
Transcriptome Analyses Show Changes in Gene Expression Triggered by a 31-bp InDel within OsSUT3 5′UTR in Rice Panicle
Abstract
:1. Introduction
2. Results
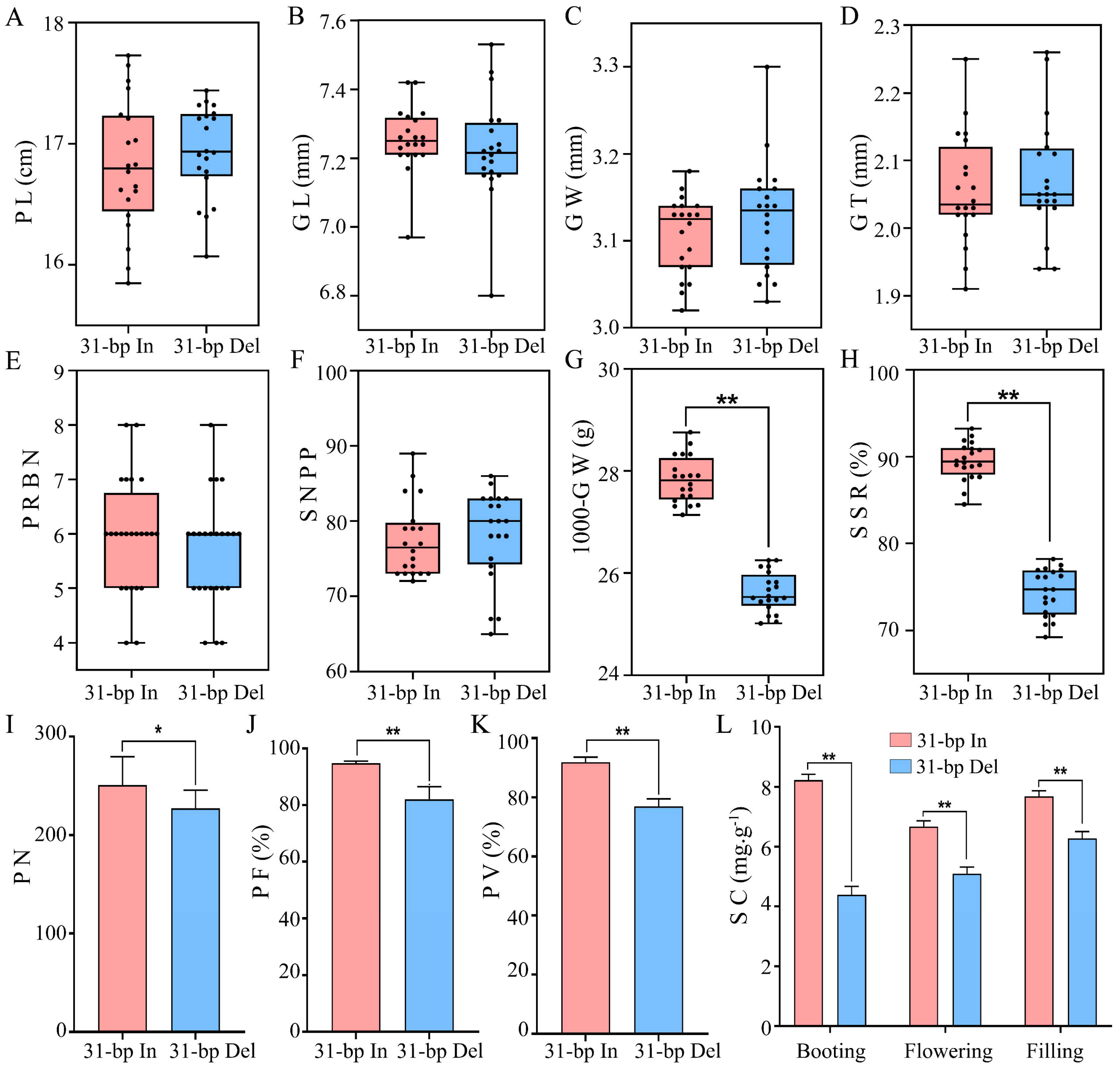
2.1. Improvement of Pollen and Panicle-Related Traits in 31-bp in Transgenic Lines
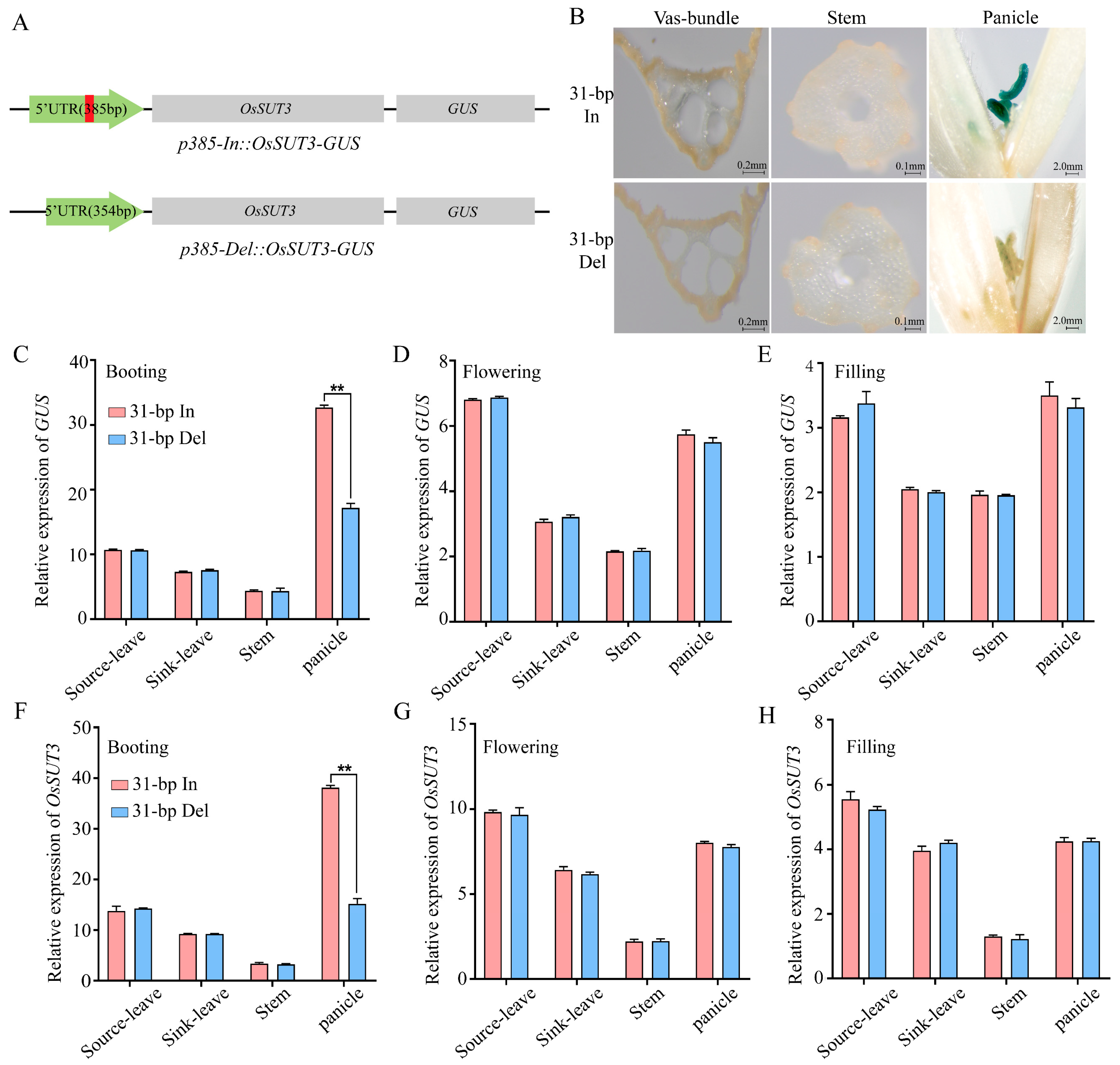
2.2. Expression Changes in OsSUT3 and GUS in Different Tissues and Different Developmental Stage of Transgenic Plants
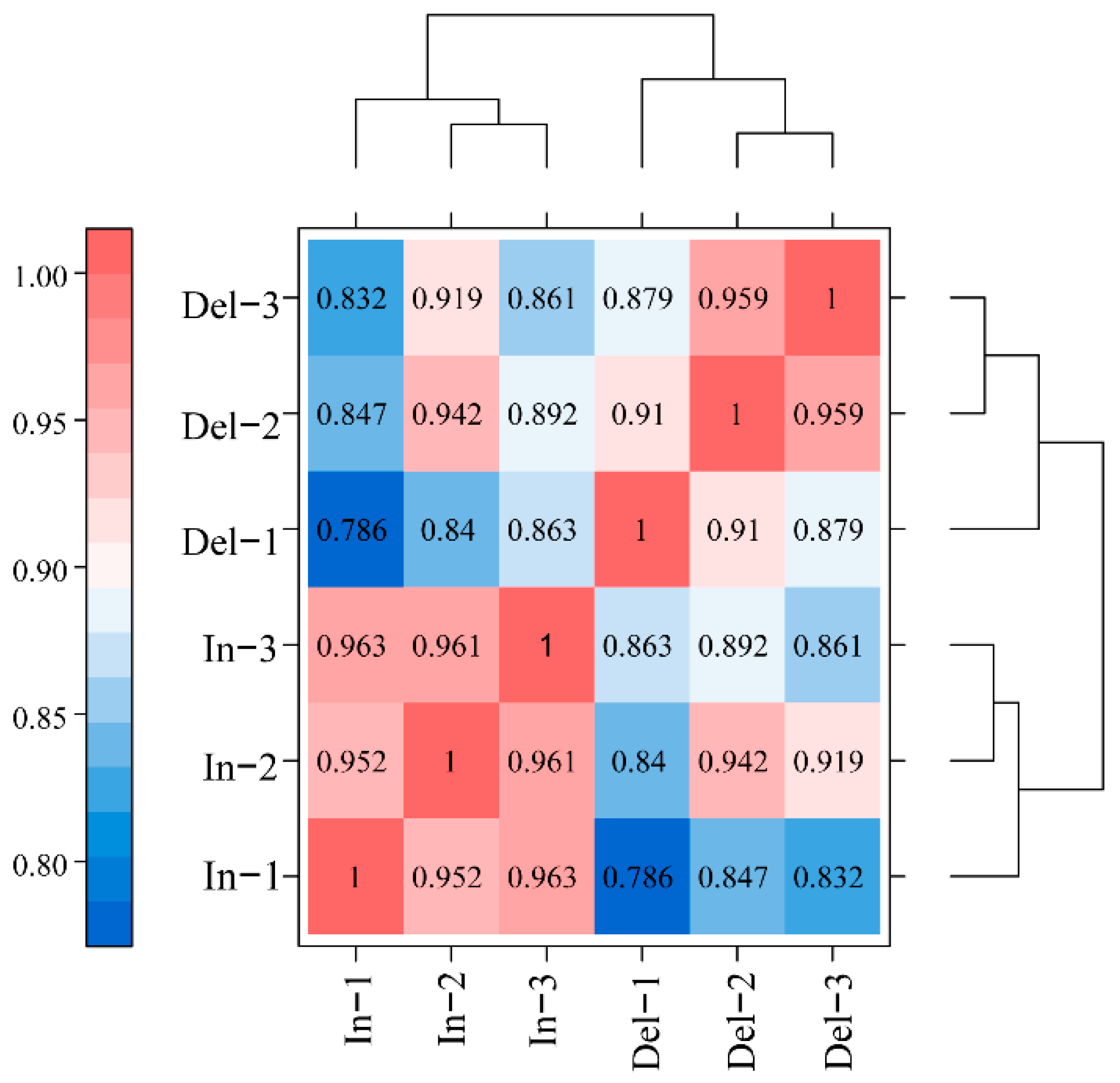
2.3. Overview of the Transcriptomic Differences to 31-bp InDel
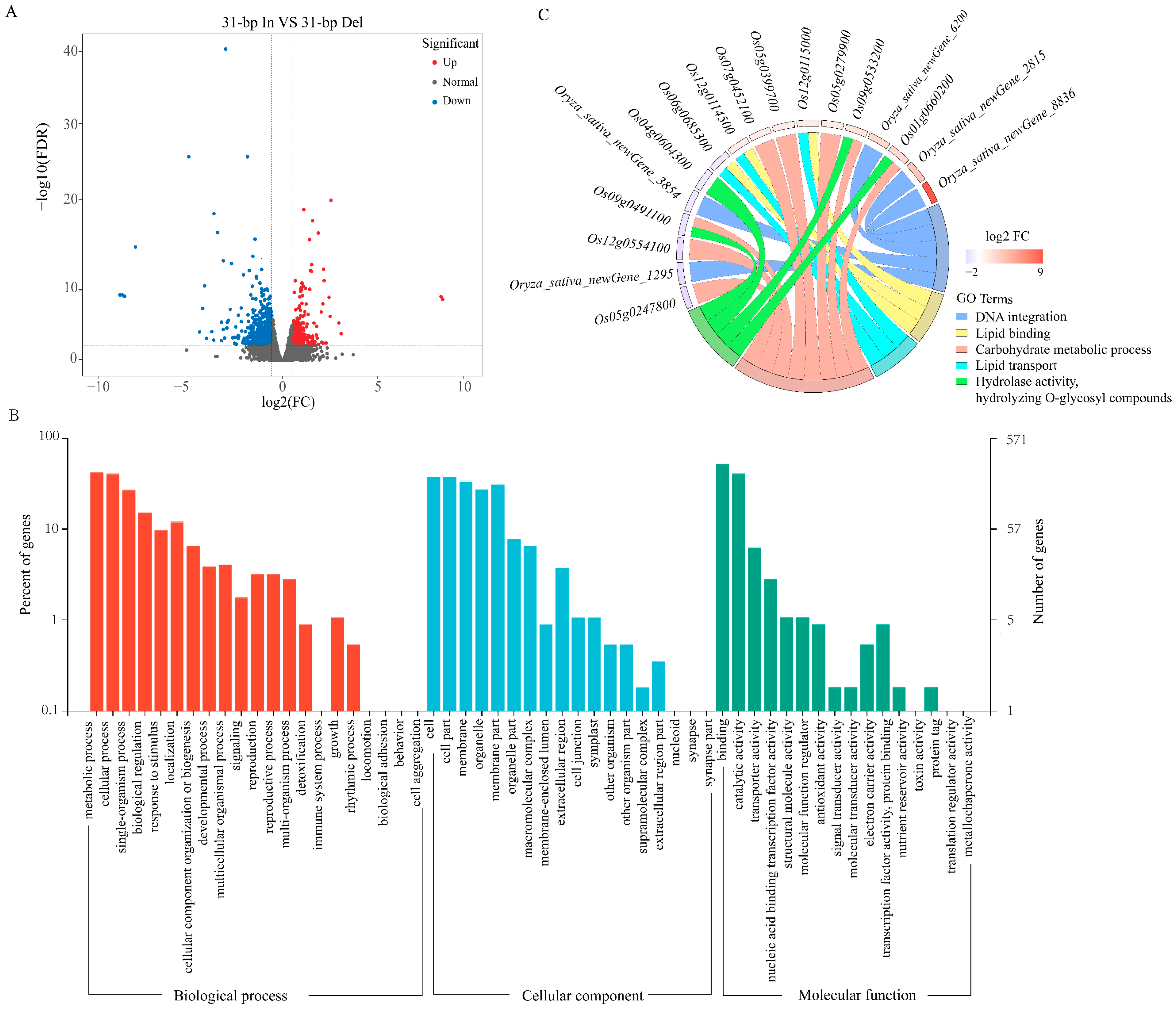
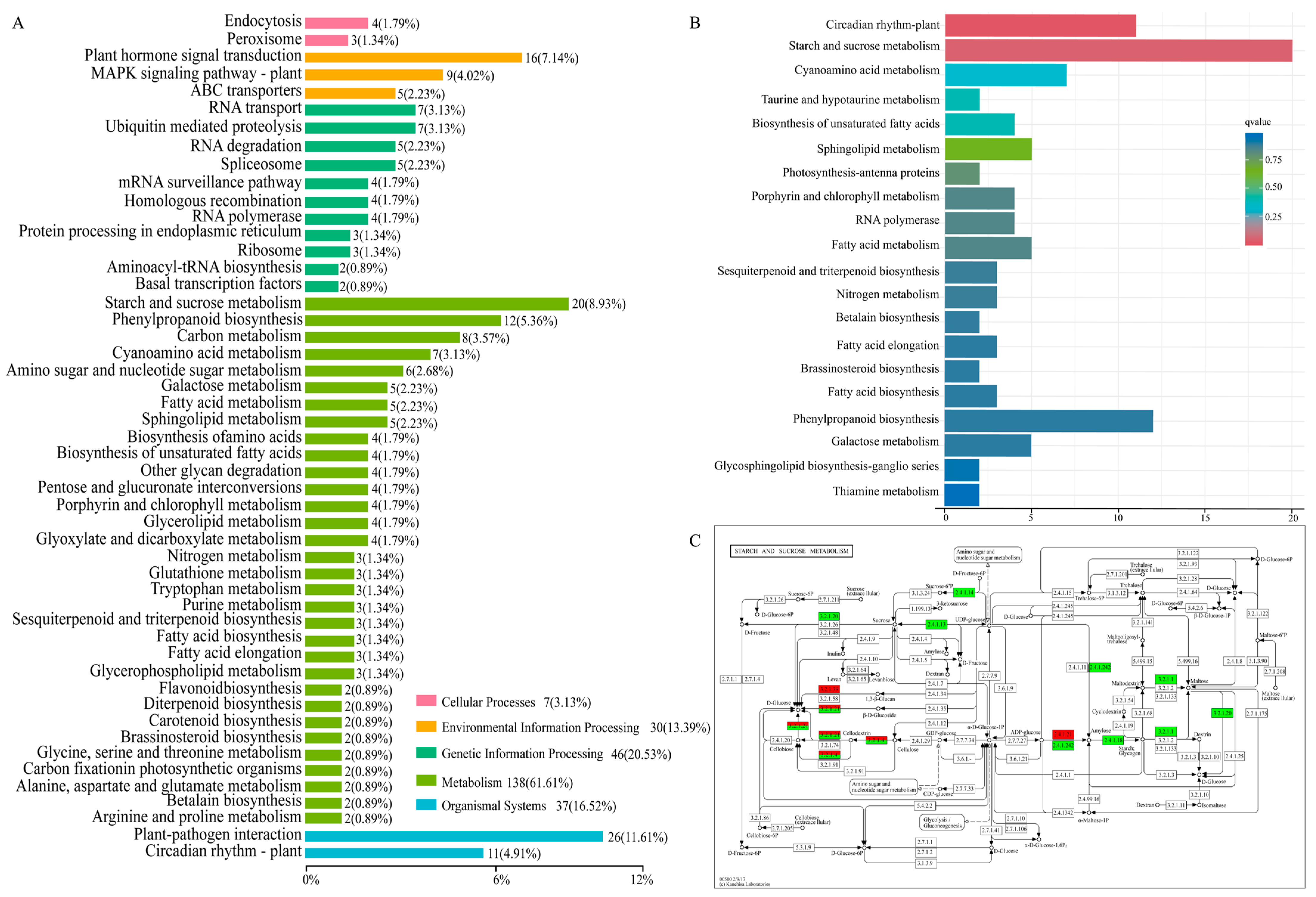
2.4. DEGs Identification and Enrichment Analysis
2.5. Enriched Metabolic Pathways of DEGs by KEGG
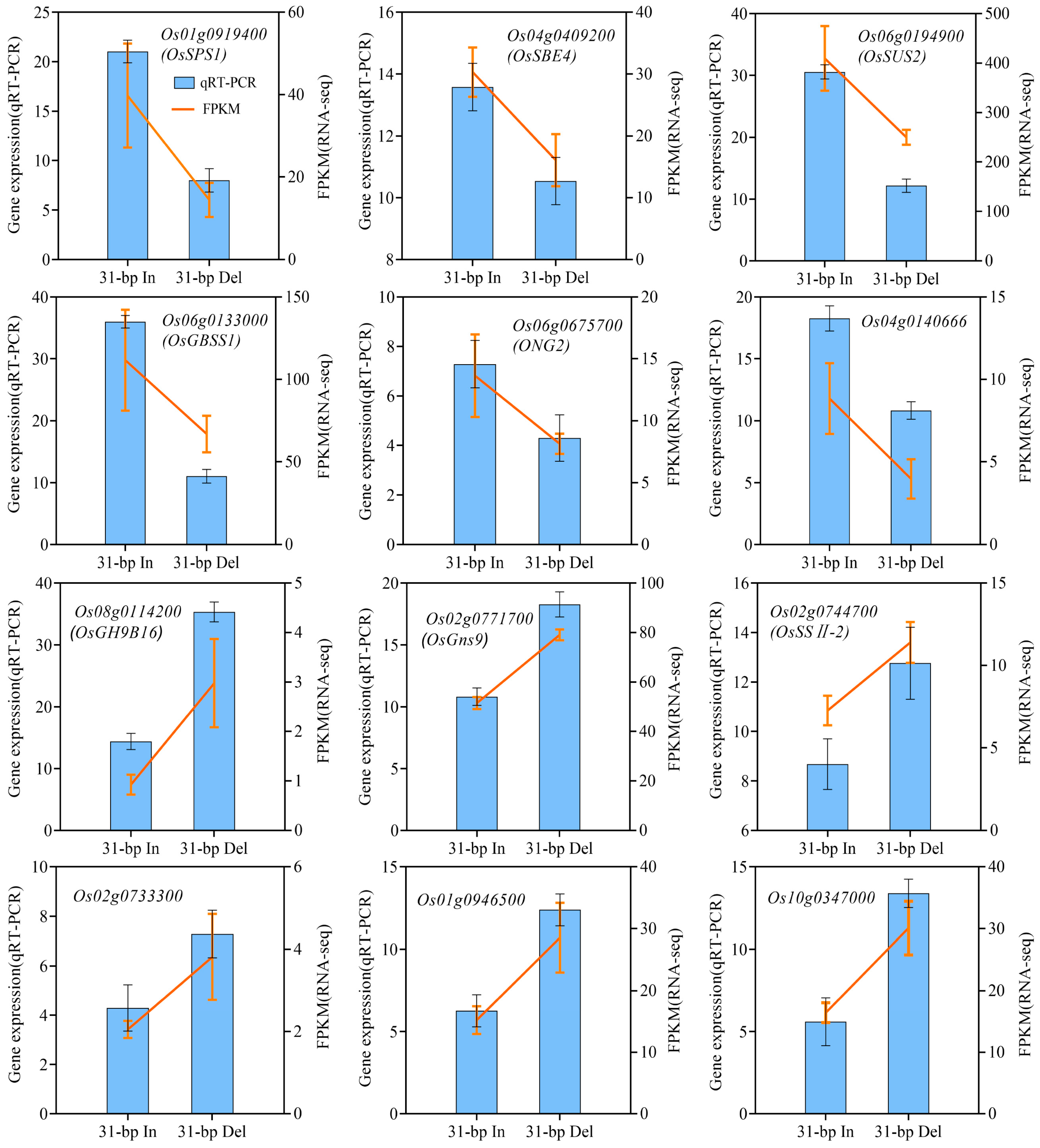
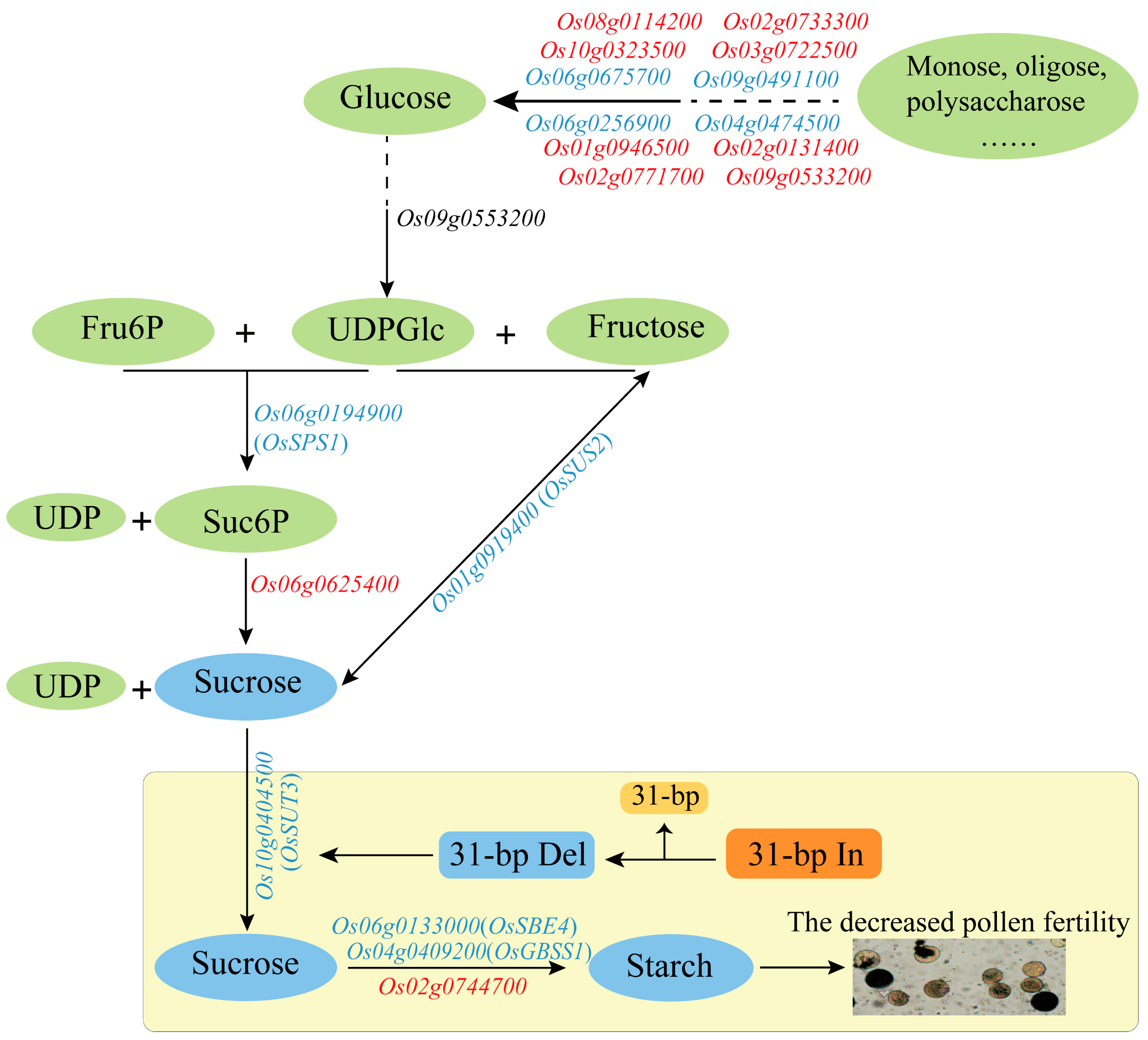
2.6. Expression Analysis of Sucrose and Starch Biosynthesis-Related Genes under 31-bp Deletion
3. Discussion
4. Materials and Methods
4.1. Plasmid Construction and Transformation
4.2. Plant Materials and Growth Conditions
4.3. GUS Staining
4.4. Total RNA Extraction and qRT-PCR Analysis of Gene Expression
4.5. Detection of Pollen Fertility and Viability
4.6. Determination of Sucrose Content
4.7. Determination of Panicle Character
4.8. Library Preparation for Transcriptome Sequencing
4.9. Data Filtering, Reads Mapping and RNA-Seq Data Analysis
4.10. Transcriptomic Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tran, T.M.; Hampton, C.S.; Brossard, T.W.; Harmata, M.; Robertson, J.D.; Jurisson, S.S.; Braun, D.M. In vivo transport of three radioactive [18F]-fluorinated deoxysucrose analogs by the maize sucrose transporter ZmSUT1. Plant Physiol. Bioch. 2017, 115, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kühn, C.; Grof, C.P. Sucrose transporters of higher plants. Curr. Opin. Plant Biol. 2010, 13, 288–298. [Google Scholar] [CrossRef]
- Hu, Z.; Tang, Z.; Zhang, Y.; Niu, L.; Yang, F.; Zhang, D.; Hu, Y. Rice SUT and SWEET transporters. Int. J. Mol. Sci. 2021, 22, 11198. [Google Scholar] [CrossRef] [PubMed]
- Aoki, N.; Hirose, T.; Scofield, G.N.; Whitfeld, P.R.; Furbank, R.T. The sucrose transporter gene family in rice. Plant Cell Physiol. 2003, 44, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, T.; Zhang, Z.; Miyao, A.; Hirochika, H.; Ohsugi, R.; Terao, T. Disruption of a gene for rice sucrose transporter, OsSUT1, impairs pollen function but pollen maturation is unaffected. J. Exp. Bot. 2010, 61, 3639–3646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, T.; Toyofuku, K.; Sobolewska, A.; Matsukura, C.; Yamaguchi, J. Sugar transporters involved in flowering and grain development of rice. J. Plant Physlol. 2001, 158, 465–470. [Google Scholar] [CrossRef]
- Li, D.D.; Xu, R.C.; Lv, D.; Zhang, C.L.; Yang, H.; Zhang, J.B.; Wen, J.C.; Li, C.Y.; Tan, X.L. Identification of the core pollen-specific regulation in the rice OsSUT3 promoter. Int. J. Mol. Sci. 2020, 21, 1909. [Google Scholar] [CrossRef] [Green Version]
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416. [Google Scholar] [CrossRef]
- Chen, C.H.; Lin, H.Y.; Pan, C.L.; Chen, F.C. The plausible reason why the length of 5' untranslated region is unrelated to organismal complexity. BMC Res. Notes. 2011, 4, 312. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Cheng, M.; Chen, Y.; Liu, B.J.; Wang, X.N.; Li, G.J.; Zhou, Y.H.; Luo, P.; Xi, Z.Y.; Yong, H.J.; et al. Natural variations in the non-coding region of ZmNAC080308 contributes maintaining grain yield under drought stress in maize. BMC Plant Biol. 2021, 21, 305. [Google Scholar] [CrossRef]
- Feng, Y.M.; Liu, M.; Wang, Z.; Zhao, X.L.; Han, B.; Xing, Y.P.; Wang, M.Y.; Yang, Y. A 4-bp deletion in the 5’UTR of TaAFP-B is associated with seed dormancy in common wheat (Triticum aestivum L.). BMC Plant Biol. 2019, 19, 349. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.P.; Han, Z.S.; Han, A.N.; Liu, X.J.; Zhang, S.Y.; Fu, B.Y.; Hu, J.; Su, J.P.; Li, S.Q.; Wang, S.J.; et al. Sdt97: A point mutation in the 5’untranslated region confers semidwarfism in rice. G3-Genes Genom. Genet. 2016, 6, 1491–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.L.; Li, Q.P.; Yang, H.; Wang, T.; Li, J.; Wen, J.C.; Jin, S.L.; Zhang, Z.L.; Chen, L.J.; Li, D.D. A 31-bp indel localised in the 5’untranslated region of OsSUT3 affects the gene expression and rice (Oryza sativa L.) pollen development. Czech J. Genet. Plant Breed. 2022, 58, 21–28. [Google Scholar] [CrossRef]
- Hazen, S.P.; Wu, Y.J.; Kreps, J.A. Gene expression profiling of plant responses to abiotic stress. Funct. Integr. Genomics 2003, 3, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.; Zhu, J.Y.; Wang, Z.Y. Interaction between BZR1 and PIF4 integrates brassinosteroid and environmental responses. Nat. Cell Biol. 2012, 14, 802–809. [Google Scholar] [CrossRef] [Green Version]
- Luo, M.M.; Liu, X.X.; Su, H.Y.; Li, M.L.; Li, M.F.; Wei, J.H. Regulatory networks of flowering genes in angelica sinensis during vernalization. Plants 2022, 11, 1355. [Google Scholar] [CrossRef]
- Wu, Y.F.; Lee, S.K.; Yoo, Y.; Wei, J.H.; Kwon, S.Y.; Lee, S.W.; Jeon, J.S.; An, G. Rice transcription factor OsDOF11 modulates sugar transport by promoting expression of sucrose transporter and SWEET genes. Mol. Plant. 2018, 11, 833–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.K.; Lee, J.; Jo, M.; Jeon, J.S. exploration of sugar and starch metabolic pathway crucial for pollen fertility in rice. Int. J. Mol. Sci. 2022, 23, 14091. [Google Scholar] [CrossRef]
- Li, J.B.; Kim, Y.J.; Zhang, D.B. Source-to-sink transport of sugar and its role in male reproductive development. Genes 2022, 13, 1323. [Google Scholar] [CrossRef]
- Jansing, J.; Buyel, J.F. The correlation between DsRed mRNA levels and transient dsred protein expression in plants depends on leaf age and the 5’Untranslated region. Biotechnol. J. 2019, 14, e1800075. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.C.; Yang, G.P.; He, Y.; Zhang, S.M.; Chen, H.Y.; Shen, P.; Chen, X.D.; Huang, Y.P. Nucleotides flanking the start codon in hsp70 mRNAs with very short 5’UTRs greatly affect gene expression in haloarchaea. PLoS ONE 2015, 10, e0138473. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Lee, G.; Jeon, E.; Sohn, E.J.; Lee, Y.; Kang, H.; Lee, D.W.; Kim, D.H.; Hwang, I. The immediate upstream region of the 5’UTR from the AUG start codon has a pronounced effect on the translational efficiency in Arabidopsis thaliana. Nucleic Acids Res. 2014, 42, 485–498. [Google Scholar] [CrossRef]
- Datta, R.; Chamusco, K.C.; Chourey, P.S. Starch biosynthesis during pollen maturation is associated with altered patterns of gene expression in maize. Plant Physiol. 2002, 130, 1645–1656. [Google Scholar] [CrossRef] [Green Version]
- Mu, H.; Ke, J.H.; Liu, W.; Zhuang, C.X.; Yip, W.K. UDP-glucose pyrophosphorylase2 (OsUgp2), a pollen-preferential gene in rice, plays a critical role in starch accumulation during pollen maturation. Chin. Sci. Bull. 2009, 2, 234–243. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.Y.; Gan, Z.S.; He, Y.S.; Li, Y.H.; Liu, X.D.; Mu, H. Functional analysis of a rice late pollen-abundant UDP-glucose pyrophosphorylase (OsUgp2) promoter. Mol. Biol. Rep. 2011, 38, 4291–4302. [Google Scholar] [CrossRef] [PubMed]
- Brenig, B.; Duan, Y.Y.; Xing, Y.Y.; Ding, N.S.; Huang, L.S.; Schütz, E. Porcine SOX9 gene expression is influenced by an 18 bp Indel in the 5’-Untranslated region. PLoS ONE 2015, 10, e0139583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simsek, D.; Tiu, G.C.; Flynn, R.A.; Byeon, G.W.; Leppek, K.; Xu, A.F.; Chang, H.Y.; Barna, M. The mammalian ribo-interactome reveals ribosome functional diversity and heterogeneity. Cell 2017, 169, 1051–1057. [Google Scholar] [CrossRef] [Green Version]
- Ren, T.H.; Yang, Y.; Lin, W.J.; Li, W.Y.; Xian, M.J.; Fu, R.; Zhang, Z.H.; Mo, G.D.; Luo, W.; Zhang, X.Q. A 31-bp indel in the 5’UTR region of GNB1L is significantly associated with chicken body weight and carcass traits. BMC Genet. 2020, 21, 91. [Google Scholar] [CrossRef]
- Seung, D. Amylose in starch: Towards an understanding of biosynthesis, structure and function. New Phytol. 2020, 228, 1490–1504. [Google Scholar] [CrossRef]
- Chávez-Bárcenas, A.T.; Valdez-Alarcón, J.J.; Martínez-Trujillo, M.; Chen, L.; Xoconostle-Cázares, B.; Lucas, W.J.; Herrera-Estrella, L. Tissue-specific and developmental pattern of expression of the rice sps1 gene. Plant Physiol. 2000, 124, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.F.; Wang, G.Y.; Wang, Y.M.; Zhang, R.; Wang, Y.T.; Feng, S.Q.; Luo, K.M.; Peng, L.C. Sucrose synthase enhances hull size and grain weight by regulating cell division and starch accumulation in transgenic rice. Int. J. Mol. Sci. 2019, 20, 4971. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Hashida, Y.; Aoki, N.; Okamura, M.; Yonekura, M.; Ohto, C.; Terao, T.; Ohsugi, R. Analysis of gene-disruption mutants of a sucrose phosphate synthase gene in rice, OsSPS1, shows the importance of sucrose synthesis in pollen germination. Plant Sci. 2014, 225, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Sugiyama, T.; Minamikawa, T.; Akazawa, T. Enzymic mechanism of starch synthesis in ripening rice grains. 3. Mechanism of the sucrose-starch conversion. Arch. Biochem. Biophys. 1966, 113, 34–44. [Google Scholar] [CrossRef]
- Shapter, F.M.; Eggler, P.; Lee, L.S.; Henry, R.J. Variation in Granule Bound Starch Synthase I (GBSSI) loci amongst Australian wild cereal relatives (Poaceae). J. Cereal Sci. 2009, 49, 4–11. [Google Scholar] [CrossRef]
- Sano, Y. Differential regulation of waxy gene expression in rice endosperm. Theor. Appl. Genet. 1984, 68, 467–473. [Google Scholar] [CrossRef]
- Saha, D.; Kumar, V.; Bhat, S.R.; Srinivasan, R. Upstream sequences of the LOJ gene leads to identification of a novel enhancer element conferring lateral organ junction-specific expression in Arabidopsis thaliana. Plant Mol. Biol. Rep. 2011, 29, 265–277. [Google Scholar] [CrossRef]
- Arocho, A.; Chen, B.Y.; Ladanyi, M.; Pan, Q.L. Validation of the 2−ΔΔCt calculation as an alternate method of data analysis for quantitative PCR of BCR-ABL P210 transcripts. Diagn. Mol. Pathol. 2006, 15, 56–61. [Google Scholar] [CrossRef]
- Zhang, D.B.; Luo, X.; Zhu, L. Cytological analysis and genetic control of rice anther development. J. Genet. Genomics 2011, 38, 379–390. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.Z.; Cai, T.; Olyarchuk, J.G.; Wei, L.P. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Clean Reads | Clean Bases | Q30 (%) | GC Content (%) |
|---|---|---|---|---|
| 31-bp In | 49,827,635 | 7,437,804,261 | 92.72 | 48.68 |
| 31-bp Del | 44,718,088 | 6,667,648,177 | 92.50 | 48.54 |
| Samples | Total Clean Reads | Clean Read Mapped (%) | Uniquely Mapped (%) | Multiple Mapped (%) | Reads Map to Sense Chain (%) | Reads Map to Antisense Chain (%) |
|---|---|---|---|---|---|---|
| 31-bp In | 48,100,141 | 96.54 | 92.90 | 3.63 | 48.03 | 48.12 |
| 31-bp Del | 43,067,200 | 96.32 | 92.93 | 3.38 | 47.96 | 48.03 |
| Pathway | Gene ID | Gene Description | In VS Del | PCC |
|---|---|---|---|---|
| Starch and sucrose metabolism | Os01g0919400 | Sucrose phosphate synthase, sucrose synthesis in pollen germination | down | 0.865 |
| Os04g0140666 | Similar to OSIGBa0124C14.6 protein | down | 0.849 | |
| Os04g0409200 | Starch branching enzyme IIa, starch biosynthesis | down | 0.890 | |
| Os06g0133000 | Granule-bound starch synthase, synthesis of amylose in endosperm | down | 0.815 | |
| Os06g0194900 | Sucrose synthase 2 | down | 0.912 | |
| Os06g0675700 | Alpha-glucosidase, α-glucosidase in rice seeds | down | 0.831 | |
| Os08g0114200 | Glycoside hydrolase family 9 subclass B16 | up | −0.855 | |
| Os10g0347000 | X8 domain containing protein | up | −0.892 | |
| Os01g0946500 | Similar to Glucan endo-1,3-beta- glucosidase GV | up | −0.881 | |
| Os02g0733300 | Glycoside hydrolase family 9 subclass B1 | up | −0.865 | |
| Os02g0744700 | Similar to starch synthase isoform zSTSII-2 | up | −0.934 | |
| Os02g0771700 | Glycoside hydrolase, family 17 protein | up | −0.990 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Q.; Zhang, C.; Wen, J.; Chen, L.; Shi, Y.; Yang, Q.; Li, D. Transcriptome Analyses Show Changes in Gene Expression Triggered by a 31-bp InDel within OsSUT3 5′UTR in Rice Panicle. Int. J. Mol. Sci. 2023, 24, 10640. https://doi.org/10.3390/ijms241310640
Li Q, Zhang C, Wen J, Chen L, Shi Y, Yang Q, Li D. Transcriptome Analyses Show Changes in Gene Expression Triggered by a 31-bp InDel within OsSUT3 5′UTR in Rice Panicle. International Journal of Molecular Sciences. 2023; 24(13):10640. https://doi.org/10.3390/ijms241310640
Chicago/Turabian StyleLi, Qiuping, Chunlong Zhang, Jiancheng Wen, Lijuan Chen, Yitong Shi, Qinghui Yang, and Dandan Li. 2023. "Transcriptome Analyses Show Changes in Gene Expression Triggered by a 31-bp InDel within OsSUT3 5′UTR in Rice Panicle" International Journal of Molecular Sciences 24, no. 13: 10640. https://doi.org/10.3390/ijms241310640