chuang, Y.S.; Wei, L.N. Cholesterol regulation of receptor-interacting protein 140 via microRNA-33 in inflammatory cytokine production. FASEB J. 2011, 25, 1758–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, N.L.; Singh, A.K.; Rotllan, N.; Goedeke, L.; Wing, A.; Canfrán-Duque, A.; Diaz-Ruiz, A.; Araldi, E.; Baldán, Á.; Camporez, J.P.; et al. Genetic Ablation of miR-33 Increases Food Intake, Enhances Adipose Tissue Expansion, and Promotes Obesity and Insulin Resistance. Cell Rep. 2018, 22, 2133–2145. [Google Scholar] [CrossRef] [Green Version]
- Shahouzehi, B.; Eghbalian, M.; Fallah, H.; Aminizadeh, S.; Masoumi-Ardakani, Y. Serum microRNA-33 levels in pre-diabetic and diabetic patients. Mol. Biol. Rep. 2021, 48, 4121–4128. [Google Scholar] [CrossRef]
- Rayner, K.J.; Sheedy, F.J.; Esau, C.C.; Hussain, F.N.; Temel, R.E.; Parathath, S.; van Gils, J.M.; Rayner, A.J.; Chang, A.N.; Suarez, Y.; et al. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J. Clin. Investig. 2011, 121, 2921–2931. [Google Scholar] [CrossRef] [Green Version]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Näär, A.M. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef] [Green Version]
- Koyama, S.; Horie, T.; Nishino, T.; Baba, O.; Sowa, N.; Miyasaka, Y.; Kuwabara, Y.; Nakao, T.; Nishiga, M.; Nishi, H.; et al. Identification of Differential Roles of MicroRNA-33a and -33b During Atherosclerosis Progression With Genetically Modified Mice. J. Am. Heart Assoc. 2019, 8, e012609. [Google Scholar] [CrossRef] [PubMed]
- Criado-Mesas, L.; Ballester, M.; Crespo-Piazuelo, D.; Passols, M.; Castelló, A.; Sánchez, A.; Folch, J.M. Expression analysis of porcine miR-33a/b in liver, adipose tissue and muscle and its potential role in fatty acid metabolism. PLoS ONE 2021, 16, e0245858. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, C.M.; Goedeke, L.; Rotllan, N.; Yoon, J.H.; Cirera-Salinas, D.; Mattison, J.A.; Suárez, Y.; de Cabo, R.; Gorospe, M.; Fernández-Hernando, C. MicroRNA 33 regulates glucose metabolism. Mol. Cell. Biol. 2013, 33, 2891–2902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, L.; Azzam, K.M.; Lin, W.C.; Rai, P.; Lowe, J.M.; Gabor, K.A.; Madenspacher, J.H.; Aloor, J.J.; Parks, J.S.; Näär, A.M.; et al. MicroRNA-33 Regulates the Innate Immune Response via ATP Binding Cassette Transporter-mediated Remodeling of Membrane Microdomains. J. Biol. Chem. 2016, 291, 19651–19660. [Google Scholar] [CrossRef] [Green Version]
- Tarling, E.J.; Ahn, H.; de Aguiar Vallim, T.Q. The nuclear receptor FXR uncouples the actions of miR-33 from SREBP-2. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Marquart, T.J.; Allen, R.M.; Ory, D.S.; Baldán, A. miR-33 links SREBP-2 induction to repression of sterol transporters. Proc. Natl. Acad. Sci. USA 2010, 107, 12228–12232. [Google Scholar] [CrossRef] [Green Version]
- Gerin, I.; Clerbaux, L.A.; Haumont, O.; Lanthier, N.; Das, A.K.; Burant, C.F.; Leclercq, I.A.; MacDougald, O.A.; Bommer, G.T. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J. Biol. Chem. 2010, 285, 33652–33661. [Google Scholar] [CrossRef] [Green Version]
- Goedeke, L.; Salerno, A.; Ramírez, C.M.; Guo, L.; Allen, R.M.; Yin, X.; Langley, S.R.; Esau, C.; Wanschel, A.; Fisher, E.A.; et al. Long-term therapeutic silencing of miR-33 increases circulating triglyceride levels and hepatic lipid accumulation in mice. EMBO Mol. Med. 2014, 6, 1133–1141. [Google Scholar] [CrossRef]
- Tokarz, V.L.; MacDonald, P.E.; Klip, A. The cell biology of systemic insulin function. J. Cell Biol. 2018, 217, 2273–2289. [Google Scholar] [CrossRef] [Green Version]
- da Silva Rosa, S.C.; Nayak, N.; Caymo, A.M.; Gordon, J.W. Mechanisms of muscle insulin resistance and the cross-talk with liver and adipose tissue. Physiol. Rep. 2020, 8, e14607. [Google Scholar] [CrossRef]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Hernando, C.; Ramírez, C.M.; Goedeke, L.; Suárez, Y. MicroRNAs in metabolic disease. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dávalos, A.; Goedeke, L.; Smibert, P.; Ramírez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C.; et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.Y.; Man, X.F.; Guo, Y.; Tang, H.N.; Tang, J.; Zhou, C.L.; Tan, S.W.; Wang, M.; Zhou, H.D. IRS-2 Partially Compensates for the Insulin Signal Defects in IRS-1−/− Mice Mediated by miR-33. Mol. Cells 2017, 40, 123–132. [Google Scholar] [CrossRef]
- Nakao, T.; Horie, T.; Baba, O.; Nishiga, M.; Nishino, T.; Izuhara, M.; Kuwabara, Y.; Nishi, H.; Usami, S.; Nakazeki, F.; et al. Genetic Ablation of MicroRNA-33 Attenuates Inflammation and Abdominal Aortic Aneurysm Formation via Several Anti-Inflammatory Pathways. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2161–2170. [Google Scholar] [CrossRef] [Green Version]
- Barna, B.P.; McPeek, M.; Malur, A.; Fessler, M.B.; Wingard, C.J.; Dobbs, L.; Verbanac, K.M.; Bowling, M.; Judson, M.A.; Thomassen, M.J. Elevated MicroRNA-33 in Sarcoidosis and a Carbon Nanotube Model of Chronic Granulomatous Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 865–871. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Parhofer, K.G. Diabetic dyslipidemia. Metabolism 2014, 63, 1469–1479. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, Z.W.; Zeng, P.H.; Zhou, Y.J.; Yin, W.J. Molecular mechanisms for ABCA1-mediated cholesterol efflux. Cell Cycle 2022, 21, 1121–1139. [Google Scholar] [CrossRef]
- Wang, S.; Smith, J.D. ABCA1 and nascent HDL biogenesis. Biofactors 2014, 40, 547–554. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, M.L.; Mujawar, Z.; Tamehiro, N. ABC transporters, atherosclerosis and inflammation. Atherosclerosis 2010, 211, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Ramasamy, I. Update on the molecular biology of dyslipidemias. Clin. Chim. Acta 2016, 454, 143–185. [Google Scholar] [CrossRef] [PubMed]
- Canfrán-Duque, A.; Ramírez, C.M.; Goedeke, L.; Lin, C.S.; Fernández-Hernando, C. microRNAs and HDL life cycle. Cardiovasc. Res. 2014, 103, 414–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, T.; Ono, K.; Horiguchi, M.; Nishi, H.; Nakamura, T.; Nagao, K.; Kinoshita, M.; Kuwabara, Y.; Marusawa, H.; Iwanaga, Y.; et al. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 17321–17326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouimet, M.; Hennessy, E.J.; van Solingen, C.; Koelwyn, G.J.; Hussein, M.A.; Ramkhelawon, B.; Rayner, K.J.; Temel, R.E.; Perisic, L.; Hedin, U.; et al. miRNA Targeting of Oxysterol-Binding Protein-Like 6 Regulates Cholesterol Trafficking and Efflux. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 942–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, T.; Nishino, T.; Baba, O.; Kuwabara, Y.; Nakao, T.; Nishiga, M.; Usami, S.; Izuhara, M.; Nakazeki, F.; Ide, Y.; et al. MicroRNA-33b knock-in mice for an intron of sterol regulatory element-binding factor 1 (Srebf1) exhibit reduced HDL-C in vivo. Sci. Rep. 2014, 4, 5312. [Google Scholar] [CrossRef] [Green Version]
- Rayner, K.J.; Esau, C.C.; Hussain, F.N.; McDaniel, A.L.; Marshall, S.M.; van Gils, J.M.; Ray, T.D.; Sheedy, F.J.; Goedeke, L.; Liu, X.; et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL triglycerides. Nature 2011, 478, 404–407. [Google Scholar] [CrossRef] [Green Version]
- Shao, F.; Wang, X.; Yu, J.; Shen, K.; Qi, C.; Gu, Z. Expression of miR-33 from an SREBP2 intron inhibits the expression of the fatty acid oxidation-regulatory genes CROT and HADHB in chicken liver. Br. Poult. Sci. 2019, 60, 115–124. [Google Scholar] [CrossRef]
- Niesor, E.J.; Schwartz, G.G.; Perez, A.; Stauffer, A.; Durrwell, A.; Bucklar-Suchankova, G.; Benghozi, R.; Abt, M.; Kallend, D. Statin-induced decrease in ATP-binding cassette transporter A1 expression via microRNA33 induction may counteract cholesterol efflux to high-density lipoprotein. Cardiovasc. Drugs Ther. 2015, 29, 7–14. [Google Scholar] [CrossRef]
- Rottiers, V.; Najafi-Shoushtari, S.H.; Kristo, F.; Gurumurthy, S.; Zhong, L.; Li, Y.; Cohen, D.E.; Gerszten, R.E.; Bardeesy, N.; Mostoslavsky, R.; et al. MicroRNAs in metabolism and metabolic diseases. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Lee, M.S.; Chang, E.; Kim, C.T.; Choi, A.J.; Kim, I.H.; Kim, Y. High hydrostatic pressure extract of mulberry leaves ameliorates hypercholesterolemia via modulating hepatic microRNA-33 expression and AMPK activity in high cholesterol diet fed rats. Food Nutr. Res. 2021, 65, 7587. [Google Scholar] [CrossRef] [PubMed]
- Martino, F.; Carlomosti, F.; Avitabile, D.; Persico, L.; Picozza, M.; Barillà, F.; Arca, M.; Montali, A.; Martino, E.; Zanoni, C.; et al. Circulating miR-33a and miR-33b are up-regulated in familial hypercholesterolaemia in paediatric age. Clin. Sci. 2015, 129, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Gjorgjieva, M.; Sobolewski, C.; Dolicka, D.; Correia de Sousa, M.; Foti, M. miRNAs and NAFLD: From pathophysiology to therapy. Gut 2019, 68, 2065–2079. [Google Scholar] [CrossRef]
- Fujii, H.; Kawada, N.; Japan Study Group of NAFLD (JSG-NAFLD). The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef]
- Lian, C.Y.; Zhai, Z.Z.; Li, Z.F.; Wang, L. High fat diet-triggered non-alcoholic fatty liver disease: A review of proposed mechanisms. Chem. Biol. Interact. 2020, 330, 109199. [Google Scholar] [CrossRef] [PubMed]
- Recena Aydos, L.; Aparecida do Amaral, L.; Serafim de Souza, R.; Jacobowski, A.C.; Freitas Dos Santos, E.; Rodrigues Macedo, M.L. Nonalcoholic Fatty Liver Disease Induced by High-Fat Diet in C57bl/6 Models. Nutrients 2019, 11, 3067. [Google Scholar] [CrossRef] [Green Version]
- Horie, T.; Nishino, T.; Baba, O.; Kuwabara, Y.; Nakao, T.; Nishiga, M.; Usami, S.; Izuhara, M.; Sowa, N.; Yahagi, N.; et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat. Commun. 2013, 4, 2883. [Google Scholar] [CrossRef] [Green Version]
- Su, D.; Zhang, R.; Hou, F.; Chi, J.; Huang, F.; Yan, S.; Liu, L.; Deng, Y.; Wei, Z.; Zhang, M. Lychee pulp phenolics ameliorate hepatic lipid accumulation by reducing miR-33 and miR-122 expression in mice fed a high-fat diet. Food Funct. 2017, 8, 808–815. [Google Scholar] [CrossRef]
- Tao, Y.; Xu, S.; Wang, J.; Xu, L.; Zhang, C.; Chen, K.; Lian, Z.; Zhou, J.; Xie, H.; Zheng, S.; et al. Delivery of microRNA-33 Antagomirs by Mesoporous Silica Nanoparticles to Ameliorate Lipid Metabolic Disorders. Front. Pharmacol. 2020, 11, 921. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, J.; Yang, S.; Hua, Y.; Su, J.; Shang, Y.; Wang, Z.; Feng, K.; Zhang, J.; Yang, X.; et al. Astragalus Flavone Ameliorates Atherosclerosis and Hepatic Steatosis Via Inhibiting Lipid-Disorder and Inflammation in apoE−/− Mice. Front. Pharmacol. 2020, 11, 610550. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.H.; Cha, H.; Tang, J.; Lee, S.; Lee, S.H.; Le, B.; Redding, M.C.; Kim, S.; Batish, M.; Kong, B.C.; et al. The role of microRNA-33 as a key regulator in hepatic lipogenesis signaling and a potential serological biomarker for NAFLD with excessive dietary fructose consumption in C57BL/6N mice. Food Funct. 2021, 12, 656–667. [Google Scholar] [CrossRef]
- Sud, N.; Zhang, H.; Pan, K.; Cheng, X.; Cui, J.; Su, Q. Aberrant expression of microRNA induced by high-fructose diet: Implications in the pathogenesis of hyperlipidemia and hepatic insulin resistance. J. Nutr. Biochem. 2017, 43, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Hanousková, B.; Neprašová, B.; Skálová, L.; Maletínská, L.; Zemanová, K.; Ambrož, M.; Matoušková, P. High-fructose drinks affect microRNAs expression differently in lean and obese mice. J. Nutr. Biochem. 2019, 68, 42–50. [Google Scholar] [CrossRef]
- Chen, L.; Chen, X.W.; Huang, X.; Song, B.L.; Wang, Y.; Wang, Y. Regulation of glucose and lipid metabolism in health and disease. Sci. China Life Sci. 2019, 62, 1420–1458. [Google Scholar] [CrossRef]
- Lin, X.; Xing, Y.; Zhang, Y.; Dong, B.; Zhao, M.; Wang, J.; Geng, T.; Gong, D.; Zheng, Y.; Liu, L. Glucose participates in the formation of goose fatty liver by regulating the expression of miRNA-33/CROT. Anim. Sci. J. 2021, 92, e13674. [Google Scholar] [CrossRef] [PubMed]
- Ghareghani, P.; Shanaki, M.; Ahmadi, S.; Khoshdel, A.R.; Rezvan, N.; Meshkani, R.; Delfan, M.; Gorgani-Firuzjaee, S. Aerobic endurance training improves nonalcoholic fatty liver disease (NAFLD) features via miR-33 dependent autophagy induction in high fat diet fed mice. Obes. Res. Clin. Pract. 2018, 12 (Suppl. 2), 80–89. [Google Scholar] [CrossRef]
- Vega-Badillo, J.; Gutiérrez-Vidal, R.; Hernández-Pérez, H.A.; Villamil-Ramírez, H.; León-Mimila, P.; Sánchez-Muñoz, F.; Morán-Ramos, S.; Larrieta-Carrasco, E.; Fernández-Silva, I.; Méndez-Sánchez, N.; et al. Hepatic miR-33a/miR-144 and their target gene ABCA1 are associated with steatohepatitis in morbidly obese subjects. Liver Int. 2016, 36, 1383–1391. [Google Scholar] [CrossRef]
- Price, N.L.; Zhang, X.; Fernández-Tussy, P.; Singh, A.K.; Burnap, S.A.; Rotllan, N.; Goedeke, L.; Sun, J.; Canfrán-Duque, A.; Aryal, B.; et al. Loss of hepatic miR-33 improves metabolic homeostasis and liver function without altering body weight or atherosclerosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2006478118. [Google Scholar] [CrossRef] [PubMed]
- WHO. Obesity and Overweight: World Health Organization. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 9 June 2021).
- Boutari, C.; Mantzoros, C.S. A 2022 update on the epidemiology of obesity and a call to action: As its twin COVID-19 pandemic appears to be receding, the obesity and dysmetabolism pandemic continues to rage on. Metabolism 2022, 133, 155217. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Zatterale, F.; Naderi, J.; Parrillo, L.; Formisano, P.; Raciti, G.A.; Beguinot, F.; Miele, C. Adipose Tissue Dysfunction as Determinant of Obesity-Associated Metabolic Complications. Int. J. Mol. Sci. 2019, 20, 2358. [Google Scholar] [CrossRef] [Green Version]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Kunz, H.E.; Hart, C.R.; Gries, K.J.; Parvizi, M.; Laurenti, M.; Dalla Man, C.; Moore, N.; Zhang, X.; Ryan, Z.; Polley, E.C.; et al. Adipose tissue macrophage populations and inflammation are associated with systemic inflammation and insulin resistance in obesity. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E105–E121. [Google Scholar] [CrossRef]
- Karunakaran, D.; Richards, L.; Geoffrion, M.; Barrette, D.; Gotfrit, R.J.; Harper, M.E.; Rayner, K.J. Therapeutic Inhibition of miR-33 Promotes Fatty Acid Oxidation but Does Not Ameliorate Metabolic Dysfunction in Diet-Induced Obesity. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2536–2543. [Google Scholar] [CrossRef] [Green Version]
- Price, N.L.; Rotllan, N.; Canfrán-Duque, A.; Zhang, X.; Pati, P.; Arias, N.; Moen, J.; Mayr, M.; Ford, D.A.; Baldán, Á.; et al. Genetic Dissection of the Impact of miR-33a and miR-33b during the Progression of Atherosclerosis. Cell Rep. 2017, 21, 1317–1330. [Google Scholar] [CrossRef] [Green Version]
- Gancheva, S.; Jelenik, T.; Álvarez-Hernández, E.; Roden, M. Interorgan Metabolic Crosstalk in Human Insulin Resistance. Physiol. Rev. 2018, 98, 1371–1415. [Google Scholar] [CrossRef] [Green Version]
- Lair, B.; Laurens, C.; Van Den Bosch, B.; Moro, C. Novel Insights and Mechanisms of Lipotoxicity-Driven Insulin Resistance. Int. J. Mol. Sci. 2020, 21, 6358. [Google Scholar] [CrossRef]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar]
- Wijesekara, N.; Zhang, L.H.; Kang, M.H.; Abraham, T.; Bhattacharjee, A.; Warnock, G.L.; Verchere, C.B.; Hayden, M.R. miR-33a modulates ABCA1 expression, cholesterol accumulation, and insulin secretion in pancreatic islets. Diabetes 2012, 61, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Rottiers, V.; Obad, S.; Petri, A.; McGarrah, R.; Lindholm, M.W.; Black, J.C.; Sinha, S.; Goody, R.J.; Lawrence, M.S.; deLemos, A.S.; et al. Pharmacological inhibition of a microRNA family in nonhuman primates by a seed-targeting 8-mer antimiR. Sci. Transl. Med. 2013, 5, 212ra162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corona-Meraz, F.I.; Vázquez-Del Mercado, M.; Ortega, F.J.; Ruiz-Quezada, S.L.; Guzmán-Ornelas, M.O.; Navarro-Hernández, R.E. Ageing influences the relationship of circulating miR-33a and miR- 33b levels with insulin resistance and adiposity. Diab Vasc. Dis. Res. 2019, 16, 244–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, S.; EBRAHIMI, M.M.; Arefhosseini, S.R.; Fallah, P.; ASGHARI, J.M.; Zununi, S.; Soleimani, M.; BANITALEBI, D.M.; Ghanbarian, H. Dietary regulation of miR-33b and miR-29a in relationship to metabolic biomarkers of glucose and lipids in obese diabetic women: A randomized clinical controlled study. Iran. Red. Crescent Med. J. 2017, 19, e37521. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Tamasawa, N.; Matsumura, K.; Murakami, H.; Yamashita, M.; Matsuki, K.; Tanabe, J.; Murakami, H.; Matsui, J.; Daimon, M. Clinical Significance of Determining Plasma MicroRNA33b in Type 2 Diabetic Patients with Dyslipidemia. J. Atheroscler. Thromb. 2016, 23, 1276–1285. [Google Scholar] [CrossRef] [Green Version]
- Smith, N.K.; Hackett, T.A.; Galli, A.; Flynn, C.R. GLP-1: Molecular mechanisms and outcomes of a complex signaling system. Neurochem. Int. 2019, 128, 94–105. [Google Scholar] [CrossRef]
- Meier, J.J. GLP-1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 728–742. [Google Scholar] [CrossRef]
- Li, P.; Gao, X.; Sun, X.; Li, W.; Yi, B.; Zhu, L. A novel epigenetic mechanism of FXR inhibiting GLP-1 secretion via miR-33 and its downstream targets. Biochem. Biophys. Res. Commun. 2019, 517, 629–635. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, H.; Li, Q. Glucagon-like peptide-1 effects lipotoxic oxidative stress by regulating the expression of microRNAs. Biochem. Biophys. Res. Commun. 2017, 482, 1462–1468. [Google Scholar] [CrossRef]
- Cao, M. miRNA-33 expression and its mechanism in patients and model rats with type 2 diabetic nephropathy. Int. J. Clin. Exp. Med. 2018, 11, 1661–1668. [Google Scholar]
- Tian, K.; Liu, Z.; Wang, J.; Xu, S.; You, T.; Liu, P. Sirtuin-6 inhibits cardiac fibroblasts differentiation into myofibroblasts via inactivation of nuclear factor κB signaling. Transl. Res. 2015, 165, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaddaj Mallat, R.; Mathew John, C.; Kendrick, D.J.; Braun, A.P. The vascular endothelium: A regulator of arterial tone and interface for the immune system. Crit. Rev. Clin. Lab. Sci. 2017, 54, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [PubMed]
- Schnoor, M.; Alcaide, P.; Voisin, M.B.; van Buul, J.D. Crossing the Vascular Wall: Common and Unique Mechanisms Exploited by Different Leukocyte Subsets during Extravasation. Mediators Inflamm. 2015, 2015, 946509. [Google Scholar] [CrossRef] [Green Version]
- Ishizuka, T.; Takamizawa-Matsumoto, M.; Suzuki, K.; Kurita, A. Endothelin-1 enhances vascular cell adhesion molecule-1 expression in tumor necrosis factor alpha-stimulated vascular endothelial cells. Eur. J. Pharmacol. 1999, 369, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Colin, S.; Chinetti-Gbaguidi, G.; Staels, B. Macrophage phenotypes in atherosclerosis. Immunol. Rev. 2014, 262, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhao, W.S.; Xu, L.; Wang, X.; Li, X.L.; Yang, X.C. Endothelium-specific endothelin-1 expression promotes pro-inflammatory macrophage activation by regulating miR-33/NR4A axis. Exp. Cell Res. 2021, 399, 112443. [Google Scholar] [CrossRef]
- Zhao, Y.; Bruemmer, D. NR4A Orphan Nuclear Receptors in Cardiovascular Biology. Drug. Discov. Today Dis. Mech. 2009, 6, e43–e48. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Crean, D. Molecular Interactions between NR4A Orphan Nuclear Receptors and NF-κB Are Required for Appropriate Inflammatory Responses and Immune Cell Homeostasis. Biomolecules 2015, 5, 1302–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geovanini, G.R.; Libby, P. Atherosclerosis and inflammation: Overview and updates. Clin. Sci. 2018, 132, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M. ABCA1 and ABCG1 as potential therapeutic targets for the prevention of atherosclerosis. J. Pharmacol. Sci. 2022, 148, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.A.; Wyatt, H.; Susser, L.; Geoffrion, M.; Rasheed, A.; Duchez, A.C.; Cottee, M.L.; Afolayan, E.; Farah, E.; Kahiel, Z.; et al. Delivery of MicroRNAs by Chitosan Nanoparticles to Functionally Alter Macrophage Cholesterol Efflux in Vitro and in Vivo. ACS Nano 2019, 13, 6491–6505. [Google Scholar] [CrossRef]
- Mao, M.; Lei, H.; Liu, Q.; Chen, Y.; Zhao, L.; Li, Q.; Luo, S.; Zuo, Z.; He, Q.; Huang, W.; et al. Effects of miR-33a-5P on ABCA1/G1-mediated cholesterol efflux under inflammatory stress in THP-1 macrophages. PLoS ONE 2014, 9, e109722. [Google Scholar] [CrossRef] [Green Version]
- Rotllan, N.; Ramírez, C.M.; Aryal, B.; Esau, C.C.; Fernández-Hernando, C. Therapeutic silencing of microRNA-33 inhibits the progression of atherosclerosis in Ldlr−/− mice--brief report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1973–1977. [Google Scholar] [CrossRef] [Green Version]
- Anagnostakos, J.; Lal, B.K. Abdominal aortic aneurysms. Prog. Cardiovasc. Dis. 2021, 65, 34–43. [Google Scholar] [CrossRef]
- Haque, K.; Bhargava, P. Abdominal Aortic Aneurysm. Am. Fam. Physician. 2022, 106, 165–172. [Google Scholar]
- Zhao, L.; Huang, J.; Zhu, Y.; Han, S.; Qing, K.; Wang, J.; Feng, Y. miR-33-5p knockdown attenuates abdominal aortic aneurysm progression via promoting target adenosine triphosphate-binding cassette transporter A1 expression and activating the PI3K/Akt signaling pathway. Perfusion 2020, 35, 57–65. [Google Scholar] [CrossRef]
- Yamasaki, T.; Horie, T.; Koyama, S.; Nakao, T.; Baba, O.; Kimura, M.; Sowa, N.; Sakamoto, K.; Yamazaki, K.; Obika, S.; et al. Inhibition of microRNA-33b specifically ameliorates abdominal aortic aneurysm formation via suppression of inflammatory pathways. Sci. Rep. 2022, 12, 11984. [Google Scholar] [CrossRef]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef] [PubMed]
- Price, N.L.; Holtrup, B.; Kwei, S.L.; Wabitsch, M.; Rodeheffer, M.; Bianchini, L.; Suárez, Y.; Fernández-Hernando, C. SREBP-1c/MicroRNA 33b Genomic Loci Control Adipocyte Differentiation. Mol. Cell. Biol. 2016, 36, 1180–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, G.; Malmuthuge, N.; McFadden, T.B.; Bao, H.; Griebel, P.J.; Stothard, P.; Guan, L.L. Potential regulatory role of microRNAs in the development of bovine gastrointestinal tract during early life. PLoS ONE 2014, 9, e92592. [Google Scholar] [CrossRef] [Green Version]
- Chang, M.; Qiao, L.; Li, B.; Wang, J.; Zhang, G.; Shi, W.; Liu, Z.; Gu, N.; Di, Z.; Wang, X.; et al. Suppression of SIRT6 by miR-33a facilitates tumor growth of glioma through apoptosis and oxidative stress resistance. Oncol. Rep. 2017, 38, 1251–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirera-Salinas, D.; Pauta, M.; Allen, R.M.; Salerno, A.G.; Ramírez, C.M.; Chamorro-Jorganes, A.; Wanschel, A.C.; Lasuncion, M.A.; Morales-Ruiz, M.; Suarez, Y.; et al. Mir-33 regulates cell proliferation and cell cycle progression. Cell Cycle 2012, 11, 922–933. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Xu, D.; Xie, H.; Tang, J.; Liu, R.; Li, J.; Wang, S.; Chen, X.; Su, J.; Zhou, X.; et al. miR-33a functions as a tumor suppressor in melanoma by targeting HIF-1α. Cancer Biol. Ther. 2015, 16, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Moradi-Chaleshtori, M.; Bandehpour, M.; Heidari, N.; Mohammadi-Yeganeh, S.; Mahmoud Hashemi, S. Exosome-mediated miR-33 transfer induces M1 polarization in mouse macrophages and exerts antitumor effect in 4T1 breast cancer cell line. Int. Immunopharmacol. 2021, 90, 107198. [Google Scholar] [CrossRef]
- Yu, B.; Li, W.; Al, F.; Chen, Z. MicroRNA-33a deficiency inhibits proliferation and fibrosis through inactivation of TGF-β/Smad pathway in human cardiac fibroblasts. Pharmazie 2017, 72, 456–460. [Google Scholar]
- Ono, K. Functions of microRNA-33a/b and microRNA therapeutics. J. Cardiol 2016, 67, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar] [CrossRef]
- Patrick, D.M.; Montgomery, R.L.; Qi, X.; Obad, S.; Kauppinen, S.; Hill, J.A.; van Rooij, E.; Olson, E.N. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J. Clin. Investig. 2010, 120, 3912–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Täubel, J.; Hauke, W.; Rump, S.; Viereck, J.; Batkai, S.; Poetzsch, J.; Rode, L.; Weigt, H.; Genschel, C.; Lorch, U.; et al. Novel antisense therapy targeting microRNA-132 in patients with heart failure: Results of a first-in-human Phase 1b randomized, double-blind, placebo-controlled study. Eur. Heart J. 2021, 42, 178–188. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhu, X.; Zhang, X.; Liu, B.; Huang, L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Mol. Ther. 2010, 18, 1650–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, R.; Liu, B.; Persaud, S.J. Effects of miR-33 Deficiency on Metabolic and Cardiovascular Diseases: Implications for Therapeutic Intervention. Int. J. Mol. Sci. 2023, 24, 10777. https://doi.org/10.3390/ijms241310777
Ortega R, Liu B, Persaud SJ. Effects of miR-33 Deficiency on Metabolic and Cardiovascular Diseases: Implications for Therapeutic Intervention. International Journal of Molecular Sciences. 2023; 24(13):10777. https://doi.org/10.3390/ijms241310777
Chicago/Turabian StyleOrtega, Rebeca, Bo Liu, and Shanta J. Persaud. 2023. "Effects of miR-33 Deficiency on Metabolic and Cardiovascular Diseases: Implications for Therapeutic Intervention" International Journal of Molecular Sciences 24, no. 13: 10777. https://doi.org/10.3390/ijms241310777