
Histone Maps in Gossypium darwinii Reveal Epigenetic Regulation Drives Subgenome Divergence and Cotton Domestication
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Genome–Wide Distribution of H3K4me3, H3K27me3, H3K36me3, H3K27ac, and H3K9ac in the Cotton Genome
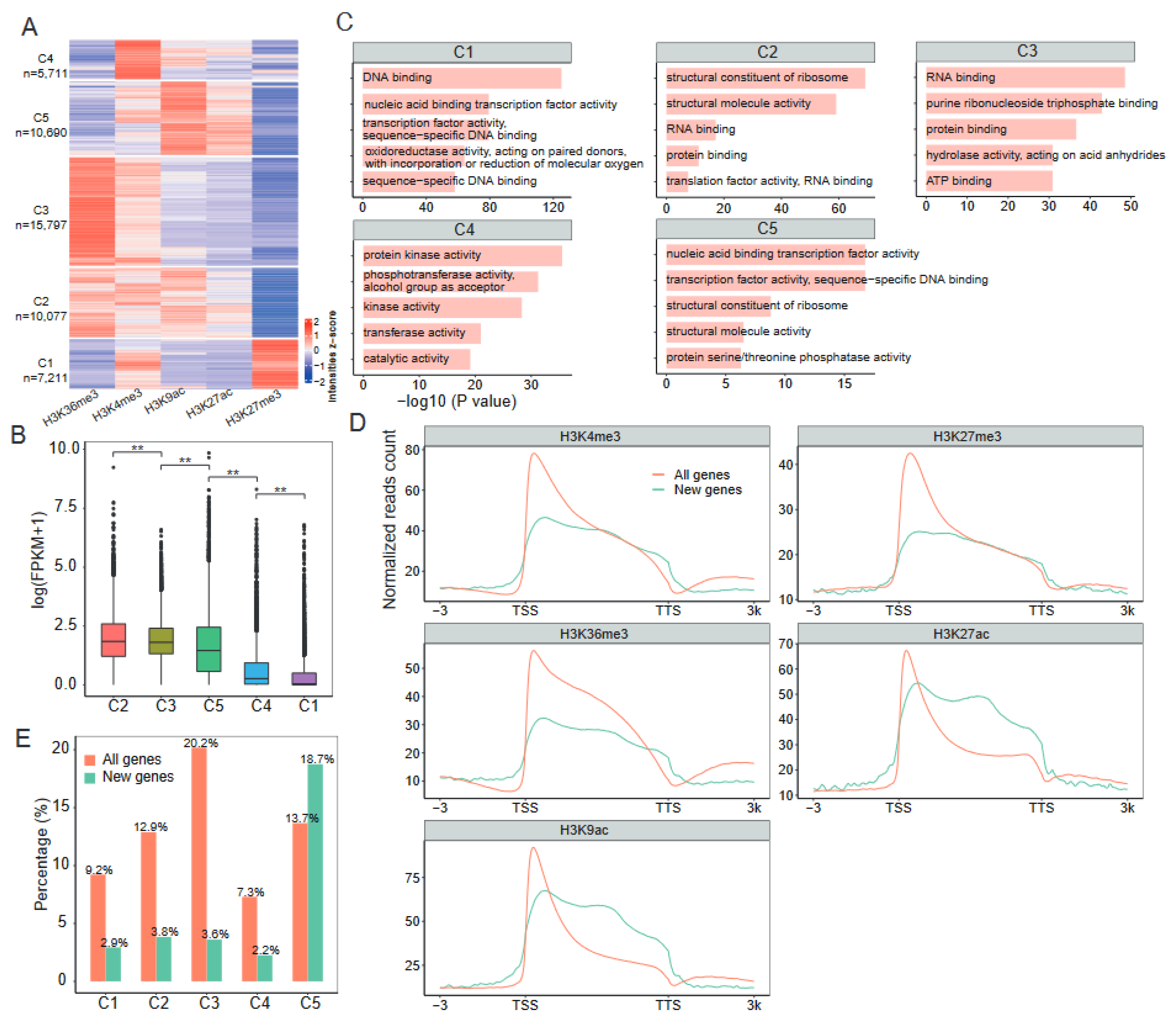
2.2. Distinct Combinatorial Patterns of Histone Modifications on Genes
2.3. Epigenetic Regulation of Newly Generated Genes during Cotton Allopolyploidization
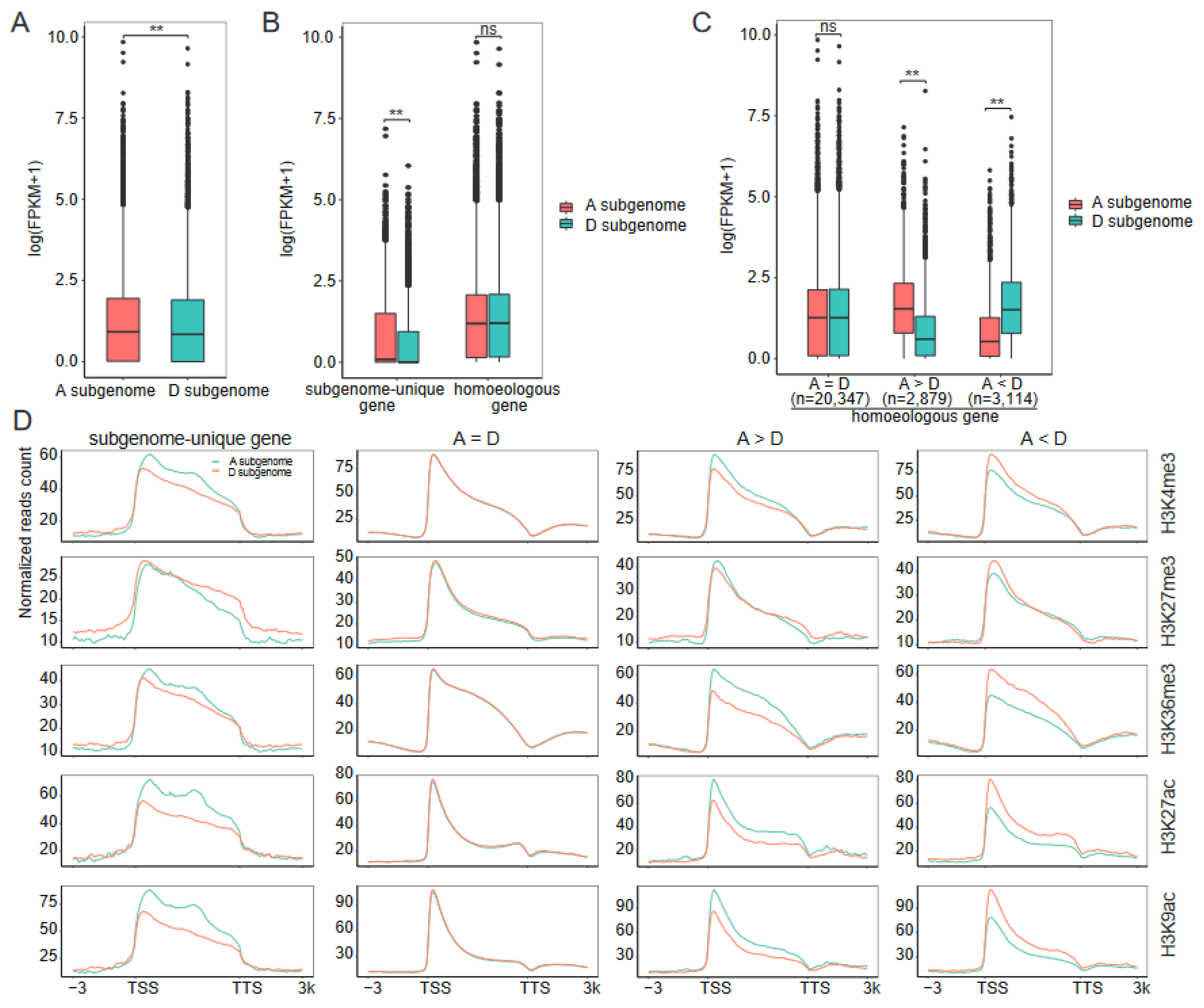
2.4. Association between Subgenome Bias in Transcription and Histone Modifications
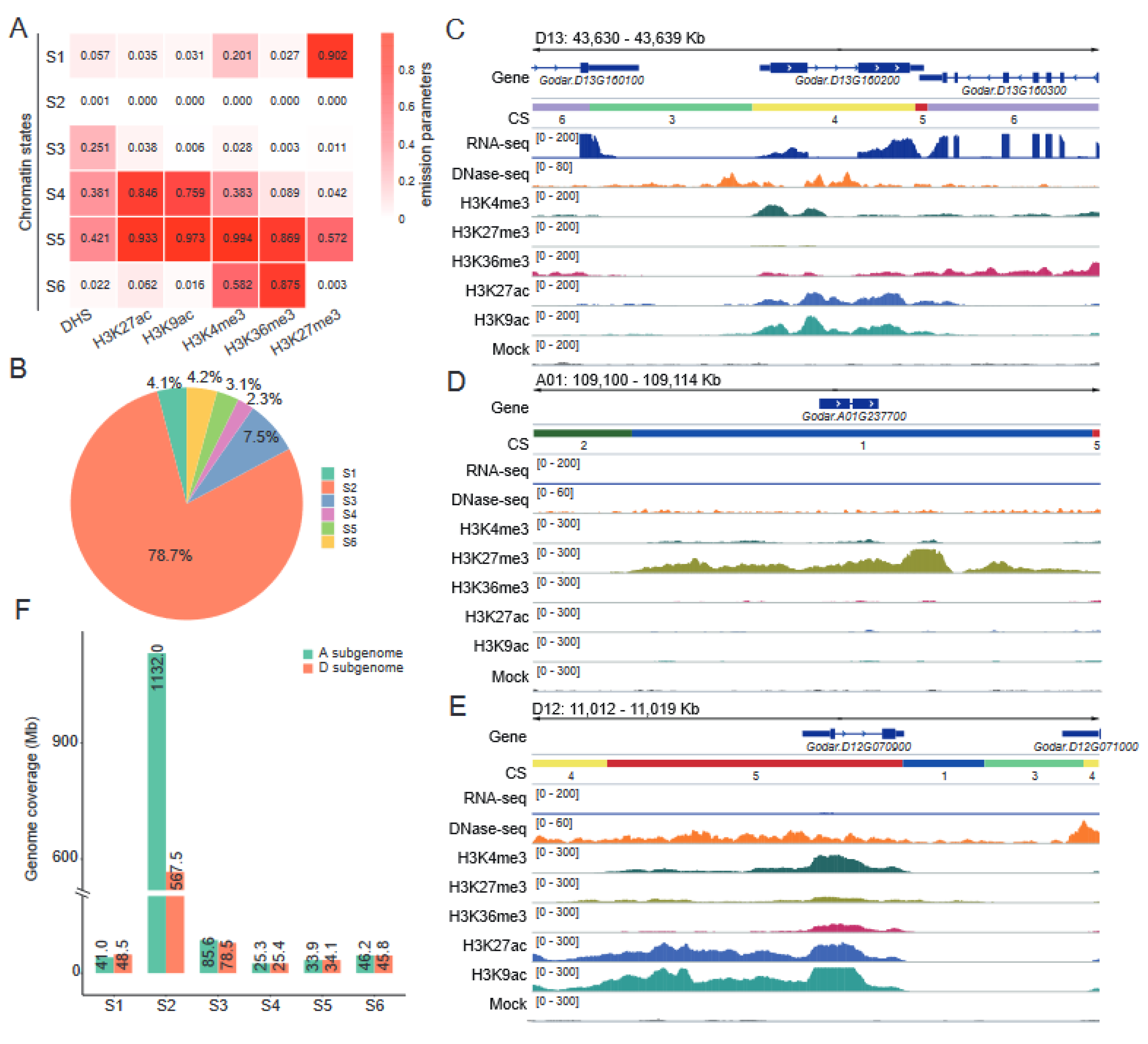
2.5. Defining Chromatin States in the Cotton Genome
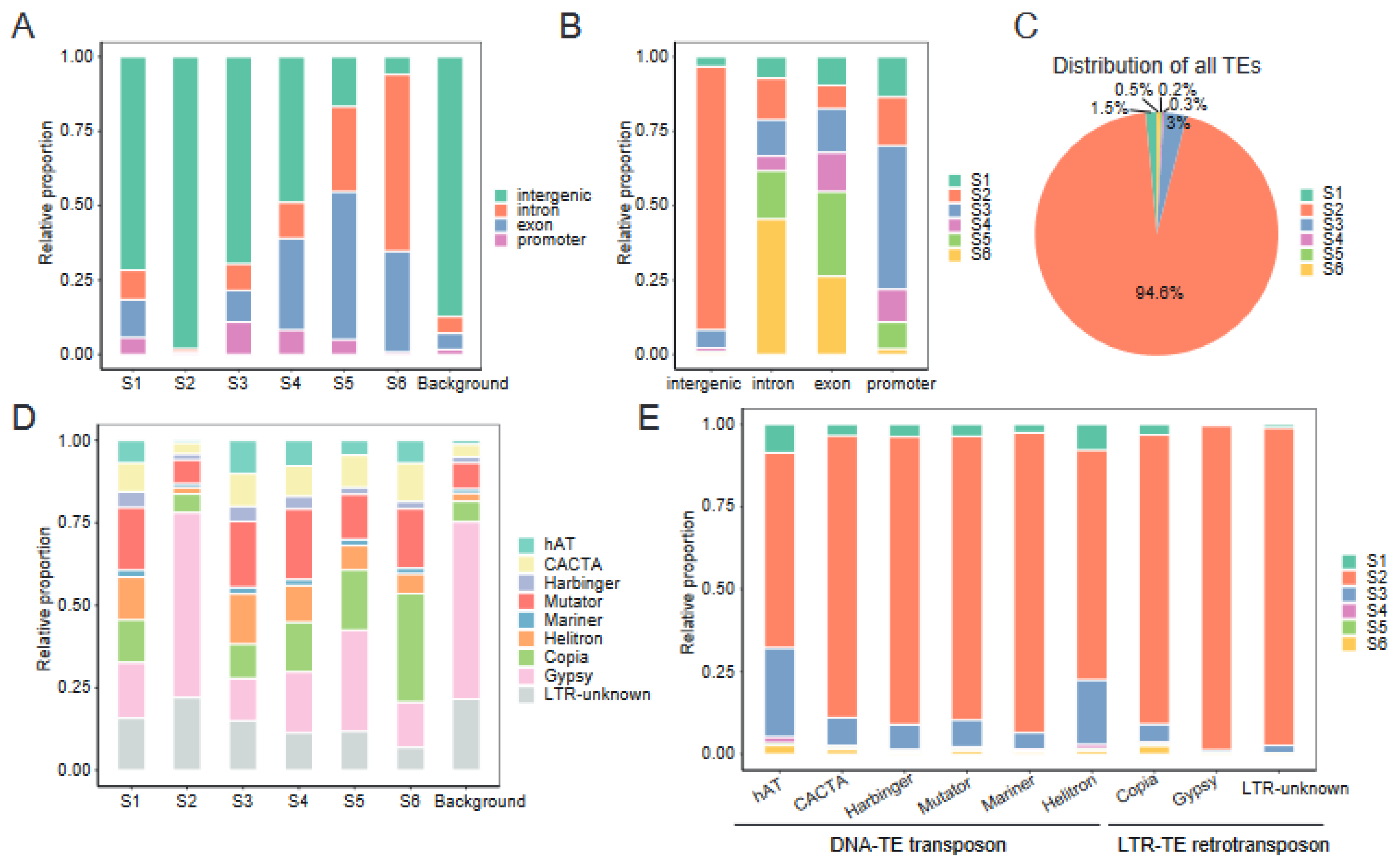
2.6. Chromatin States Associate with Genomic Elements and TEs Landscape
2.7. Domestication Has a Dramatic Impact on Chromatin States
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. ChIP–Seq and Data Analysis
4.3. RNA–Seq and Data Analysis
4.4. TE Analysis
4.5. Definition of Homoeologous Gene Pairs and Subgenome–Unique Genes
4.6. DNase–Seq Data Analysis
4.7. Chromatin States Analysis
4.8. Data Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Booth, L.N.; Brunet, A. The Aging Epigenome. Mol. Cell 2016, 62, 728–744. [Google Scholar] [CrossRef] [Green Version]
- Guarino, F.; Cicatelli, A.; Castiglione, S.; Agius, D.R.; Orhun, G.E.; Fragkostefanakis, S.; Leclercq, J.; Dobránszki, J.; Kaiserli, E.; Lieberman-Lazarovich, M.; et al. An Epigenetic Alphabet of Crop Adaptation to Climate Change. Front. Genet. 2022, 13, 818727. [Google Scholar] [CrossRef]
- Nunez-Vazquez, R.; Desvoyes, B.; Gutierrez, C. Histone variants and modifications during abiotic stress response. Front. Plant Sci. 2022, 13, 984702. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, J.; Ghoshal, B.; Wang, M.; Jacobsen, S.E. CRISPR-Cas-mediated transcriptional control and epi-mutagenesis. Plant Physiol. 2022, 188, 1811–1824. [Google Scholar] [CrossRef]
- Chen, Z.J.; Mas, P. Interactive roles of chromatin regulation and circadian clock function in plants. Genome Biol. 2019, 20, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antunez-Sanchez, J.; Naish, M.; Ramirez-Prado, J.S.; Ohno, S.; Huang, Y.; Dawson, A.; Opassathian, K.; Manza-Mianza, D.; Ariel, F.; Raynaud, C.; et al. A new role for histone demethylases in the maintenance of plant genome integrity. eLife 2020, 9, e58533. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Chen, D.; Fang, J.; Wang, P.; Deng, X.; Chu, C. Understanding the genetic and epigenetic architecture in complex network of rice flowering pathways. Protein Cell 2014, 5, 889–898. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Lin, J.; Liu, X.; Chu, W.; Li, J.; Gao, Y.; An, K.; Song, W.; Xin, M.; Yao, Y.; et al. Histone acetyltransferase TaHAG1 acts as a crucial regulator to strengthen salt tolerance of hexaploid wheat. Plant Physiol. 2021, 186, 1951–1969. [Google Scholar] [CrossRef]
- Sanders, D.; Qian, S.; Fieweger, R.; Lu, L.; Dowell, J.A.; Denu, J.M.; Zhong, X. Histone Lysine-to-Methionine Mutations Reduce Histone Methylation and Cause Developmental Pleiotropy. Plant Physiol. 2017, 173, 2243–2252. [Google Scholar] [CrossRef] [Green Version]
- Deevy, O.; Bracken, A.P. PRC2 functions in development and congenital disorders. Dev. (Camb. Engl.) 2019, 146, dev181354. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Kumar, A.; Hamada, K.; Okada, C.; Oguni, A.; Machiyama, A.; Sakuraba, S.; Nishizawa, T.; Nureki, O.; Kono, H.; et al. Structural basis of the regulation of the normal and oncogenic methylation of nucleosomal histone H3 Lys36 by NSD2. Nat. Commun. 2021, 12, 6605. [Google Scholar] [CrossRef] [PubMed]
- Veluchamy, A.; Rastogi, A.; Lin, X.; Lombard, B.; Murik, O.; Thomas, Y.; Dingli, F.; Rivarola, M.; Ott, S.; Liu, X.; et al. An integrative analysis of post-translational histone modifications in the marine diatom Phaeodactylum tricornutum. Genome Biol. 2015, 16, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Guan, P.; Zhao, L.; Ma, M.; Xie, L.; Yue, L.; Zheng, R.; Ouyang, W.; Wang, S.; Li, H.; et al. Asymmetric epigenome maps of subgenomes reveal imbalanced transcription and distinct evolutionary trends in Brassica napus. Mol. Plant 2021, 14, 604–619. [Google Scholar] [CrossRef]
- Li, Z.; Wang, M.; Lin, K.; Xie, Y.; Guo, J.; Ye, L.; Zhuang, Y.; Teng, W.; Ran, X.; Tong, Y.; et al. The bread wheat epigenomic map reveals distinct chromatin architectural and evolutionary features of functional genetic elements. Genome Biol. 2019, 20, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.; Koch, Z.; Fiziev, P.; Ernst, J. A framework for group-wise summarization and comparison of chromatin state annotations. Bioinformatics 2023, 39, btac722. [Google Scholar] [CrossRef]
- Yen, A.; Kellis, M. Systematic chromatin state comparison of epigenomes associated with diverse properties including sex and tissue type. Nat. Commun. 2015, 6, 7973. [Google Scholar] [CrossRef] [Green Version]
- Ernst, J.; Kellis, M. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 2012, 9, 215–216. [Google Scholar] [CrossRef] [Green Version]
- Gorkin, D.U.; Barozzi, I.; Zhao, Y.; Zhang, Y.; Huang, H.; Lee, A.Y.; Li, B.; Chiou, J.; Wildberg, A.; Ding, B.; et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature 2020, 583, 744–751. [Google Scholar] [CrossRef]
- Zhang, C.; Macchi, F.; Magnani, E.; Sadler, K.C. Chromatin states shaped by an epigenetic code confer regenerative potential to the mouse liver. Nat. Commun. 2021, 12, 4110. [Google Scholar] [CrossRef]
- Roudier, F.; Ahmed, I.; Bérard, C.; Sarazin, A.; Mary-Huard, T.; Cortijo, S.; Bouyer, D.; Caillieux, E.; Duvernois-Berthet, E.; Al-Shikhley, L.; et al. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 2011, 30, 1928–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.Y.; Shang, G.D.; Wang, F.X.; Gao, J.; Wan, M.C.; Xu, Z.G.; Wang, J.W. Dynamic chromatin state profiling reveals regulatory roles of auxin and cytokinin in shoot regeneration. Dev. Cell 2022, 57, 526–542.e7. [Google Scholar] [CrossRef]
- Zhao, L.; Xie, L.; Zhang, Q.; Ouyang, W.; Deng, L.; Guan, P.; Ma, M.; Li, Y.; Zhang, Y.; Xiao, Q.; et al. Integrative analysis of reference epigenomes in 20 rice varieties. Nat. Commun. 2020, 11, 2658. [Google Scholar] [CrossRef] [PubMed]
- Foroozani, M.; Zahraeifard, S.; Oh, D.H.; Wang, G.; Dassanayake, M.; Smith, A.P. Low-Phosphate Chromatin Dynamics Predict a Cell Wall Remodeling Network in Rice Shoots. Plant Physiol. 2020, 182, 1494–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Peer, Y.; Mizrachi, E.; Marchal, K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017, 18, 411–424. [Google Scholar] [CrossRef]
- Wendel, J.F.; Jackson, S.A.; Meyers, B.C.; Wing, R.A. Evolution of plant genome architecture. Genome Biol. 2016, 17, 37. [Google Scholar] [CrossRef] [Green Version]
- Yuan, D.; Grover, C.E.; Hu, G.; Pan, M.; Miller, E.R.; Conover, J.L.; Hunt, S.P.; Udall, J.A.; Wendel, J.F. Parallel and Intertwining Threads of Domestication in Allopolyploid Cotton. Adv. Sci. (Weinh) 2021, 8, 2003634. [Google Scholar] [CrossRef]
- He, P.; Zhang, Y.; Xiao, G. Origin of a Subgenome and Genome Evolution of Allotetraploid Cotton Species. Mol. Plant 2020, 13, 1238–1240. [Google Scholar] [CrossRef]
- Chen, Z.J.; Sreedasyam, A.; Ando, A.; Song, Q.; De Santiago, L.M.; Hulse-Kemp, A.M.; Ding, M.; Ye, W.; Kirkbride, R.C.; Jenkins, J.; et al. Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 2020, 52, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Wendel, J.F.; Cronn, R.C. Polyploidy and the evolutionary history of cotton. Adv. Agron. 2003, 78, 139–186. [Google Scholar]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Lopez-Arredondo, D.; Yu, G.; Wang, Y.; Wang, B.; Wall, S.B.; Zhang, X.; Fang, H.; Barragán-Rosillo, A.C.; Pan, X.; et al. Genome-wide chromatin accessibility analysis unveils open chromatin convergent evolution during polyploidization in cotton. Proc. Natl. Acad. Sci. USA 2022, 119, e2209743119. [Google Scholar] [CrossRef] [PubMed]
- Lafos, M.; Kroll, P.; Hohenstatt, M.L.; Thorpe, F.L.; Clarenz, O.; Schubert, D. Dynamic regulation of H3K27 trimethylation during Arabidopsis differentiation. PLoS Genet. 2011, 7, e1002040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringer, J.M.; Forster, S.C.; Qu, Z.; Prokopuk, L.; O’Bryan, M.K.; Gardner, D.K.; White, S.J.; Adelson, D.; Western, P.S. Reduced PRC2 function alters male germline epigenetic programming and paternal inheritance. BMC Biol. 2018, 16, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Umer, M.J.; Liu, F.; Cai, X.; Zheng, J.; Xu, Y.; Hou, Y.; Zhou, Z. Genome-Wide Identification and Characterization of CPR5 Genes in Gossypium Reveals Their Potential Role in Trichome Development. Front. Genet. 2022, 13, 921096. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Peng, Z.; Li, H.; Qin, G.; Jia, Y.; Pan, Z.; He, S.; Qayyum, A.; Du, X. Genome wide analysis of IQD gene family in diploid and tetraploid species of cotton (Gossypium spp.). Int. J. Biol. Macromol. 2021, 184, 1035–1061. [Google Scholar] [CrossRef] [PubMed]
- Maryum, Z.; Luqman, T.; Nadeem, S.; Khan, S.; Wang, B.; Ditta, A.; Khan, M.K.R. An overview of salinity stress, mechanism of salinity tolerance and strategies for its management in cotton. Front. Plant Sci. 2022, 13, 907937. [Google Scholar] [CrossRef]
- Paparelli, E.; Gonzali, S.; Parlanti, S.; Novi, G.; Giorgi, F.M.; Licausi, F.; Kosmacz, M.; Feil, R.; Lunn, J.E.; Brust, H.; et al. Misexpression of a chloroplast aspartyl protease leads to severe growth defects and alters carbohydrate metabolism in Arabidopsis. Plant Physiol. 2012, 160, 1237–1250. [Google Scholar] [CrossRef] [Green Version]
- Sundaram, V.; Cheng, Y.; Ma, Z.; Li, D.; Xing, X.; Edge, P.; Snyder, M.P.; Wang, T. Widespread contribution of transposable elements to the innovation of gene regulatory networks. Genome Res. 2014, 24, 1963–1976. [Google Scholar] [CrossRef] [Green Version]
- Song, Q.; Zhang, T.; Stelly, D.M.; Chen, Z.J. Epigenomic and functional analyses reveal roles of epialleles in the loss of photoperiod sensitivity during domestication of allotetraploid cottons. Genome Biol. 2017, 18, 99. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Chen, Z.J. Epigenetic perspectives on the evolution and domestication of polyploid plant and crops. Curr. Opin. Plant Biol. 2018, 42, 37–48. [Google Scholar] [CrossRef]
- Gilsbach, R.; Schwaderer, M.; Preissl, S.; Grüning, B.A.; Kranzhöfer, D.; Schneider, P.; Nührenberg, T.G.; Mulero-Navarro, S.; Weichenhan, D.; Braun, C.; et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018, 9, 391. [Google Scholar] [CrossRef] [Green Version]
- Ngo, V.; Chen, Z.; Zhang, K.; Whitaker, J.W.; Wang, M.; Wang, W. Epigenomic analysis reveals DNA motifs regulating histone modifications in human and mouse. Proc. Natl. Acad. Sci. USA 2019, 116, 3668–3677. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Willson, T.A.; Wakefield, M.J.; Trounson, E.; Hilton, D.J.; Blewitt, M.E.; Oshlack, A.; Majewski, I.J. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 2011, 39, 7415–7427. [Google Scholar] [CrossRef]
- Arvanitis, M.; Tampakakis, E.; Zhang, Y.; Wang, W.; Auton, A.; Dutta, D.; Glavaris, S.; Keramati, A.; Chatterjee, N.; Chi, N.C.; et al. Genome-wide association and multi-omic analyses reveal ACTN2 as a gene linked to heart failure. Nat. Commun. 2020, 11, 1122. [Google Scholar] [CrossRef] [Green Version]
- Olsen, K.M.; Wendel, J.F. Crop plants as models for understanding plant adaptation and diversification. Front. Plant Sci. 2013, 4, 290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.; Su, W.; Liao, Y.; Chougule, K.; Agda, J.R.A.; Hellinga, A.J.; Lugo, C.S.B.; Elliott, T.A.; Ware, D.; Peterson, T.; et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 2019, 20, 275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Yu, G.; Zhang, X.; Dai, Y.; Zhang, H.; Zhang, B.; Wang, K. Histone Maps in Gossypium darwinii Reveal Epigenetic Regulation Drives Subgenome Divergence and Cotton Domestication. Int. J. Mol. Sci. 2023, 24, 10607. https://doi.org/10.3390/ijms241310607
Han J, Yu G, Zhang X, Dai Y, Zhang H, Zhang B, Wang K. Histone Maps in Gossypium darwinii Reveal Epigenetic Regulation Drives Subgenome Divergence and Cotton Domestication. International Journal of Molecular Sciences. 2023; 24(13):10607. https://doi.org/10.3390/ijms241310607
Chicago/Turabian StyleHan, Jinlei, Guangrun Yu, Xin Zhang, Yan Dai, Hui Zhang, Baohong Zhang, and Kai Wang. 2023. "Histone Maps in Gossypium darwinii Reveal Epigenetic Regulation Drives Subgenome Divergence and Cotton Domestication" International Journal of Molecular Sciences 24, no. 13: 10607. https://doi.org/10.3390/ijms241310607