
Dynamics of Ex Vivo Mesenchymal Stromal Cell Potency under Continuous Perfusion

Abstract
:1. Introduction
2. Results
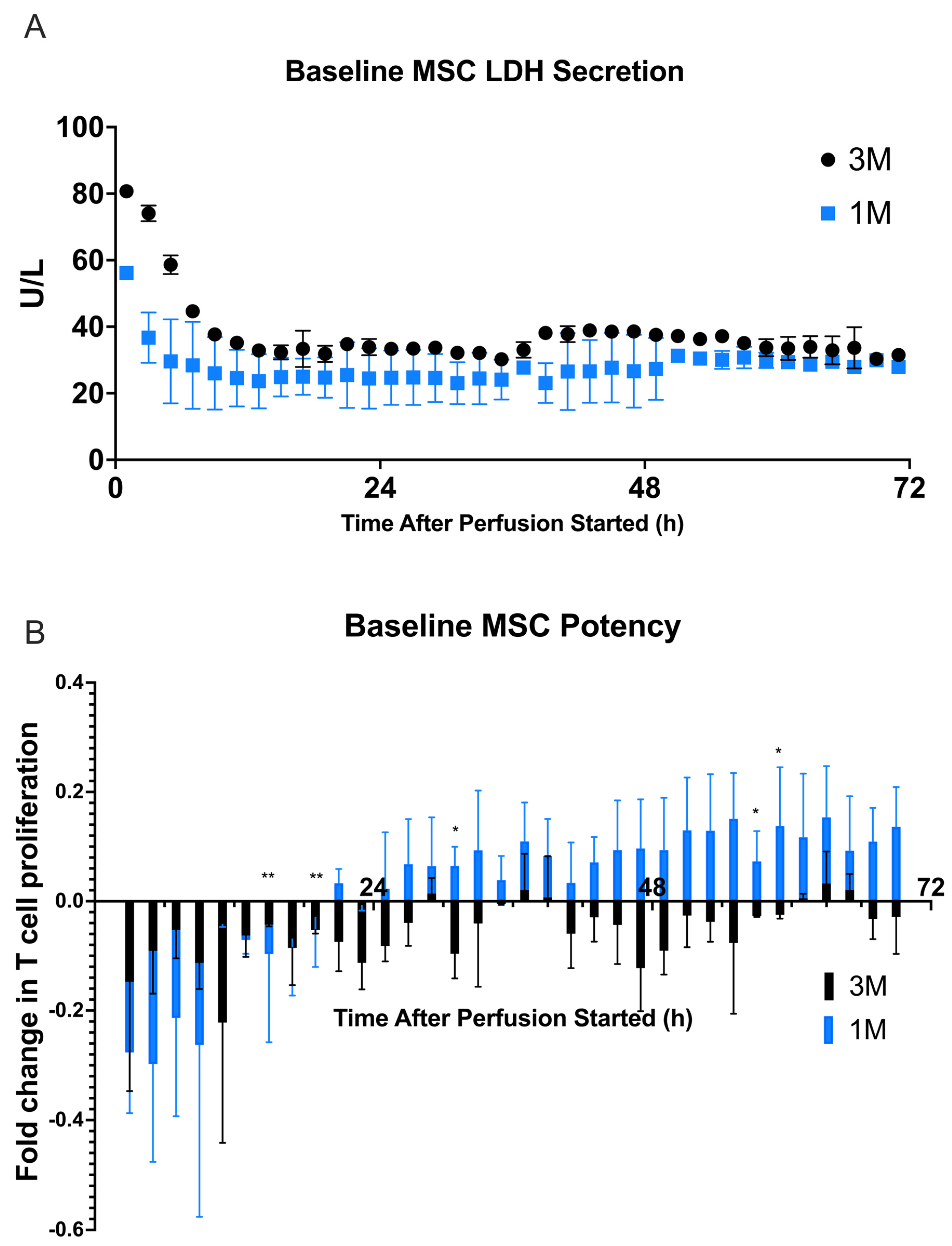
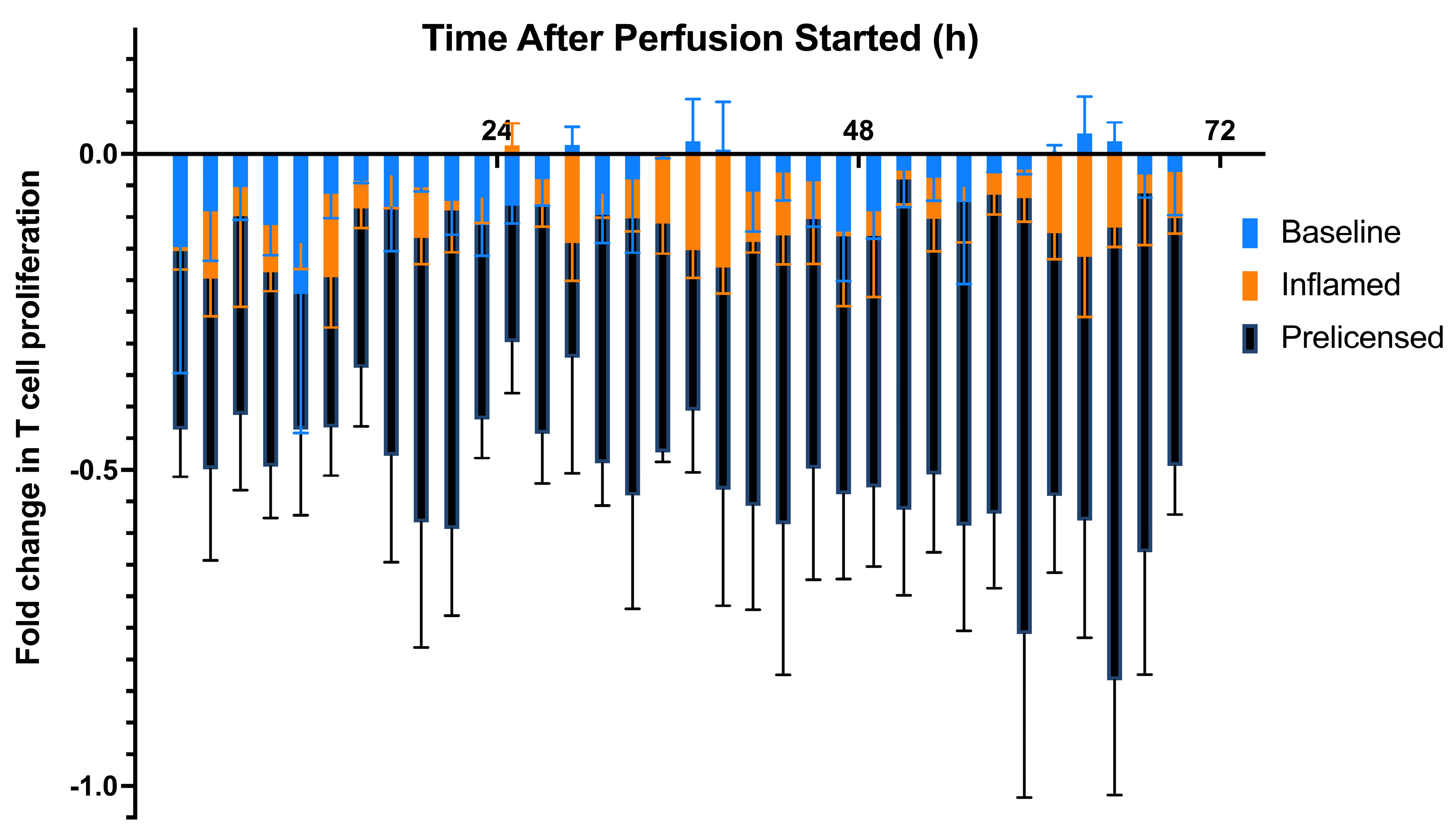
2.1. Baseline Viability, Potency, and Secreted Factor Release of Immobilized MSCs in the Continuous Perfusion System
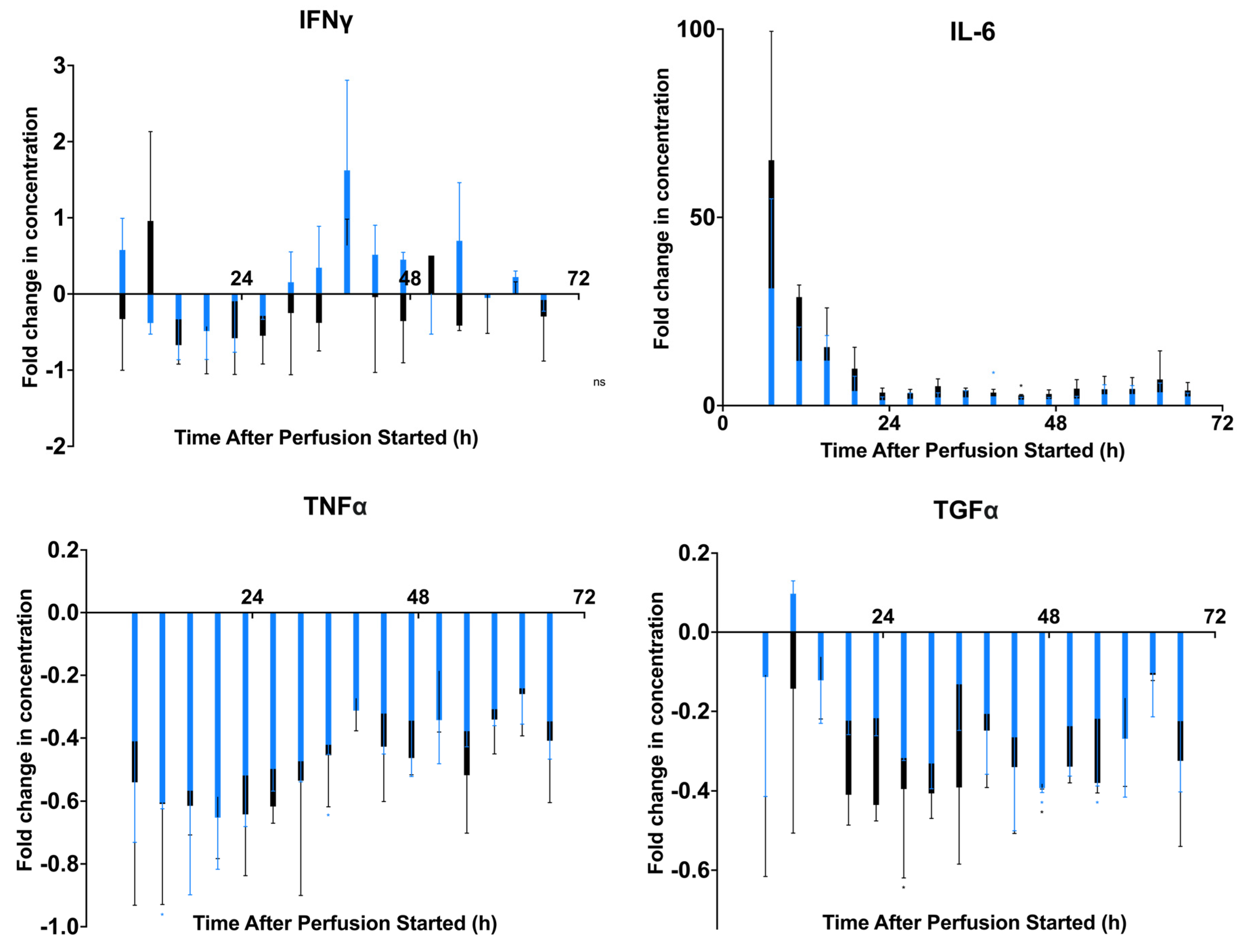
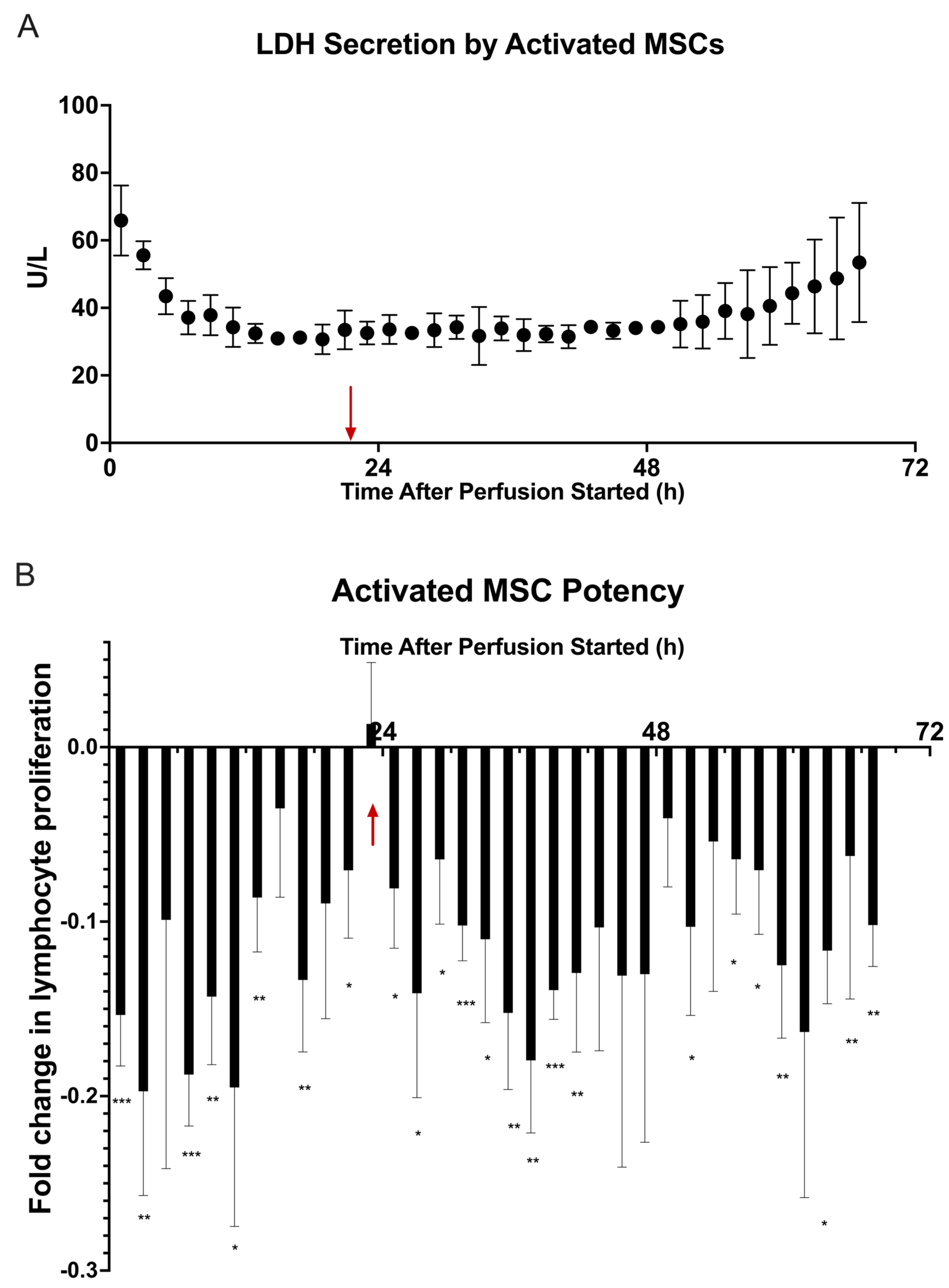
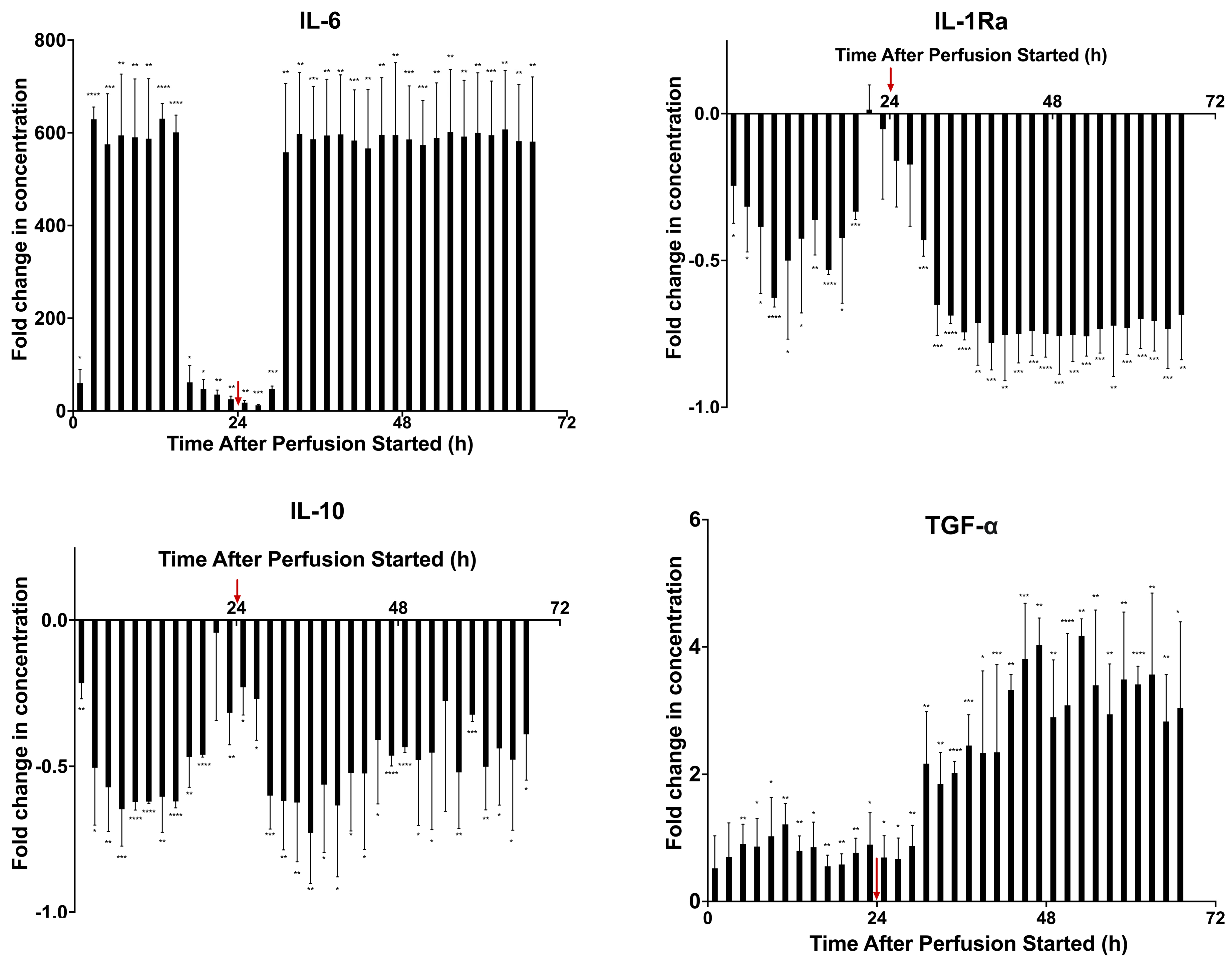
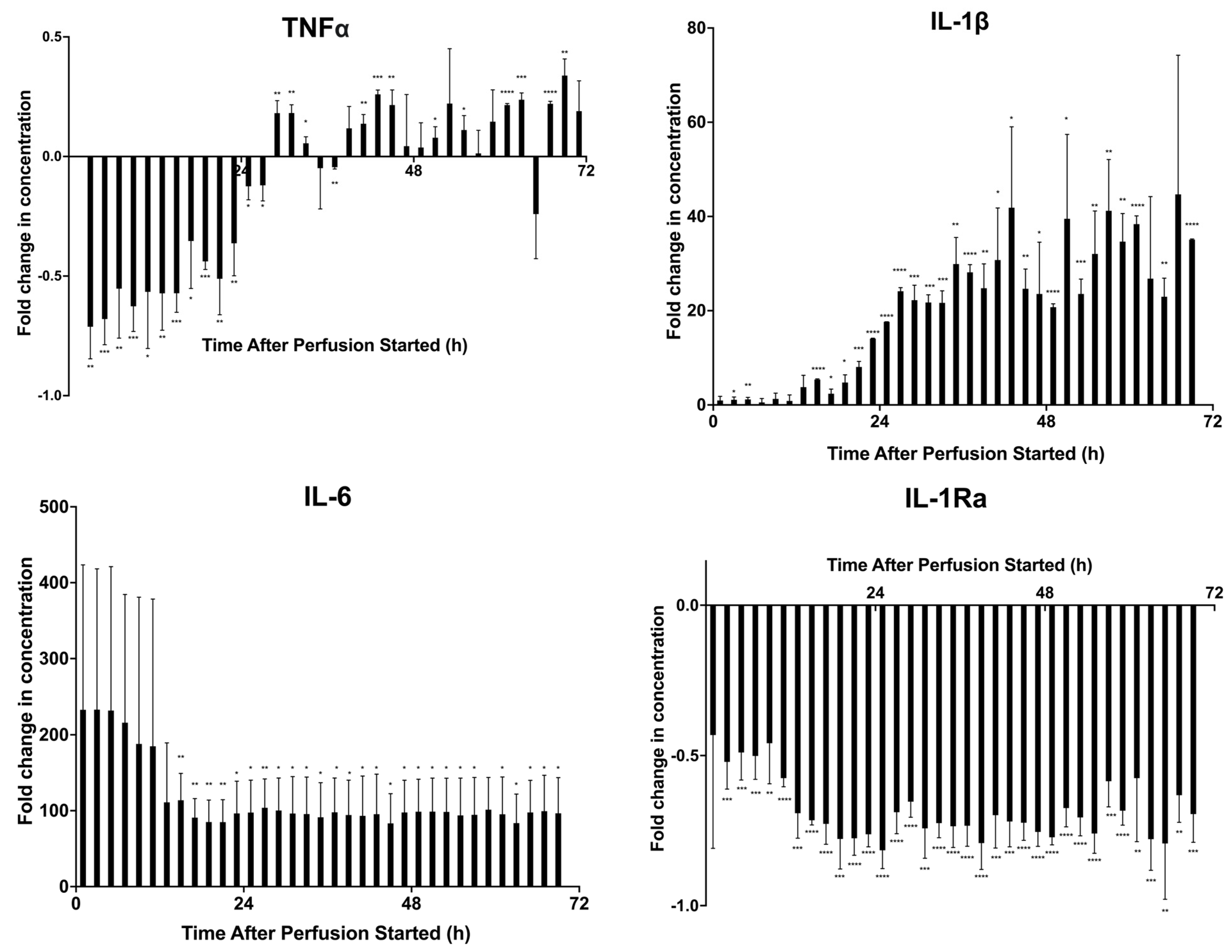
2.2. Modeling Inflammation: Viability, Potency, and Secreted Factor Release of MSCs Introduced to Proinflammatory Cytokine Medium
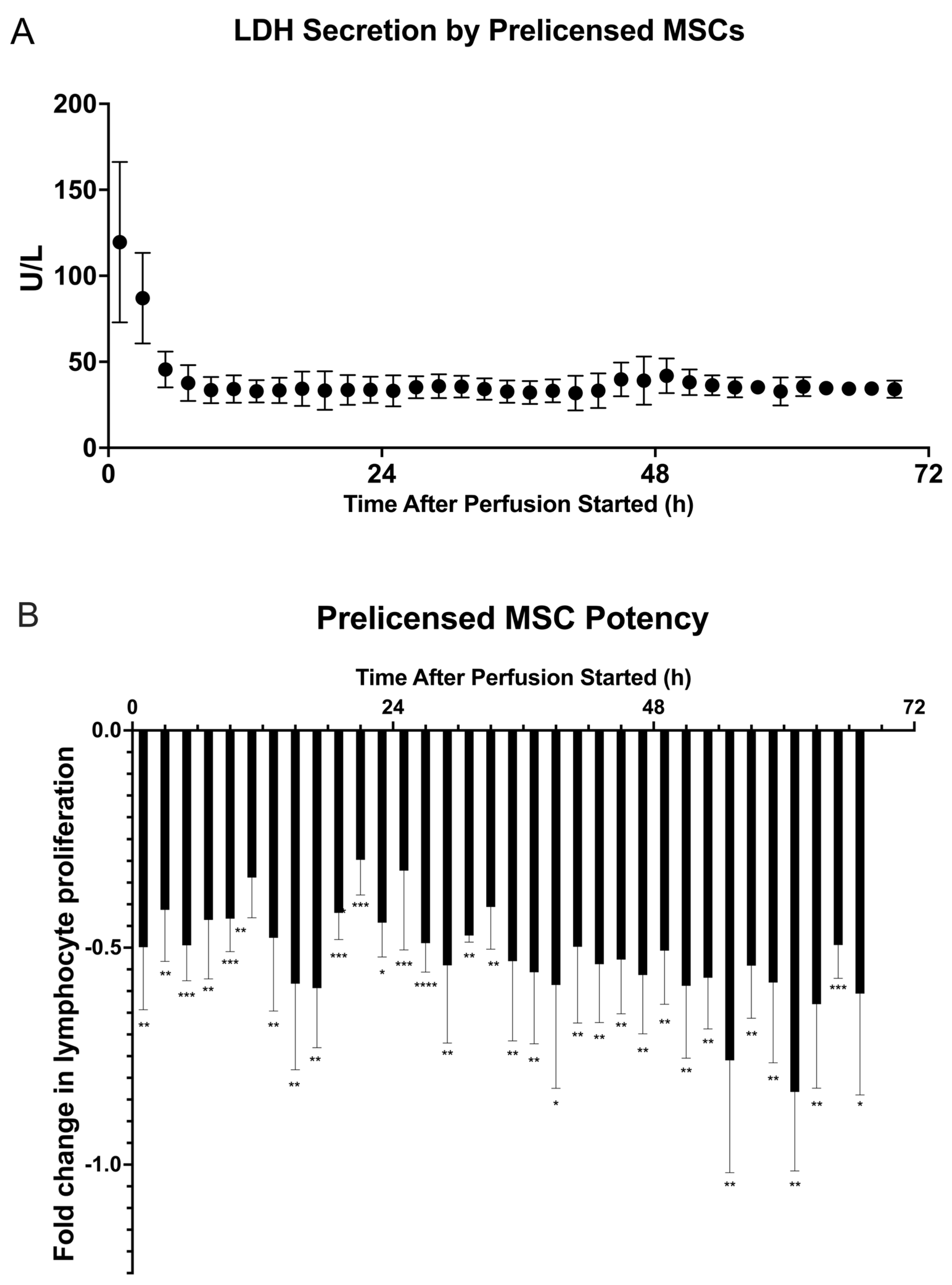
2.3. Prelicensing: Viability, Potency, and Secreted Factor Release of Prelicensed MSCs
3. Discussion
4. Materials and Methods
4.1. Human Cell Cultures
4.2. Media Preparation
4.3. Bioreactor MSC Seeding
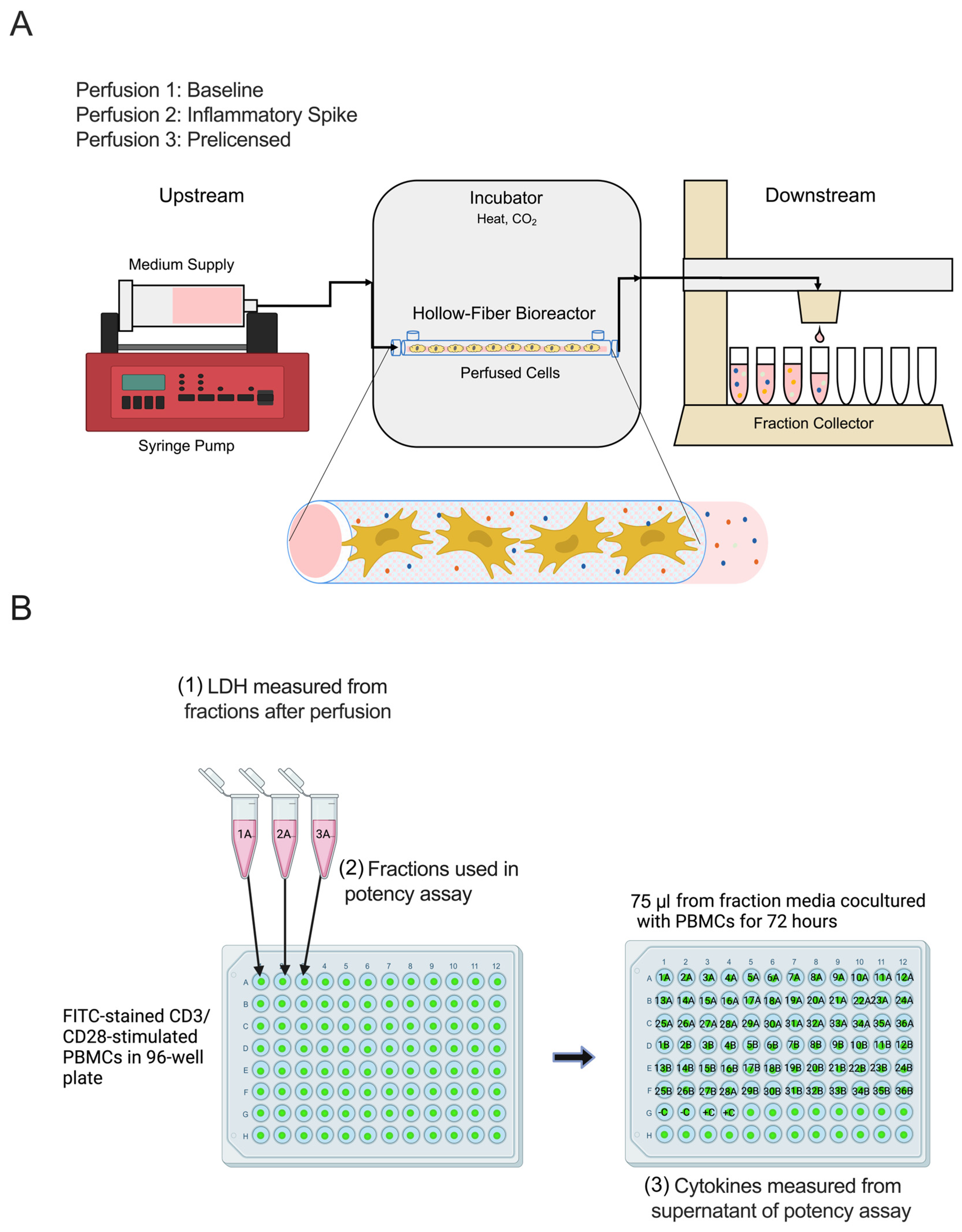
4.4. Perfusion and Fraction Collection
4.5. Cytokine Activation
4.6. Prelicensing
4.7. Measurement of LDH Production
4.8. PBMC Isolation
4.9. CFSE Labeling for PBMC Proliferation
4.10. PBMC Stimulation
4.11. Flow Cytometric Analysis
4.12. Analysis of Cytokine Production
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murphy, M.B.; Moncivais, K.; Caplan, A.I. Mesenchymal stem cells: Environmentally responsive therapeutics for regenerative medicine. Exp. Mol. Med. 2013, 45, e54. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, H.M.; Koc, O.N.; Devine, S.M.; Curtin, P.; Maziarz, R.T.; Holland, H.K.; Shpall, E.J.; McCarthy, P.; Atkinson, K.; Cooper, B.W.; et al. Cotransplantation of HLA-identical sibling culture-expanded mesenchymal stem cells and hematopoietic stem cells in hematologic malignancy patients. Biol. Blood Marrow Transplant. 2005, 11, 389–398. [Google Scholar] [CrossRef]
- Mazzini, L.; Fagioli, F.; Boccaletti, R.; Mareschi, K.; Oliveri, G.; Olivieri, C.; Pastore, I.; Marasso, R.; Madon, E. Stem cell therapy in amyotrophic lateral sclerosis: A methodological approach in humans. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2003, 4, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Fujita, R.; Asai, M.; Kaneda, Y.; Tamai, K. Receptor for Advanced Glycation End Products-Mediated Signaling Impairs the Maintenance of Bone Marrow Mesenchymal Stromal Cells in Diabetic Model Mice. Stem Cells Dev. 2016, 25, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-B.; Huang, H.; Sun, P.; Ma, S.-Z.; Liu, A.-H.; Xue, J.; Fu, J.-H.; Liang, Y.-Q.; Liu, B.; Wu, D.-Y.; et al. Human Umbilical Cord-Derived Mesenchymal Stromal Cells Improve Left Ventricular Function, Perfusion, and Remodeling in a Porcine Model of Chronic Myocardial Ischemia. Stem Cells Transl. Med. 2016, 5, 1004–1013. [Google Scholar] [CrossRef]
- Parekkadan, B.; Milwid, J.M. Mesenchymal stem cells as therapeutics. Annu. Rev. Biomed. Eng. 2010, 12, 87–117. [Google Scholar] [CrossRef]
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal Stromal Cells: Sensors and Switchers of Inflammation. Cell Stem Cell 2013, 13, 392–402. [Google Scholar] [CrossRef]
- Fan, X.-L.; Zhang, Y.; Li, X.; Fu, Q.-L. Mechanisms underlying the protective effects of mesenchymal stem cell-based therapy. Cell. Mol. Life Sci. 2020, 77, 2771–2794. [Google Scholar] [CrossRef]
- Cuerquis, J.; Romieu-Mourez, R.; Francois, M.; Routy, J.P.; Young, Y.K.; Zhao, J.; Eliopoulos, N. Human mesenchymal stromal cells transiently increase cytokine production by activated T cells before suppressing T-cell proliferation: Effect of interferon-gamma and tumor necrosis factor-alpha stimulation. Cytotherapy 2014, 16, 191–202. [Google Scholar] [CrossRef]
- Boland, L.K.; Burand, A.J.; Brown, A.J.; Boyt, D.T.; Lira, V.A.; Ankrum, J.A. IFN-γ and TNF-α Pre-licensing Protects Mesenchymal Stromal Cells from the Pro-inflammatory Effects of Palmitate. Mol. Ther. 2018, 26, 860–873. [Google Scholar] [CrossRef]
- Boyt, D.T.; Boland, L.K.; Burand, A.J., Jr.; Brown, A.J.; Ankrum, J.A. Dose and duration of interferon γ prelicensing interact with donor characteristics to influence the expression and function of indoleamine-2,3-dioxygenase in mesenchymal stromal cells. J. R. Soc. Interface 2020, 17, 20190815. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhang, L.; Zhao, X.; Xu, G.; Zhang, Y.; Roberts, A.I.; Zhao, R.C.; Shi, Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell 2008, 2, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, J.; Luc, S.B. Mesenchymal stromal cells: Clinical challenges and therapeutic opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Eggenhofer, E.; Benseler, V.; Kroemer, A.; Popp, F.; Geissler, E.; Schlitt, H.; Baan, C.; Dahlke, M.; Hoogduijn, M. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Front. Immunol. 2012, 3, 297. [Google Scholar] [CrossRef]
- Moll, G.; Ignatowicz, L.; Catar, R.; Luecht, C.; Sadeghi, B.; Hamad, O.; Jungebluth, P.; Dragun, D.; Schmidtchen, A.; Ringden, O. Different Procoagulant Activity of Therapeutic Mesenchymal Stromal Cells Derived from Bone Marrow and Placental Decidua. Stem Cells Dev. 2015, 24, 2269–2279. [Google Scholar] [CrossRef]
- Coppin, L.; Sokal, E.; Stephenne, X. Thrombogenic Risk Induced by Intravascular Mesenchymal Stem Cell Therapy: Current Status and Future Perspectives. Cells 2019, 8, 1160. [Google Scholar] [CrossRef] [PubMed]
- Silachev, D.N.; Goryunov, K.V.; Shpilyuk, M.A.; Beznoschenko, O.S.; Morozova, N.Y.; Kraevaya, E.E.; Popkov, V.A.; Pevzner, I.B.; Zorova, L.D.; Evtushenko, E.A.; et al. Effect of MSCs and MSC-Derived Extracellular Vesicles on Human Blood Coagulation. Cells 2019, 8, 258. [Google Scholar] [CrossRef]
- Baranovskii, D.S.; Klabukov, I.D.; Arguchinskaya, N.V.; Yakimova, A.O.; Kisel, A.A.; Yatsenko, E.M.; Ivanov, S.A.; Shegay, P.V.; Kaprin, A.D. Adverse events, side effects and complications in mesenchymal stromal cell-based therapies. Stem Cell. Investig. 2022, 9, 7. [Google Scholar] [CrossRef]
- Wang, Y.; Han, Z.-b.; Song, Y.-p.; Han, Z.C. Safety of Mesenchymal Stem Cells for Clinical Application. Stem Cells Int. 2012, 2012, 652034. [Google Scholar] [CrossRef]
- Parekkadan, B.; van Poll, D.; Suganuma, K.; Carter, E.A.; Berthiaume, F.; Tilles, A.W.; Yarmush, M.L. Mesenchymal stem cell-derived molecules reverse fulminant hepatic failure. PLoS ONE 2007, 2, e941. [Google Scholar] [CrossRef]
- Osugi, M.; Katagiri, W.; Yoshimi, R.; Inukai, T.; Hibi, H.; Ueda, M. Conditioned media from mesenchymal stem cells enhanced bone regeneration in rat calvarial bone defects. Tissue Eng. Part A 2012, 18, 1479–1489. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, M.; Kopyt, N.; Atta, M.G.; Radhakrishnan, J.; Umanath, K.; Nguyen, S.; O’Rourke, B.; Allen, A.; Vaninov, N.; Tilles, A.; et al. Pharmacological effects of ex vivo mesenchymal stem cell immunotherapy in patients with acute kidney injury and underlying systemic inflammation. Stem Cells Transl. Med. 2021, 10, 1588–1601. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.; Vaninov, N.; Li, M.; Nguyen, S.; Singh, M.; Igo, P.; Tilles, A.W.; O’Rourke, B.; Miller, B.L.K.; Parekkadan, B.; et al. Mesenchymal Stromal Cell Bioreactor for Ex Vivo Reprogramming of Human Immune Cells. Sci. Rep. 2020, 10, 10142. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Tilles, A.W.; Milwid, J.M.; Hammad, M.; Lee, J.; Yarmush, M.L.; Parekkadan, B. Phenotypic and functional characterization of human bone marrow stromal cells in hollow-fibre bioreactors. J. Tissue Eng. Regen. Med. 2012, 6, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Erickson, P.; Houwayek, T.; Burr, A.; Teryek, M.; Parekkadan, B. A continuous flow cell culture system for precision cell stimulation and time-resolved profiling of cell secretion. Anal. Biochem. 2021, 625, 114213. [Google Scholar] [CrossRef] [PubMed]
- Shelke, G.V.; Lasser, C.; Gho, Y.S.; Lotvall, J. Importance of exosome depletion protocols to eliminate functional and RNA-containing extracellular vesicles from fetal bovine serum. J. Extracell. Vesicles 2014, 3, 24783. [Google Scholar] [CrossRef]
- Chan, F.K.-M.; Moriwaki, K.; De Rosa, M.J. Detection of Necrosis by Release of Lactate Dehydrogenase Activity. In Immune Homeostasis: Methods and Protocols; Snow, A.L., Lenardo, M.J., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 65–70. [Google Scholar]
- Khong, D.; Li, M.; Singleton, A.; Chin, L.-Y.; Munkundan, S.; Parekkadan, B. Orthogonal Potency Analysis of Mesenchymal Stromal Cell Function During Ex Vivo Expansion. Exp. Cell. Res. 2018, 362, 102–110. [Google Scholar] [CrossRef]
- Krampera, M. Mesenchymal stromal cell ‘licensing’: A multistep process. Leukemia 2011, 25, 1408–1414. [Google Scholar] [CrossRef]
- Christy, B.A.; Herzig, M.C.; Delavan, C.P.; Abaasah, I.; Cantu, C.; Salgado, C.; Lovelace, S.; Garcia, L.; Jensen, K.; Montgomery, R.; et al. Use of multiple potency assays to evaluate human mesenchymal stromal cells. J. Trauma Acute Care Surg. 2020, 89, S109–S117. [Google Scholar] [CrossRef]
- Erickson, P.; Doshi, A.; Jetley, G.; Amin, P.; Mejevdiwala, A.; Patel, A.; Bento, R.; Parekkadan, B. Multi-Stream Perfusion Bioreactor Integrated with Outlet Fractionation for Dynamic Cell Culture. J. Vis. Exp. 2022, 185, e63935. [Google Scholar] [CrossRef]
- Caplan, H.; Olson, S.D.; Kumar, A.; George, M.; Prabhakara, K.S.; Wenzel, P.; Bedi, S.; Toledano-Furman, N.E.; Triolo, F.; Kamhieh-Milz, J.; et al. Mesenchymal Stromal Cell Therapeutic Delivery: Translational Challenges to Clinical Application. Front. Immunol. 2019, 10, 1645. [Google Scholar] [CrossRef]
- Caplan, A.I. New MSC: MSCs as pericytes are Sentinels and gatekeepers. J. Orthop. Res. 2017, 35, 1151–1159. [Google Scholar] [CrossRef]
- Kulakov, A.; Kogan, E.; Brailovskaya, T.; Vedyaeva, A.; Zharkov, N.; Krasilnikova, O.; Krasheninnikov, M.; Baranovskii, D.; Rasulov, T.; Klabukov, I. Mesenchymal Stromal Cells Enhance Vascularization and Epithelialization within 7 Days after Gingival Augmentation with Collagen Matrices in Rabbits. Dent. J. 2021, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Guillamat-Prats, R. The Role of MSC in Wound Healing, Scarring and Regeneration. Cells 2021, 10, 1729. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Zhao, G.; Liu, L.; Liu, F.; Gong, W.; Liu, X.; Yang, L.; Wang, J.; Hou, Y. Pre-treatment with IL-1beta enhances the efficacy of MSC transplantation in DSS-induced colitis. Cell. Mol. Immunol. 2012, 9, 473–481. [Google Scholar] [CrossRef] [PubMed]



 ). (A) LDH (U/L) secretion was quantified from each fraction by Cedex Bio Analyzer. (B) Fractions were cocultured with CD3/CD28–stimulated and CFSE–stained PBMCs for 72 h before being analyzed via flow cytometry for T cell proliferation. Fold change in T cell proliferation for each 3 M cell dose coculture compared to a negative control dose of 0 M was quantified in FlowJo. The figure shows the dynamics of T cell proliferation per fraction coculture with standard deviations for each. A multiple unpaired student’s t-test was conducted on the two cell dosage groups for all timepoints, showing a significant difference in cell dosage potency between 0 M and 3 M cell groups. Statistical significance indicated by: * = p-value < 0.05, ** = p-value < 0.01, *** = p-value < 0.001. A table of p-values can be found in Table S4.
). (A) LDH (U/L) secretion was quantified from each fraction by Cedex Bio Analyzer. (B) Fractions were cocultured with CD3/CD28–stimulated and CFSE–stained PBMCs for 72 h before being analyzed via flow cytometry for T cell proliferation. Fold change in T cell proliferation for each 3 M cell dose coculture compared to a negative control dose of 0 M was quantified in FlowJo. The figure shows the dynamics of T cell proliferation per fraction coculture with standard deviations for each. A multiple unpaired student’s t-test was conducted on the two cell dosage groups for all timepoints, showing a significant difference in cell dosage potency between 0 M and 3 M cell groups. Statistical significance indicated by: * = p-value < 0.05, ** = p-value < 0.01, *** = p-value < 0.001. A table of p-values can be found in Table S4.
). (A) LDH (U/L) secretion was quantified from each fraction by Cedex Bio Analyzer. (B) Fractions were cocultured with CD3/CD28–stimulated and CFSE–stained PBMCs for 72 h before being analyzed via flow cytometry for T cell proliferation. Fold change in T cell proliferation for each 3 M cell dose coculture compared to a negative control dose of 0 M was quantified in FlowJo. The figure shows the dynamics of T cell proliferation per fraction coculture with standard deviations for each. A multiple unpaired student’s t-test was conducted on the two cell dosage groups for all timepoints, showing a significant difference in cell dosage potency between 0 M and 3 M cell groups. Statistical significance indicated by: * = p-value < 0.05, ** = p-value < 0.01, *** = p-value < 0.001. A table of p-values can be found in Table S4.
). (A) LDH (U/L) secretion was quantified from each fraction by Cedex Bio Analyzer. (B) Fractions were cocultured with CD3/CD28–stimulated and CFSE–stained PBMCs for 72 h before being analyzed via flow cytometry for T cell proliferation. Fold change in T cell proliferation for each 3 M cell dose coculture compared to a negative control dose of 0 M was quantified in FlowJo. The figure shows the dynamics of T cell proliferation per fraction coculture with standard deviations for each. A multiple unpaired student’s t-test was conducted on the two cell dosage groups for all timepoints, showing a significant difference in cell dosage potency between 0 M and 3 M cell groups. Statistical significance indicated by: * = p-value < 0.05, ** = p-value < 0.01, *** = p-value < 0.001. A table of p-values can be found in Table S4.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0–24 h | 25–48 h | 49–72 h | |
|---|---|---|---|
| 0 M vs. 1 M | p = 0.0170 | p < 0.0001 | p < 0.0001 |
| 0 M vs. 3 M | p = 0.0003 | p < 0.0266 | p = 0.0932 |
| 1 M vs. 3 M | p = 0.7862 | p < 0.0001 | p < 0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doshi, A.; Erickson, P.; Teryek, M.; Parekkadan, B. Dynamics of Ex Vivo Mesenchymal Stromal Cell Potency under Continuous Perfusion. Int. J. Mol. Sci. 2023, 24, 9602. https://doi.org/10.3390/ijms24119602
Doshi A, Erickson P, Teryek M, Parekkadan B. Dynamics of Ex Vivo Mesenchymal Stromal Cell Potency under Continuous Perfusion. International Journal of Molecular Sciences. 2023; 24(11):9602. https://doi.org/10.3390/ijms24119602
Chicago/Turabian StyleDoshi, Aneesha, Patrick Erickson, Matthew Teryek, and Biju Parekkadan. 2023. "Dynamics of Ex Vivo Mesenchymal Stromal Cell Potency under Continuous Perfusion" International Journal of Molecular Sciences 24, no. 11: 9602. https://doi.org/10.3390/ijms24119602