
Unveiling the Stereoselectivity and Regioselectivity of the [3+2] Cycloaddition Reaction between N-methyl-C-4-methylphenyl-nitrone and 2-Propynamide from a MEDT Perspective

 , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
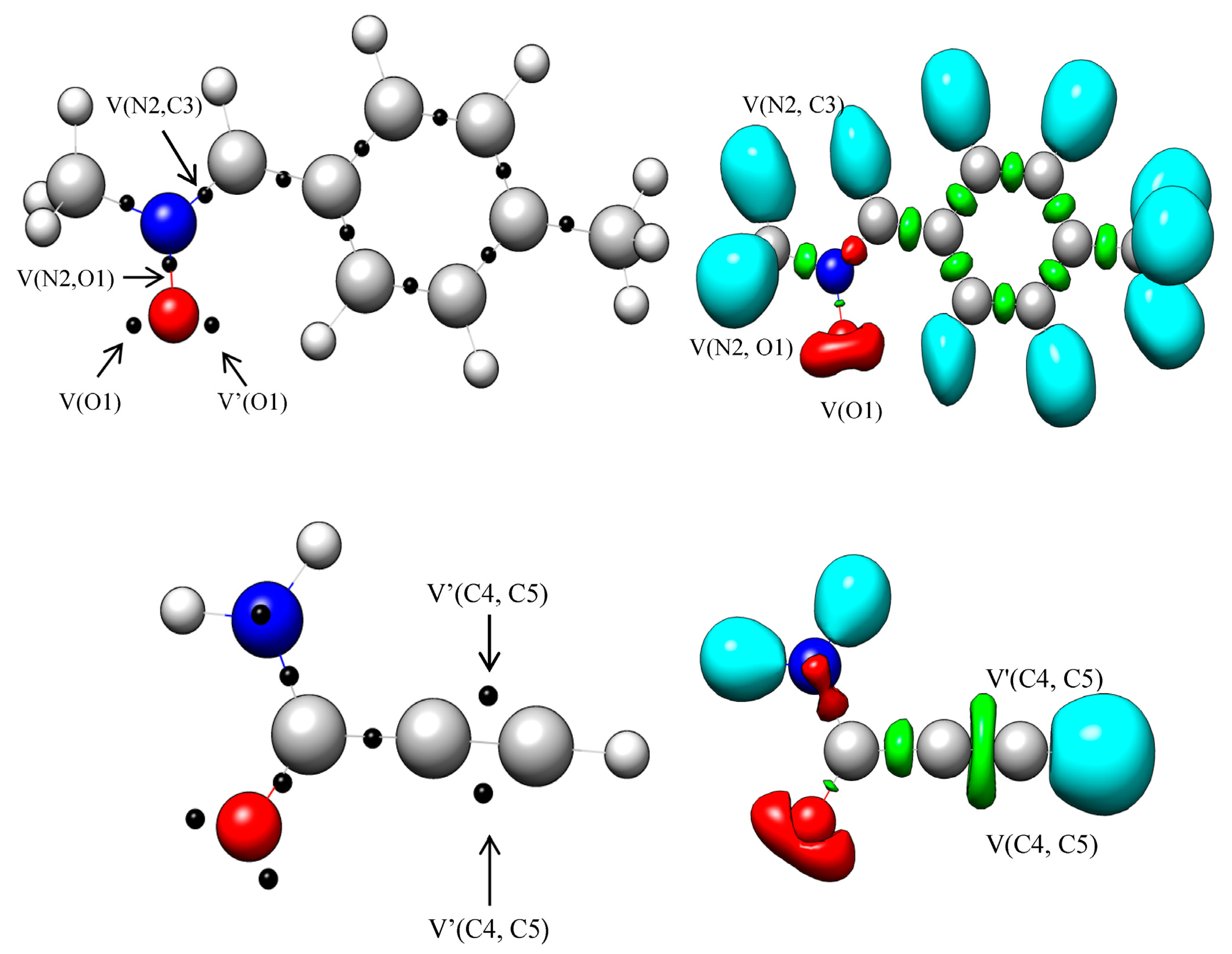
2.1. Analysis of the ELF Topology of the Reactants N-methyl-C-4-methylphenyl-nitrone 1 and 2-Propynamide 2
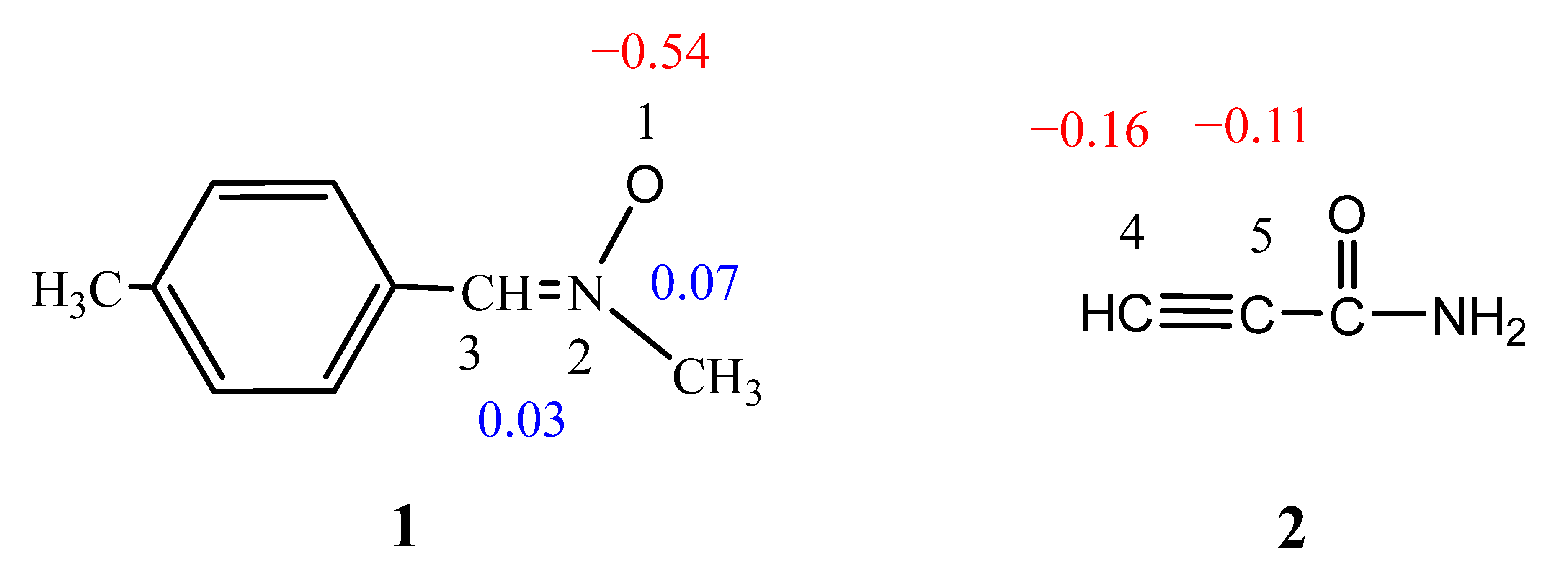
2.2. Analysis of the CDFT Indices
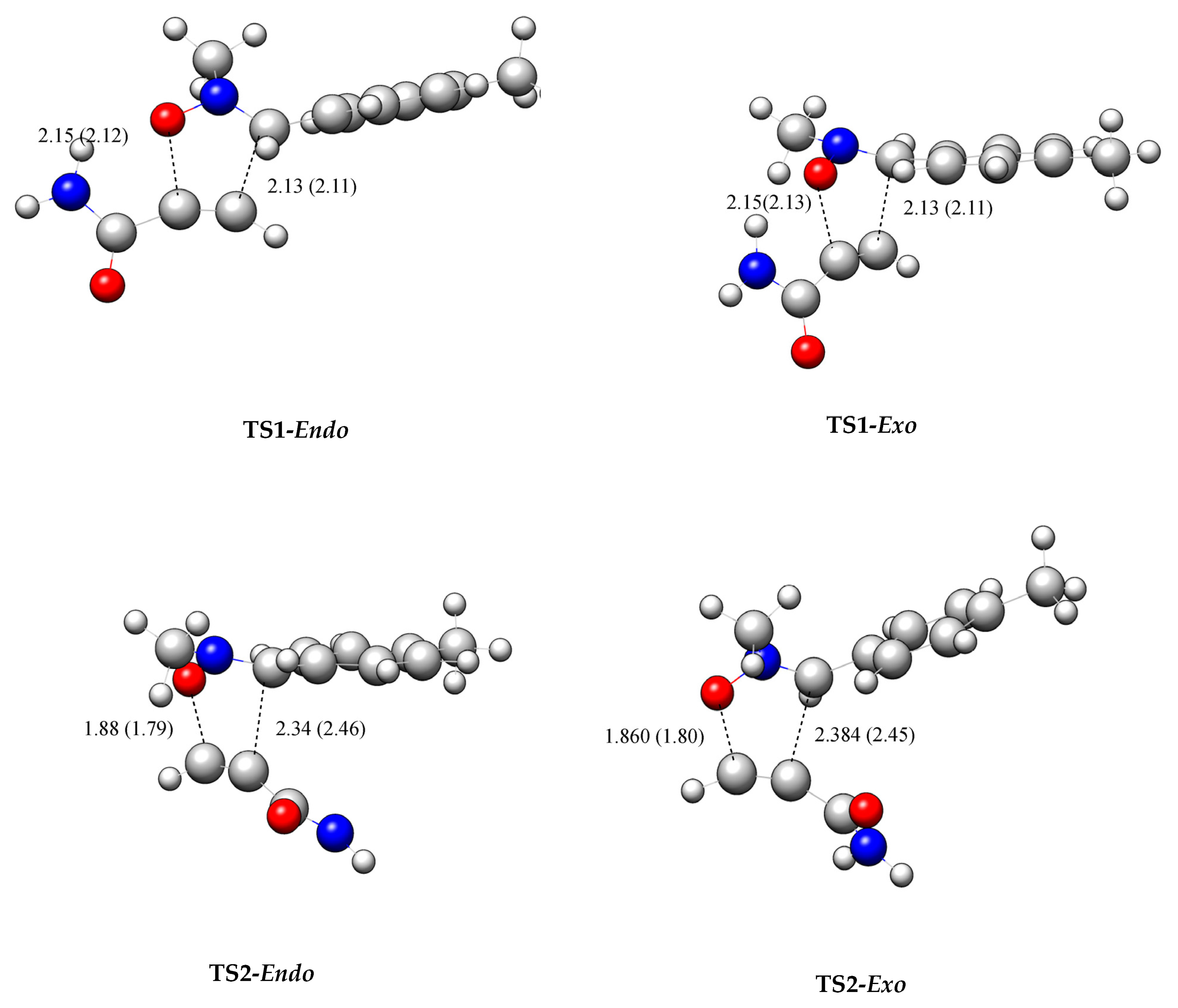
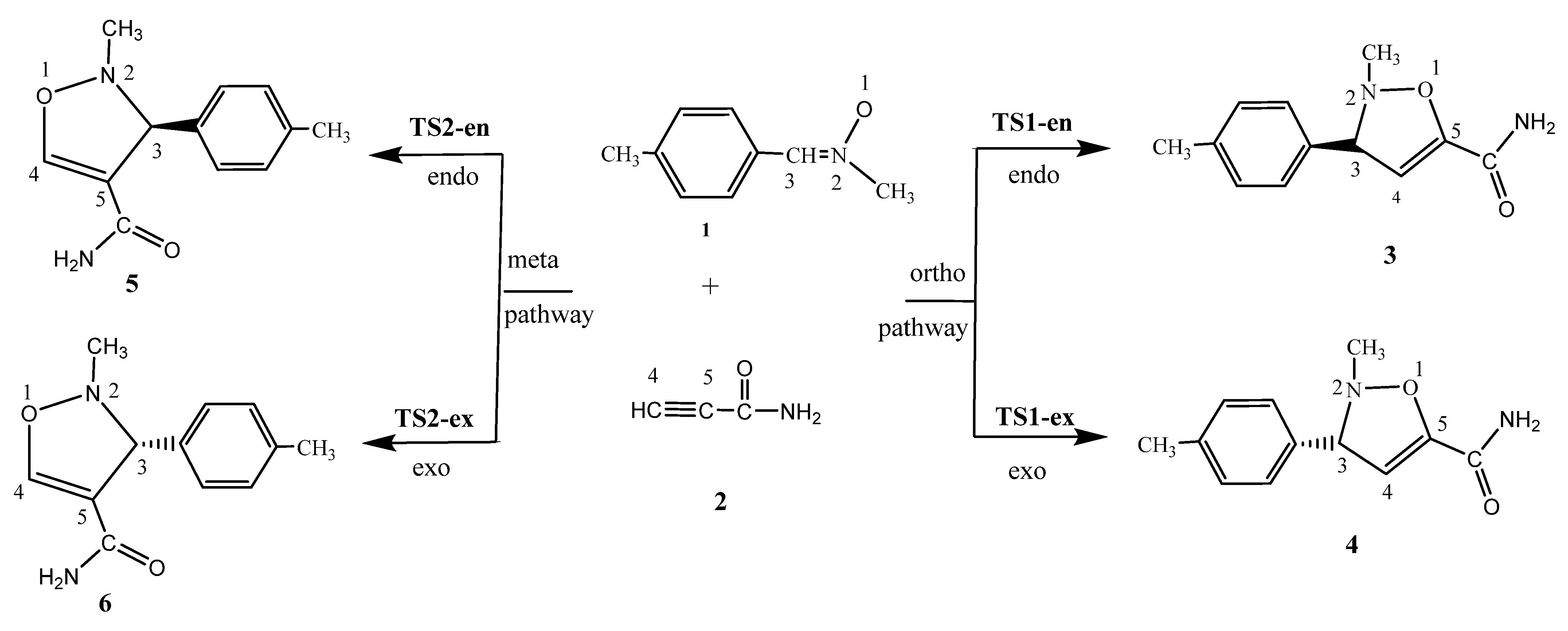
2.3. Analysis of the Potential Energy Surface along the Feasible Regioisomeric Pathways
- (i)
- The 32CA reaction of N-methyl-C-4-methylphenyl-nitrone 1 and 2-propynamide 2 shows negative reaction free energies from −45.96 to −79.36 kJ mol−1, including zero-point energy (ZPE) (see Supplementary Materials file), suggesting thermodynamic control and, hence, irreversibility.
- (ii)
- The ΔE of TS2-en is lower than that of TS1-en by 5.86, 6.07, 6.26, 7.14, and 6.9, in the gas phase, toluene, benzene, THF, and dichloromethane, respectively, suggesting exclusive TS2-en selectivity, in complete agreement with the experimental findings [38].
- (iii)
- TS1-en has an activation enthalpy of 80.52 kJ mol−1 in the gas phase, which increases to 90.12 kJ mol−1 in toluene, 87.50 kJ mol−1 in benzene, 94.29 kJ mol−1 in THF, and 94.82 kJ mol−1 in dichloroethane. This indicates an increase of 14.3 kJ mol−1 from the gas phase to dichloromethane, making the reaction energetically feasible in low-polarity solvents.
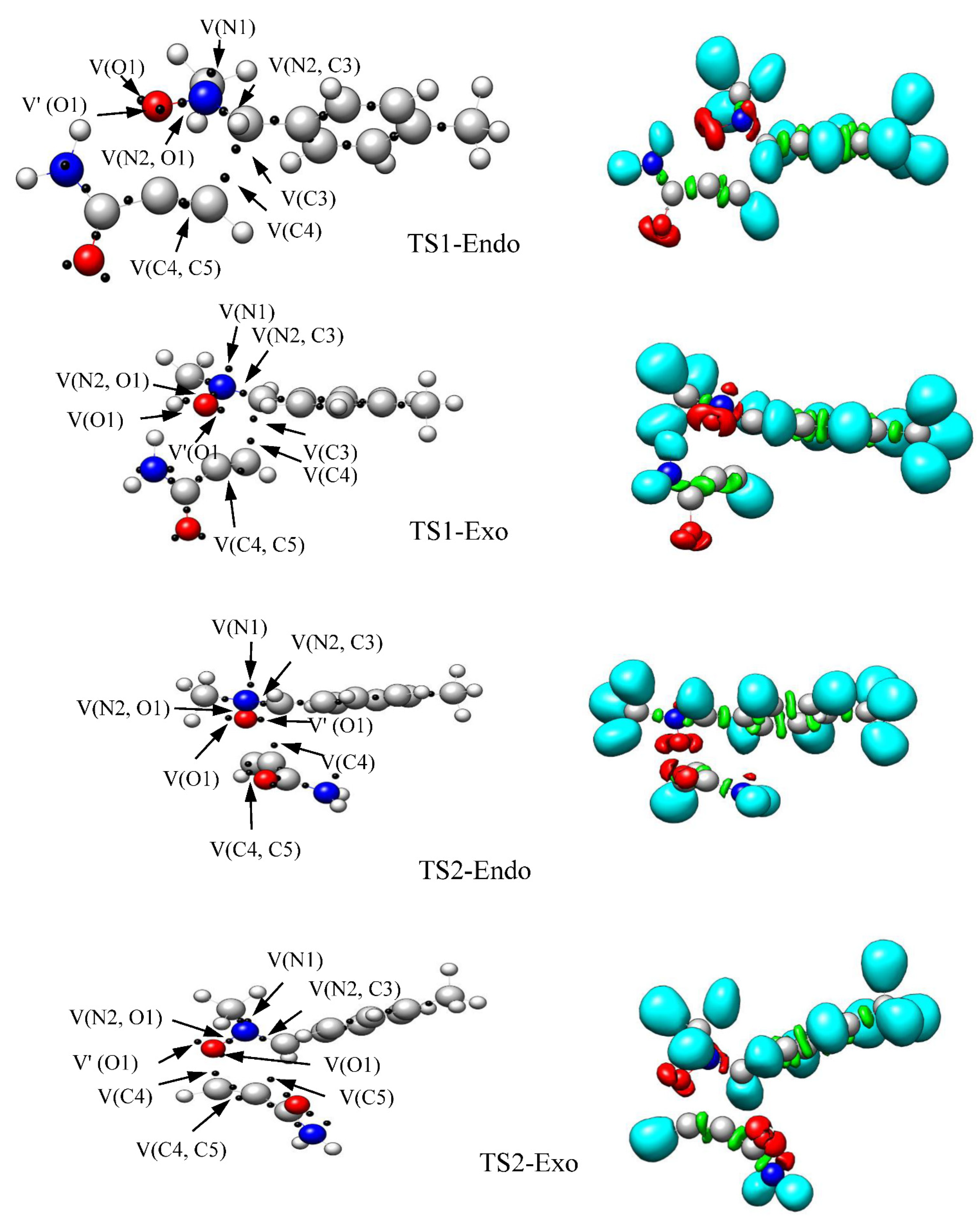
2.4. Topological Analysis of the ELF at the TSs
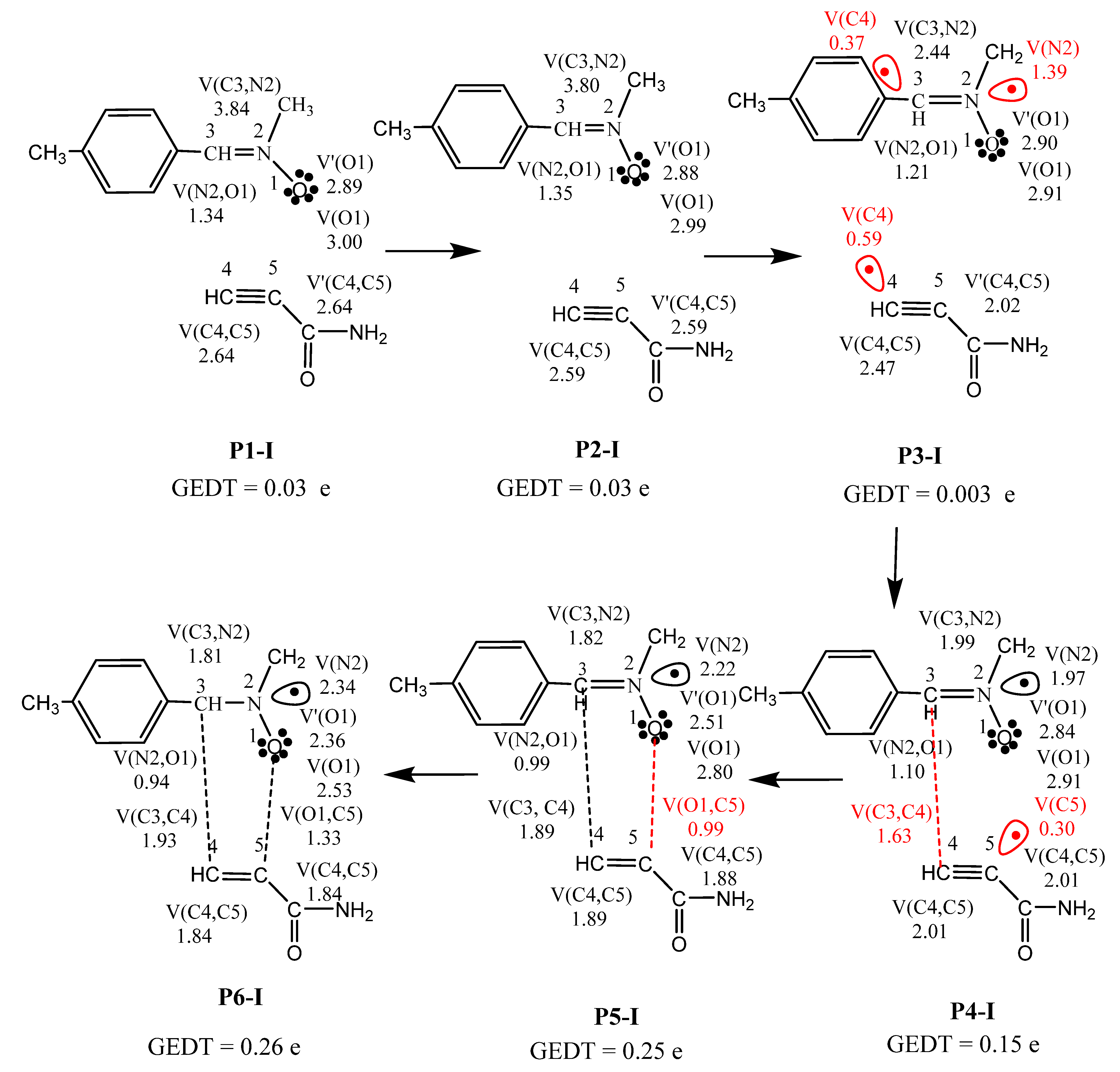
2.5. BET Study along the Favored Regiochemical Pathway
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feuer, H. Nitrile Oxides, Nitrones and Nitronates in Organic Synthesis: Novel Strategies in Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed]
- Mohammad-Salim, H.A.; Acharjee, N.; Abdallah, H. Insights into the mechanism and regioselectivity of the [3+2] cycloaddition reactions of cyclic nitrone to nitrile functions with a molecular electron density theory perspective. Theor. Chem. Acc. 2021, 140, 1. [Google Scholar] [CrossRef]
- Mohammad-Salim, H.A.; Basheer, H.A.; Abdallah, H.H.; Zeroual, A.; Jamil, L.A. A molecular electron density theory study for [3+2] cycloaddition reactions of N-benzylcyclohexylnitrone with methyl-3-butenoate. New J. Chem. 2021, 45, 262–267. [Google Scholar] [CrossRef]
- Mohammad-Salim, H.A.; Acharjee, N.; Domingo, L.R.; Abdallah, H.H. A molecular electron density theory study for [3+2] cycloaddition reactions of 1-pyrroline-1-oxide with disubstituted acetylenes leading to bicyclic 4-isoxazolines. Int. J. Quantum Chem. 2021, 121, e26503. [Google Scholar] [CrossRef]
- Acharjee, N.; Mohammad-Salim, H.; Chakraborty, M. Unveiling [3+2] cycloaddition reactions of benzonitrile oxide and diphenyl diazomethane to cyclopentene and norbornene: A molecular electron density theory perspective. Theor. Chem. Acc. 2021, 140, 113. [Google Scholar] [CrossRef]
- Acharjee, N.; Mohammad-Salim, H.A.; Chakraborty, M. Unveiling the regioselective synthesis of antiviral 5-isoxazol-5-yl-2′-deoxyuridines from the perspective of a molecular electron density theory. J. Serb. Chem. Soc. 2022, 87, 707–721. [Google Scholar] [CrossRef]
- Mohammad-Salim, H.A. A molecular electron density theory study of the [3+2] cycloaddition reaction of nitronic ester with methyl acrylate. Theor. Chem. Acc. 2021, 140, 83. [Google Scholar] [CrossRef]
- Acharjee, N.; Mohammad-Salim, H.A.; Chakraborty, M. Unveiling the synthesis of spirocyclic, tricyclic, and bicyclic triazolooxazines from intramolecular [3+2] azide-alkyne cycloadditions with a molecular electron density theory perspective. Struct. Chem. 2022, 33, 555–570. [Google Scholar] [CrossRef]
- Pérez, P.; Domingo, L.R.; Aurell, M.J.; Contreras, R. Quantitative characterization of the global electrophilicity pattern of some reagents involved in 1, 3-dipolar cycloaddition reactions. Tetrahedron 2003, 59, 3117–3125. [Google Scholar] [CrossRef]
- Domingo, L.R.; Saéz, J.A.; Zaragozá, R.J.; Arnó, M. Understanding the participation of quadricyclane as nucleophile in polar [2σ+ 2σ+ 2π] cycloadditions toward electrophilic π molecules. J. Org. Chem. 2008, 73, 8791–8799. [Google Scholar] [CrossRef]
- Domingo, L.R.; Aurell, M.; Pérez, P.; Contreras, R. Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels–Alder reactions. Tetrahedron 2002, 58, 4417–4423. [Google Scholar] [CrossRef]
- Jasiński, R.; Ziółkowska, M.; Demchuk, O.M.; Maziarka, A. Regio-and stereoselectivity of polar [2+3] cycloaddition reactions between (Z)-C-(3, 4, 5-trimethoxyphenyl)-N-methylnitrone and selected (E)-2-substituted nitroethenes. Cent. Eur. J. Chem. 2014, 12, 586–593. [Google Scholar] [CrossRef]
- Ríos-Gutiérrez, M.; Darù, A.; Tejero, T.; Domingo, L.R.; Merino, P. A molecular electron density theory study of the [3+2] cycloaddition reaction of nitrones with ketenes. Org. Biomol. Chem. 2017, 15, 1618–1627. [Google Scholar] [CrossRef]
- Domingo, L.R. Molecular electron density theory: A modern view of reactivity in organic chemistry. Molecules 2016, 21, 1319. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M. A molecular electron density theory study of the reactivity of azomethine imine in [3+2] cycloaddition reactions. Molecules 2017, 22, 750. [Google Scholar] [CrossRef]
- Nasri, L.; Ríos-Gutiérrez, M.; Nacereddine, A.K.; Djerourou, A.; Domingo, L.R. A molecular electron density theory study of [3+2] cycloaddition reactions of chiral azomethine ylides with β-nitrostyrene. Theor. Chem. Acc. 2017, 136, 104. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P.; Sáez, J. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, D.; Zhang, W.; Wang, Y.; Zhu, Y.; Jiab, Y.; Tang, M. A theoretical study on the mechanisms of the reactions between 1, 3-dialkynes and ammonia derivatives for the formation of five-membered N-heterocycles. Org. Biomol. Chem. 2014, 12, 7503–7514. [Google Scholar] [CrossRef]
- Jasiński, R. Competition between one-step and two-step mechanism in polar [3+2] cycloadditions of (Z)-C-(3, 4, 5-trimethoxyphenyl)-N-methyl-nitrone with (Z)-2-EWG-1-bromo-1-nitroethenes. Comput. Theor. Chem. 2018, 1125, 77–85. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A molecular electron density theory study of the reactivity and selectivities in [3 + 2] cycloaddition reactions of C, N-dialkyl nitrones with ethylene derivatives. J. Org. Chem. 2018, 83, 2182–2197. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Acharjee, N. A molecular electron density theory study of the chemoselectivity, regioselectivity, and diastereofacial selectivity in the synthesis of an anticancer spiroisoxazoline derived from α-santonin. Molecules 2019, 24, 832. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Parr, R.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Moss, S.; Coady, C. Potential-energy surfaces and transition-state theory. J. Chem. Educ. 1983, 60, 455. [Google Scholar] [CrossRef]
- Domingo, L.R. A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv. 2014, 4, 32415–32428. [Google Scholar] [CrossRef]
- Mohammad-Salim, H.A. Understanding the Reactivity of C-Cyclopropyl-N-Methylnitrone Participating in [3+2] Cycloaddition Reactions Towards Styrene with a Molecular Electron Density Theory Perspective. J. Mex. Chem. Soc. 2021, 65, 129–140. [Google Scholar] [CrossRef]
- Mohammad-Salim, H.A.; Hassan, R.; Abdallah, H.H.; Oftadeh, M. Theoretical Study on the Mechanism of [3+2] Cycloaddition Reactions between α, β-unsaturated Selenoaldehyde with Nitrone and with Nitrile Oxide. J. Mex. Chem. Soc. 2020, 64, 147–164. [Google Scholar] [CrossRef]
- Bader, R.F. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.; Essén, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943–1960. [Google Scholar] [CrossRef]
- Krokidis, X.; Noury, S.; Silvi, B. Characterization of elementary chemical processes by catastrophe theory. J. Phys. Chem. A 1997, 101, 7277–7282. [Google Scholar] [CrossRef]
- Mohammed Salih, S.A.; Basheer, H.A.; Mohammad-Salim, H.A. Insights into the mechanism and stereoselectivity of the [3+2] cycloaddition reaction between N-methyl-C-(4-hydroxylphenyl) nitrone and maleic anhydride with a molecular electron density theory perspective. Theor. Chem. Acc. 2022, 141, 33. [Google Scholar] [CrossRef]
- De Proft, F.; Vivas, R.; Peeters, A.; Van Alsenoy, C.; Geerlings, P. Hirshfeld partitioning of the electron density: Atomic dipoles and their relation with functional group properties. J. Comput. Chem. 2003, 24, 463–470. [Google Scholar] [CrossRef]
- Domingo, L.R.; Acharjee, N. Molecular electron density theory: A new theoretical outlook on organic chemistry. Front. Comput. Chem. 2020, 5, 174–227. [Google Scholar]
- Domingo, L.R.; Acharjee, N. Unravelling the strain-promoted [3+2] cycloaddition reactions of phenyl azide with cycloalkynes from the molecular electron density theory perspective. New J. Chem. 2020, 44, 13633–13643. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef]
- Domingo, L.R.; Ríos-Gutiérrez, M.; Pérez, P. A molecular electron density theory study of the participation of tetrazines in aza-Diels–Alder reactions. RSC Adv. 2020, 10, 15394–15405. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Andrés, J.; González-Navarrete, P.; Safont, V.S.; Silvi, B. Curly arrows, electron flow, and reaction mechanisms from the perspective of the bonding evolution theory. Phys. Chem. Chem. Phys. 2017, 19, 29031–29046. [Google Scholar] [CrossRef]
- Deng, L.; Ziegler, T. The determination of intrinsic reaction coordinates by density functional theory. Int. J. Quantum Chem. 1994, 52, 731–765. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Wiberg, K.B. Ab Initio Molecular Orbital Theory by WJ Hehre, L. Radom, P. v. R. Schleyer, and JA Pople, John Wiley, New York, 548pp. (1986). J. Comput. Chem. 1986, 7, 379. [Google Scholar] [CrossRef]
- Dresler, E.; Kącka-Zych, A.; Kwiatkowska, M.; Jasiński, R. Regioselectivity, stereoselectivity, and molecular mechanism of [3+ 2] cycloaddition reactions between 2-methyl-1-nitroprop-1-ene and (Z)-C-aryl-N-phenylnitrones: A DFT computational study. J. Mol. Model. 2018, 24, 329. [Google Scholar] [CrossRef] [PubMed]
- Alnajjar, R.A.; Jasiński, R. Competition between [2+1]-and [4+1]-cycloaddition mechanisms in reactions of conjugated nitroalkenes with dichlorocarbene in the light of a DFT computational study. J. Mol. Model. 2019, 25, 157. [Google Scholar] [CrossRef] [PubMed]
- Acharjee, N.; Mohammad-Salim, H.A.; Chakraborty, M.; Rao, M.P.; Ganesh, M. Unveiling the high regioselectivity and stereoselectivity within the synthesis of spirooxindolenitropyrrolidine: A molecular electron density theory perspective. J. Phys. Org. Chem. 2021, 34, e4189. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. Reaction path following in mass-weighted internal coordinates. J. Phys. Chem. 1990, 94, 5523–5527. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Simkin, B.Y.; Sheikhet, I.I. Quantum Chemical and Statistical Theory of Solutions: A Computational Approach; Ellis Horwood: London, UK, 1995. [Google Scholar]
- Reed, A.E.; Weinstock, R.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Weitao, Y. Aspects of Atoms and Molecules. In Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Kohn, W.; Sham, L. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016; Volume 421. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Basin | 1 | 2 |
|---|---|---|
| V(O1) | 3.00 | - |
| V’(O1) | 2.89 | - |
| V(N2,O1) | 1.34 | - |
| V(N2,C3) | 3.90 | - |
| V(C4,C5) | - | 2.64 |
| V′(C4,C5) | - | 2.64 |
| Reactant | μ | η | ω | N |
|---|---|---|---|---|
| 1 | −3.71 | 4.16 | 1.65 | 3.69 |
| 2 | −4.55 | 6.30 | 1.64 | 1.79 |
| TS | Solvent | ΔE ‡ | ΔH ‡ | ΔG ‡ | GEDT | Product | ΔE | ΔH | ΔG |
|---|---|---|---|---|---|---|---|---|---|
| TS1-en | Gas phase | 82.87 | 80.52 | 133.29 | 0.003 | 3 | −132.48 | −136.48 | −78.47 |
| TS1-ex | Gas phase | 80.33 | 77.80 | 134.22 | 0.007 | 4 | −123.45 | −130.08 | −62.92 |
| TS2-en | Gas phase | 77.01 | 75.97 | 127.00 | 0.089 | 5 | −126.20 | −130.99 | −69.07 |
| TS2-ex | Gas phase | 81.18 | 79.03 | 134.54 | 0.119 | 6 | −136.06 | −140.81 | −79.36 |
| TS1-en | Toluene | 90.09 | 90.12 | 137.71 | 0.011 | 3 | −121.29 | −122.65 | −73.59 |
| TS1-ex | Toluene | 88.37 | 88.41 | 136.76 | 0.009 | 4 | −112.47 | −114.21 | −62.24 |
| TS2-en | Toluene | 84.02 | 85.44 | 130.38 | 0.098 | 5 | −116.86 | −119.01 | −65.11 |
| TS2-ex | Toluene | 87.91 | 88.55 | 134.12 | 0.127 | 6 | −126.06 | −128.09 | −75.58 |
| TS1-en | Benzene | 90.01 | 87.50 | 144.78 | 0.011 | 3 | −121.76 | −125.50 | −67.49 |
| TS1-ex | Benzene | 88.00 | 85.67 | 142.68 | 0.008 | 4 | −112.92 | −117.05 | −56.08 |
| TS2-en | Benzene | 83.75 | 82.78 | 136.76 | 0.098 | 5 | −117.24 | −121.79 | −58.80 |
| TS2-ex | Benzene | 87.66 | 85.92 | 140.23 | 0.127 | 6 | −126.47 | −130.89 | −69.30 |
| TS1-en | THF | 96.88 | 94.29 | 153.33 | 0.022 | 3 | −112.03 | −115.66 | −57.56 |
| TS1-ex | THF | 94.50 | 92.31 | 149.78 | 0.019 | 4 | −103.32 | −107.35 | −45.96 |
| TS2-en | THF | 89.74 | 88.95 | 141.86 | 0.105 | 5 | −109.05 | −113.38 | −50.19 |
| TS2-ex | THF | 92.63 | 88.80 | 151.33 | 0.133 | 6 | −117.89 | −121.98 | −61.45 |
| TS1-en | DCM | 97.13 | 94.82 | 150.53 | 0.023 | 3 | −111.18 | −114.70 | −58.81 |
| TS1-ex | DCM | 95.02 | 93.01 | 146.64 | 0.044 | 4 | −102.47 | −106.38 | −47.16 |
| TS2-en | DCM | 90.23 | 89.55 | 140.05 | 0.105 | 5 | −108.07 | −112.55 | −51.52 |
| TS2-ex | DCM | 93.24 | 91.71 | 145.45 | 0.134 | 6 | −117.16 | −121.11 | −62.79 |
| Basin | TS1-en | TS1-ex | TS2-en | TS2-ex |
|---|---|---|---|---|
| V(O1) | 2.91 | 2.91 | 2.89 | 2.86 |
| V′(O1) | 2.90 | 2.88 | 2.83 | 2.86 |
| V(N2) | 1.39 | 1.46 | 1.45 | 1.31 |
| V(N2,O1) | 1.21 | 1.20 | 1.15 | 1.18 |
| V(N2,C3) | 2.44 | 2.36 | 2.69 | 2.83 |
| V(C4,C5) | 2.47 | 2.26 | 2.25 | 2.24 |
| V′(C4,C5) | 2.02 | 2.26 | 2.25 | 2.14 |
| V(C4) | 0.59 | 0.62 | 0.73 | 0.76 |
| V(3) | 0.37 | 0.33 | ||
| V(5) | 0.06 |
| Phases | I | II | III | IV | V | VI | |
|---|---|---|---|---|---|---|---|
| Structures | P1-I | P2-I | P3-I TS-ex | P4-I | P5-I | P6-I | 3 |
| d(C3–C4) | 3.124 | 2.686 | 2.131 | 1.814 | 1.625 | 1.549 | 1.511 |
| d(O1–C5) | 3.231 | 2.656 | 2.154 | 1.892 | 1.614 | 1.458 | 1.384 |
| GEDT | 0.03 | 0.03 | 0.003 | 0.15 | 0.25 | 0.26 | 0.25 |
| V(O1) | 3.00 | 2.99 | 2.91 | 2.91 | 2.80 | 2.53 | 2.49 |
| V′(O1) | 2.89 | 2.88 | 2.90 | 2.84 | 2.51 | 2.36 | 2.36 |
| V(C3,N2) | 3.84 | 3.80 | 2.44 | 1.99 | 1.82 | 1.81 | 1.76 |
| V(N2,O1) | 1.34 | 1.35 | 1.21 | 1.10 | 0.99 | 0.94 | 0.91 |
| V(N2) | 1.39 | 1.97 | 2.22 | 2.34 | 2.38 | ||
| V(C4,C5) | 2.64 | 2.59 | 2.02 | 2.01 | 1.88 | 1.84 | 1.82 |
| V′(C4,C5) | 2.64 | 2.59 | 2.47 | 2.01 | 1.89 | 1.84 | 1.77 |
| V(C3) | 0.37 | ||||||
| V(C4) | 0.59 | ||||||
| V(C5) | 0.30 | ||||||
| V(C3,C4) | 1.63 | 1.89 | 1.93 | 1.96 | |||
| V(O1,C5) | 0.99 | 1.33 | 1.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salih, S.A.M.; Basheer, H.A.; de Julián-Ortiz, J.V.; Mohammad-Salim, H.A. Unveiling the Stereoselectivity and Regioselectivity of the [3+2] Cycloaddition Reaction between N-methyl-C-4-methylphenyl-nitrone and 2-Propynamide from a MEDT Perspective. Int. J. Mol. Sci. 2023, 24, 9102. https://doi.org/10.3390/ijms24109102
Salih SAM, Basheer HA, de Julián-Ortiz JV, Mohammad-Salim HA. Unveiling the Stereoselectivity and Regioselectivity of the [3+2] Cycloaddition Reaction between N-methyl-C-4-methylphenyl-nitrone and 2-Propynamide from a MEDT Perspective. International Journal of Molecular Sciences. 2023; 24(10):9102. https://doi.org/10.3390/ijms24109102
Chicago/Turabian StyleSalih, Sabir A. Mohammed, Huda A. Basheer, Jesus Vicente de Julián-Ortiz, and Haydar A. Mohammad-Salim. 2023. "Unveiling the Stereoselectivity and Regioselectivity of the [3+2] Cycloaddition Reaction between N-methyl-C-4-methylphenyl-nitrone and 2-Propynamide from a MEDT Perspective" International Journal of Molecular Sciences 24, no. 10: 9102. https://doi.org/10.3390/ijms24109102