
Parkinson’s Disease: Exploring Different Animal Model Systems

Abstract
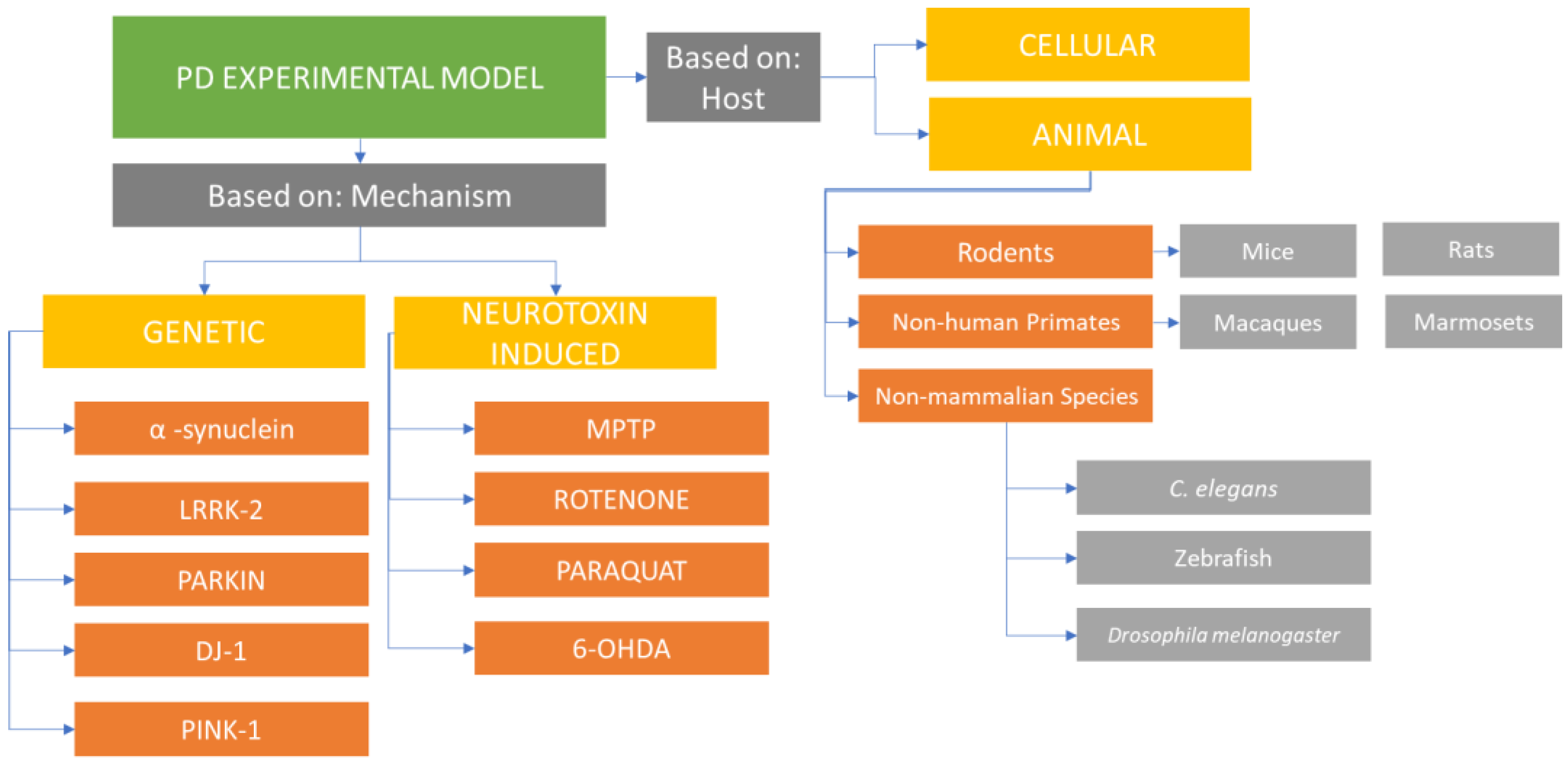
:1. Introduction
2. Parkinson’s Disease Model Systems
3. Animal Model
- The presence of a complement of DA neurons during the birth stage, with specific and gradual depletion of DA neurons as the organism progresses to adulthood. The loss in neurons should be more than 50% of the total amount and be easily noticeable via biochemistry- and neuropathology-related techniques [16];
- The model animal must be able to exhibit motor deficits observed in the disease or the expected behavioral phenotype, including slowness in movement, resting tremor, and rigidity [16];
- The presence of Lewy body and its development as an indicator of the manifestation of α-synuclein pathology [16];
- The model must also be sure to replicate the disease progression over a period of a few months allowing for a faster and less expensive screening of potential therapeutic candidates [16].
4. Common Laboratory Animals Used to Model PD
4.1. Rodents
4.2. Non-Human Primates (NHPs)
4.3. Non-Mammalian Species (NMSs)
4.3.1. Caenorhabditis elegans
4.3.2. Drosophila melanogaster
4.3.3. Zebrafish
5. PD Induction in Animal Models
5.1. PD Induction in Animal Models by Pharmacological Intervention
5.2. Commonly Used Neurotoxins to Induce PD
5.2.1. 6-OHDA (6-Hydroydopamine)
5.2.2. Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
5.2.3. Paraquat (N,N-Dimethyl-4-4-4-bipyridinium)
5.2.4. Rotenone
5.3. PD Induction in Animal Model by α-Synuclein Pre-Formed Fibril (PFF)
5.4. PD Induction in Animal Model by Genetic Manipulation
5.4.1. α-Synuclein
5.4.2. Leucine-Rich Repeat Kinase 2 (LRRK2)
5.4.3. Parkin
5.4.4. Protein Deglycase (DJ-1)
5.4.5. PINK1 (Phosphatase and Tensin Homolog—PTEN-Induced Novel Kinase 1)
5.5. PD Induction in Animal Models by Combination of Pharmacological Intervention and Genetic Manipulation
6. Recent Development in PD Model System
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 6-OHDA | 6-hydroydopamine |
| BBB | Blood–Brain Barrier |
| BDNF | Brain-derived neurotrophic factor |
| C. elegans | Caenorhabditis elegans |
| D2R | Dopamine D2 receptor |
| DA | Dopamine |
| DAT | Dopamine Transporter |
| DJ-1 | Protein Deglycase |
| GDNF | Glial cell line-derived neurotrophic factor |
| iPSC | induced pluripotent stem cell |
| IP | Intraperitoneal |
| IV | Intravenous |
| LB | Lewy body |
| LRRK2 | Leucine Rich Repeat Kinase |
| LUHMES | Lund Human Mesencephalic cells |
| MPP+ | 1-methyl-4-phenylpyridinium |
| MPTP | Methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| NGF | Nerve Growth Factor |
| NHP | Non-Human Primate |
| NMS | Non-Mammalian Species |
| NT | Neurotransmitter |
| PD | Parkinson’s disease |
| PINK1 | PTEN-induced novel kinase 1 |
| p-PKCa | Protein Kinase C |
| SNc/SNpc/SN | Substantia Nigra (pars compacta) |
| TH | Tyrosine Hydroxylase |
| VMAT | Vesicular Monoamine transporter |
| YOPD | Young-onset Parkinson’s disease |
| PQ | paraquat |
| KO | Knockout |
References
- Chia, S.J.; Tan, E.-K.; Chao, Y.-X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef]
- Parkinson, J. An Essay on the Shaking Palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet Lond. Engl. 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Gasser, T.; Hardy, J.; Mizuno, Y. Milestones in PD Genetics. Mov. Disord. 2011, 26, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-Scale Meta-Analysis of Genome-Wide Association Data Identifies Six New Risk Loci for Parkinson’s Disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. Alpha-Synuclein Overexpression in PC12 and Chromaffin Cells Impairs Catecholamine Release by Interfering with a Late Step in Exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased Expression of Alpha-Synuclein Reduces Neurotransmitter Release by Inhibiting Synaptic Vesicle Reclustering after Endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Scott, D.A.; Tabarean, I.; Tang, Y.; Cartier, A.; Masliah, E.; Roy, S. A Pathologic Cascade Leading to Synaptic Dysfunction in α-Synuclein-Induced Neurodegeneration. J. Neurosci. 2010, 30, 8083–8095. [Google Scholar] [CrossRef]
- Scott, D.; Roy, S. α-Synuclein Inhibits Intersynaptic Vesicle Mobility and Maintains Recycling-Pool Homeostasis. J. Neurosci. 2012, 32, 10129–10135. [Google Scholar] [CrossRef]
- Vargas, K.J.; Makani, S.; Davis, T.; Westphal, C.H.; Castillo, P.E.; Chandra, S.S. Synucleins Regulate the Kinetics of Synaptic Vesicle Endocytosis. J. Neurosci. 2014, 34, 9364–9376. [Google Scholar] [CrossRef]
- Wang, L.; Das, U.; Scott, D.A.; Tang, Y.; McLean, P.J.; Roy, S. α-Synuclein Multimers Cluster Synaptic-Vesicles and Attenuate Recycling. Curr. Biol. CB 2014, 24, 2319–2326. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wang, L.; Bao, H.; Premi, S.; Das, U.; Chapman, E.R.; Roy, S. Functional Cooperation of α-Synuclein and VAMP2 in Synaptic Vesicle Recycling. Proc. Natl. Acad. Sci. USA 2019, 116, 11113–11115. [Google Scholar] [CrossRef] [PubMed]
- Falkenburger, B.H.; Saridaki, T.; Dinter, E. Cellular Models for Parkinson’s Disease. J. Neurochem. 2016, 139, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Jagmag, S.A.; Tripathi, N.; Shukla, S.D.; Maiti, S.; Khurana, S. Evaluation of Models of Parkinson’s Disease. Front. Neurosci. 2016, 9, 503. [Google Scholar] [CrossRef]
- Koprich, J.B.; Kalia, L.V.; Brotchie, J.M. Animal Models of α-Synucleinopathy for Parkinson Disease Drug Development. Nat. Rev. Neurosci. 2017, 18, 515–529. [Google Scholar] [CrossRef]
- Beal, M.F. Experimental Models of Parkinson’s Disease. Nat. Rev. Neurosci. 2001, 2, 325–334. [Google Scholar] [CrossRef]
- Meredith, G.E.; Kang, U.J. Behavioral Models of Parkinson’s Disease in Rodents: A New Look at an Old Problem. Mov. Disord. 2006, 21, 1595–1606. [Google Scholar] [CrossRef]
- Sedelis, M.; Schwarting, R.K.; Huston, J.P. Behavioral Phenotyping of the MPTP Mouse Model of Parkinson’s Disease. Behav. Brain Res. 2001, 125, 109–125. [Google Scholar] [CrossRef]
- Ogawa, N.; Hirose, Y.; Ohara, S.; Ono, T.; Watanabe, Y. A Simple Quantitative Bradykinesia Test in MPTP-Treated Mice. Res. Commun. Chem. Pathol. Pharmacol. 1985, 50, 435–441. [Google Scholar]
- Kraeuter, A.-K.; Guest, P.C.; Sarnyai, Z. The Open Field Test for Measuring Locomotor Activity and Anxiety-Like Behavior. Methods Mol. Biol. 2019, 1916, 99–103. [Google Scholar] [CrossRef]
- Lalvay, L.; Lara, M.; Mora, A.; Alarcón, F.; Fraga, M.; Pancorbo, J.; Marina, J.L.; Mena, M.Á.; Lopez Sendón, J.L.; García de Yébenes, J. Quantitative Measurement of Akinesia in Parkinson’s Disease. Mov. Disord. Clin. Pract. 2017, 4, 316–322. [Google Scholar] [CrossRef]
- Leem, Y.-H.; Park, J.-S.; Park, J.-E.; Kim, D.-Y.; Kim, H.-S. Neurogenic Effects of Rotarod Walking Exercise in Subventricular Zone, Subgranular Zone, and Substantia Nigra in MPTP-Induced Parkinson’s Disease Mice. Sci. Rep. 2022, 12, 10544. [Google Scholar] [CrossRef]
- Commons, K.G.; Cholanians, A.B.; Babb, J.A.; Ehlinger, D.G. The Rodent Forced Swim Test Measures Stress-Coping Strategy, Not Depression-like Behavior. ACS Chem. Neurosci. 2017, 8, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Can, A.; Dao, D.T.; Terrillion, C.E.; Piantadosi, S.C.; Bhat, S.; Gould, T.D. The Tail Suspension Test. J. Vis. Exp. 2012, 59, e3769. [Google Scholar] [CrossRef]
- Exner, N.; Lutz, A.K.; Haass, C.; Winklhofer, K.F. Mitochondrial Dysfunction in Parkinson’s Disease: Molecular Mechanisms and Pathophysiological Consequences. EMBO J. 2012, 31, 3038–3062. [Google Scholar] [CrossRef]
- Matsui, H.; Uemura, N.; Yamakado, H.; Takeda, S.; Takahashi, R. Exploring the Pathogenetic Mechanisms Underlying Parkinson’s Disease in Medaka Fish. J. Park. Dis. 2014, 4, 301–310. [Google Scholar] [CrossRef]
- Morais, V.A.; Haddad, D.; Craessaerts, K.; De Bock, P.-J.; Swerts, J.; Vilain, S.; Aerts, L.; Overbergh, L.; Grünewald, A.; Seibler, P.; et al. PINK1 Loss-of-Function Mutations Affect Mitochondrial Complex I Activity via NdufA10 Ubiquinone Uncoupling. Science 2014, 344, 203–207. [Google Scholar] [CrossRef]
- Lachenmayer, M.L.; Yue, Z. Genetic Animal Models for Evaluating the Role of Autophagy in Etiopathogenesis of Parkinson Disease. Autophagy 2012, 8, 1837–1838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Duan, C.; Yang, H. Defective Autophagy in Parkinson’s Disease: Lessons from Genetics. Mol. Neurobiol. 2015, 51, 89–104. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Bott, L.C. The Ubiquitin-Proteasome System in Neurodegenerative Diseases: Precipitating Factor, yet Part of the Solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef]
- Joselin, A.P.; Hewitt, S.J.; Callaghan, S.M.; Kim, R.H.; Chung, Y.-H.; Mak, T.W.; Shen, J.; Slack, R.S.; Park, D.S. ROS-Dependent Regulation of Parkin and DJ-1 Localization during Oxidative Stress in Neurons. Hum. Mol. Genet. 2012, 21, 4888–4903. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Przedborski, S. Parkinson’s Disease: Animal Models and Dopaminergic Cell Vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef]
- Phillips, K.A.; Bales, K.L.; Capitanio, J.P.; Conley, A.; Czoty, P.W.; ’t Hart, B.A.; Hopkins, W.D.; Hu, S.-L.; Miller, L.A.; Nader, M.A.; et al. Why Primate Models Matter. Am. J. Primatol. 2014, 76, 801–827. [Google Scholar] [CrossRef]
- Emborg, M.E. Nonhuman Primate Models of Parkinson’s Disease. ILAR J. 2007, 48, 339–355. [Google Scholar] [CrossRef]
- Davin, A.; Chabardès, S.; Belaid, H.; Fagret, D.; Djaileb, L.; Dauvilliers, Y.; David, O.; Torres-Martinez, N.; Piallat, B. Early Onset of Sleep/Wake Disturbances in a Progressive Macaque Model of Parkinson’s Disease. Sci. Rep. 2022, 12, 17499. [Google Scholar] [CrossRef]
- Choudhury, G.R.; Daadi, M.M. Charting the Onset of Parkinson-like Motor and Non-Motor Symptoms in Nonhuman Primate Model of Parkinson’s Disease. PLoS ONE 2018, 13, e0202770. [Google Scholar] [CrossRef]
- Langston, J.W.; Forno, L.S.; Rebert, C.S.; Irwin, I. Selective Nigral Toxicity after Systemic Administration of 1-Methyl-4-Phenyl-1,2,5,6-Tetrahydropyrine (MPTP) in the Squirrel Monkey. Brain Res. 1984, 292, 390–394. [Google Scholar] [CrossRef]
- Chen, M.-K.; Kuwabara, H.; Zhou, Y.; Adams, R.J.; Brasić, J.R.; McGlothan, J.L.; Verina, T.; Burton, N.C.; Alexander, M.; Kumar, A.; et al. VMAT2 and Dopamine Neuron Loss in a Primate Model of Parkinson’s Disease. J. Neurochem. 2008, 105, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Hurley, P.J.; Elsworth, J.D.; Whittaker, M.C.; Roth, R.H.; Redmond, D.E. Aged Monkeys as a Partial Model for Parkinson’s Disease. Pharmacol. Biochem. Behav. 2011, 99, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Li, H.; Huang, B.; Rizak, J.; Li, L.; Xu, L.; Huang, T.; Liu, L.; Wu, J.; Lü, L.; et al. 1-Methyl-4-Phenylpyridinium Stereotactic Infusion Completely and Specifically Ablated the Nigrostriatal Dopaminergic Pathway in Rhesus Macaque. PLoS ONE 2015, 10, e0127953. [Google Scholar] [CrossRef]
- Eslamboli, A.; Baker, H.F.; Ridley, R.M.; Annett, L.E. Sensorimotor Deficits in a Unilateral Intrastriatal 6-OHDA Partial Lesion Model of Parkinson’s Disease in Marmoset Monkeys. Exp. Neurol. 2003, 183, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Vermilyea, S.C.; Emborg, M.E. α-Synuclein and Nonhuman Primate Models of Parkinson’s Disease. J. Neurosci. Methods 2015, 255, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Kirik, D.; Annett, L.E.; Burger, C.; Muzyczka, N.; Mandel, R.J.; Björklund, A. Nigrostriatal Alpha-Synucleinopathy Induced by Viral Vector-Mediated Overexpression of Human Alpha-Synuclein: A New Primate Model of Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2884–2889. [Google Scholar] [CrossRef] [PubMed]
- Vermilyea, S.C.; Emborg, M.E. The Role of Nonhuman Primate Models in the Development of Cell-Based Therapies for Parkinson’s Disease. J. Neural Transm. 2018, 125, 365. [Google Scholar] [CrossRef]
- Eslamboli, A.; Romero-Ramos, M.; Burger, C.; Bjorklund, T.; Muzyczka, N.; Mandel, R.J.; Baker, H.; Ridley, R.M.; Kirik, D. Long-Term Consequences of Human Alpha-Synuclein Overexpression in the Primate Ventral Midbrain. Brain J. Neurol. 2007, 130, 799–815. [Google Scholar] [CrossRef]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic Animal Models of Parkinson’s Disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Koprich, J.B.; Johnston, T.H.; Reyes, G.; Omana, V.; Brotchie, J.M. Towards a Non-Human Primate Model of Alpha-Synucleinopathy for Development of Therapeutics for Parkinson’s Disease: Optimization of AAV1/2 Delivery Parameters to Drive Sustained Expression of Alpha Synuclein and Dopaminergic Degeneration in Macaque. PLoS ONE 2016, 11, e0167235. [Google Scholar] [CrossRef]
- Lasbleiz, C.; Mestre-Francés, N.; Devau, G.; Luquin, M.-R.; Tenenbaum, L.; Kremer, E.J.; Verdier, J.-M. Combining Gene Transfer and Nonhuman Primates to Better Understand and Treat Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 10. [Google Scholar] [CrossRef]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Austrulia, 2018; ISBN 978-0-9944381-6-4. [Google Scholar]
- White, J.G.; Southgate, E.; Thomson, J.N.; Brenner, S. The Structure of the Nervous System of the Nematode Caenorhabditis Elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1986, 314, 1–340. [Google Scholar] [CrossRef]
- Martinez, B.A.; Caldwell, K.A.; Caldwell, G.A. C. Elegans as a Model System to Accelerate Discovery for Parkinson Disease. Curr. Opin. Genet. Dev. 2017, 44, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Brenner, S. The Genetics of Caenorhabditis Elegans. Genetics 1974, 77, 71–94. [Google Scholar] [CrossRef]
- Shukla, A.K.; Pragya, P.; Chaouhan, H.S.; Patel, D.K.; Abdin, M.Z.; Kar Chowdhuri, D. Mutation in Drosophila Methuselah Resists Paraquat Induced Parkinson-like Phenotypes. Neurobiol. Aging 2014, 35, e1–e2419. [Google Scholar] [CrossRef] [PubMed]
- Feany, M.B.; Bender, W.W. A Drosophila Model of Parkinson’s Disease. Nature 2000, 404, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Vaz, R.L.; Outeiro, T.F.; Ferreira, J.J. Zebrafish as an Animal Model for Drug Discovery in Parkinson’s Disease and Other Movement Disorders: A Systematic Review. Front. Neurol. 2018, 9, 347. [Google Scholar] [CrossRef]
- Zeng, X.-S.; Geng, W.-S.; Jia, J.-J. Neurotoxin-Induced Animal Models of Parkinson Disease: Pathogenic Mechanism and Assessment. ASN Neuro 2018, 10, 1759091418777438. [Google Scholar] [CrossRef]
- Shimoji, M.; Zhang, L.; Mandir, A.S.; Dawson, V.L.; Dawson, T.M. Absence of Inclusion Body Formation in the MPTP Mouse Model of Parkinson’s Disease. Mol. Brain Res. 2005, 134, 103–108. [Google Scholar] [CrossRef]
- Miyazaki, I.; Isooka, N.; Imafuku, F.; Sun, J.; Kikuoka, R.; Furukawa, C.; Asanuma, M. Chronic Systemic Exposure to Low-Dose Rotenone Induced Central and Peripheral Neuropathology and Motor Deficits in Mice: Reproducible Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 3254. [Google Scholar] [CrossRef]
- Maries, E.; Dass, B.; Collier, T.J.; Kordower, J.H.; Steece-Collier, K. The Role of Alpha-Synuclein in Parkinson’s Disease: Insights from Animal Models. Nat. Rev. Neurosci. 2003, 4, 727–738. [Google Scholar] [CrossRef]
- Alvarez-Fischer, D.; Guerreiro, S.; Hunot, S.; Saurini, F.; Marien, M.; Sokoloff, P.; Hirsch, E.C.; Hartmann, A.; Michel, P.P. Modelling Parkinson-like Neurodegeneration via Osmotic Minipump Delivery of MPTP and Probenecid. J. Neurochem. 2008, 107, 701–711. [Google Scholar] [CrossRef]
- Ungerstedt, U. 6-Hydroxy-Dopamine Induced Degeneration of Central Monoamine Neurons. Eur. J. Pharmacol. 1968, 5, 107–110. [Google Scholar] [CrossRef]
- Verhave, P.S.; Vanwersch, R.A.P.; van Helden, H.P.M.; Smit, A.B.; Philippens, I.H.C.H.M. Two New Test Methods to Quantify Motor Deficits in a Marmoset Model for Parkinson’s Disease. Behav. Brain Res. 2009, 200, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Barraud, Q.; Lambrecq, V.; Forni, C.; McGuire, S.; Hill, M.; Bioulac, B.; Balzamo, E.; Bezard, E.; Tison, F.; Ghorayeb, I. Sleep Disorders in Parkinson’s Disease: The Contribution of the MPTP Non-Human Primate Model. Exp. Neurol. 2009, 219, 574–582. [Google Scholar] [CrossRef]
- Cooper, J.F.; Van Raamsdonk, J.M. Modeling Parkinson’s Disease in C. elegans. J. Park. Dis. 2018, 8, 17–32. [Google Scholar] [CrossRef]
- Gonçalves, D.F.; Courtes, A.A.; Hartmann, D.D.; da Rosa, P.C.; Oliveira, D.M.; Soares, F.A.A.; Dalla Corte, C.L. 6-Hydroxydopamine Induces Different Mitochondrial Bioenergetics Response in Brain Regions of Rat. Neurotoxicology 2019, 70, 1–11. [Google Scholar] [CrossRef]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and New Animal Models of Parkinson’s Disease. J. Biomed. Biotechnol. 2012, 2012, 845618. [Google Scholar] [CrossRef]
- Przedborski, S.; Levivier, M.; Jiang, H.; Ferreira, M.; Jackson-Lewis, V.; Donaldson, D.; Togasaki, D.M. Dose-Dependent Lesions of the Dopaminergic Nigrostriatal Pathway Induced by Intrastriatal Injection of 6-Hydroxydopamine. Neuroscience 1995, 67, 631–647. [Google Scholar] [CrossRef] [PubMed]
- Sauer, H.; Oertel, W.H. Progressive Degeneration of Nigrostriatal Dopamine Neurons Following Intrastriatal Terminal Lesions with 6-Hydroxydopamine: A Combined Retrograde Tracing and Immunocytochemical Study in the Rat. Neuroscience 1994, 59, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Sauer, H.; Bjorklund, A. Dopaminergic Neuronal Degeneration and Motor Impairments Following Axon Terminal Lesion by Instrastriatal 6-Hydroxydopamine in the Rat. Neuroscience 1996, 72, 641–653. [Google Scholar] [CrossRef]
- Airavaara, M.; Parkkinen, I.; Konovalova, J.; Albert, K.; Chmielarz, P.; Domanskyi, A. Back and to the Future: From Neurotoxin-Induced to Human Parkinson’s Disease Models. Curr. Protoc. Neurosci. 2020, 91, e88. [Google Scholar] [CrossRef]
- Yang, S.; Huh, E.; Moon, G.H.; Ahn, J.; Woo, J.; Han, H.-S.; Lee, H.-H.; Chung, K.-S.; Lee, K.-T.; Oh, M.S.; et al. In Vitro and in Vivo Neuroprotective Effect of Novel MPGES-1 Inhibitor in Animal Model of Parkinson’s Disease. Bioorg. Med. Chem. Lett. 2022, 74, 128920. [Google Scholar] [CrossRef]
- Yu, H.; Liu, X.; Chen, B.; Vickstrom, C.R.; Friedman, V.; Kelly, T.J.; Bai, X.; Zhao, L.; Hillard, C.J.; Liu, Q.-S. The Neuroprotective Effects of the CB2 Agonist GW842166x in the 6-OHDA Mouse Model of Parkinson’s Disease. Cells 2021, 10, 3548. [Google Scholar] [CrossRef]
- Wang, J.Y.; Yang, J.Y.; Wang, F.; Fu, S.Y.; Hou, Y.; Jiang, B.; Ma, J.; Song, C.; Wu, C.F. Neuroprotective Effect of Pseudoginsenoside-F11 on a Rat Model of Parkinson’s Disease Induced by 6-Hydroxydopamine. Evid. Based Complement. Alternat. Med. 2013, 2013, e152798. [Google Scholar] [CrossRef] [PubMed]
- El Nebrisi, E.; Javed, H.; Ojha, S.K.; Oz, M.; Shehab, S. Neuroprotective Effect of Curcumin on the Nigrostriatal Pathway in a 6-Hydroxydopmine-Induced Rat Model of Parkinson’s Disease Is Mediated by A7-Nicotinic Receptors. Int. J. Mol. Sci. 2020, 21, 7329. [Google Scholar] [CrossRef]
- Masini, D.; Plewnia, C.; Bertho, M.; Scalbert, N.; Caggiano, V.; Fisone, G. A Guide to the Generation of a 6-Hydroxydopamine Mouse Model of Parkinson’s Disease for the Study of Non-Motor Symptoms. Biomedicines 2021, 9, 598. [Google Scholar] [CrossRef]
- Schober, A. Classic Toxin-Induced Animal Models of Parkinson’s Disease: 6-OHDA and MPTP. Cell Tissue Res. 2004, 318, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Rademacher, D.J. MPTP Mouse Models of Parkinson’s Disease: An Update. J. Park. Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Smeyne, R.J.; Jackson-Lewis, V. The MPTP Model of Parkinson’s Disease. Brain Res. Mol. Brain Res. 2005, 134, 57–66. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in Humans Due to a Product of Meperidine-Analog Synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Cui, M.; Aras, R.; Christian, W.V.; Rappold, P.M.; Hatwar, M.; Panza, J.; Jackson-Lewis, V.; Javitch, J.A.; Ballatori, N.; Przedborski, S.; et al. The Organic Cation Transporter-3 Is a Pivotal Modulator of Neurodegeneration in the Nigrostriatal Dopaminergic Pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 8043–8048. [Google Scholar] [CrossRef]
- Bezard, E.; Gross, C.E.; Fournier, M.C.; Dovero, S.; Bloch, B.; Jaber, M. Absence of MPTP-Induced Neuronal Death in Mice Lacking the Dopamine Transporter. Exp. Neurol. 1999, 155, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Hamre, K.; Tharp, R.; Poon, K.; Xiong, X.; Smeyne, R.J. Differential Strain Susceptibility Following 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP) Administration Acts in an Autosomal Dominant Fashion: Quantitative Analysis in Seven Strains of Mus Musculus. Brain Res. 1999, 828, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP Mouse Model of Parkinson’s Disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Sedelis, M.; Hofele, K.; Auburger, G.W.; Morgan, S.; Huston, J.P.; Schwarting, R.K. MPTP Susceptibility in the Mouse: Behavioral, Neurochemical, and Histological Analysis of Gender and Strain Differences. Behav. Genet. 2000, 30, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Merghani, M.M.; Ardah, M.T.; Al Shamsi, M.; Kitada, T.; Haque, M.E. Dose-Related Biphasic Effect of the Parkinson’s Disease Neurotoxin MPTP, on the Spread, Accumulation, and Toxicity of α-Synuclein. Neurotoxicology 2021, 84, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Schlüter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like Syndrome Induced by Continuous MPTP Infusion: Convergent Roles of the Ubiquitin-Proteasome System and α-Synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418. [Google Scholar] [CrossRef]
- Graham, J.D.; Lewis, M.J.; Williams, J. Proceedings: The Effect of Delta-1-Tetrahydrocannabinol on the Noradrenaline and Dopamine Content of the Brain and Heart of the Rat. Br. J. Pharmacol. 1974, 52, 446P. [Google Scholar]
- Rappold, P.M.; Cui, M.; Chesser, A.S.; Tibbett, J.; Grima, J.C.; Duan, L.; Sen, N.; Javitch, J.A.; Tieu, K. Paraquat Neurotoxicity Is Mediated by the Dopamine Transporter and Organic Cation Transporter-3. Proc. Natl. Acad. Sci. USA 2011, 108, 20766–20771. [Google Scholar] [CrossRef]
- Corasaniti, M.T.; Bagetta, G.; Rodinò, P.; Gratteri, S.; Nisticò, G. Neurotoxic Effects Induced by Intracerebral and Systemic Injection of Paraquat in Rats. Hum. Exp. Toxicol. 1992, 11, 535–539. [Google Scholar] [CrossRef]
- Dinis-Oliveira, R.J.; Remião, F.; Carmo, H.; Duarte, J.A.; Navarro, A.S.; Bastos, M.L.; Carvalho, F. Paraquat Exposure as an Etiological Factor of Parkinson’s Disease. Neurotoxicology 2006, 27, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- McCormack, A.L.; Thiruchelvam, M.; Manning-Bog, A.B.; Thiffault, C.; Langston, J.W.; Cory-Slechta, D.A.; Di Monte, D.A. Environmental Risk Factors and Parkinson’s Disease: Selective Degeneration of Nigral Dopaminergic Neurons Caused by the Herbicide Paraquat. Neurobiol. Dis. 2002, 10, 119–127. [Google Scholar] [CrossRef]
- Cicchetti, F.; Lapointe, N.; Roberge-Tremblay, A.; Saint-Pierre, M.; Jimenez, L.; Ficke, B.W.; Gross, R.E. Systemic Exposure to Paraquat and Maneb Models Early Parkinson’s Disease in Young Adult Rats. Neurobiol. Dis. 2005, 20, 360–371. [Google Scholar] [CrossRef]
- Niso-Santano, M.; González-Polo, R.A.; Pedro, J.M.B.-S.; Gómez-Sánchez, R.; Lastres-Becker, I.; Ortiz-Ortiz, M.A.; Soler, G.; Morán, J.M.; Cuadrado, A.; Fuentes, J.M. Activation of Apoptosis Signal-Regulating Kinase 1 Is a Key Factor in Paraquat-Induced Cell Death: Modulation by the Nrf2/Trx Axis. Free Radic. Biol. Med. 2010, 48, 1370–1381. [Google Scholar] [CrossRef]
- Ossowska, K.; Wardas, J.; Śmiałowska, M.; Kuter, K.; Lenda, T.; Wierońska, J.M.; Zięba, B.; Nowak, P.; Dąbrowska, J.; Bortel, A.; et al. A Slowly Developing Dysfunction of Dopaminergic Nigrostriatal Neurons Induced by Long-Term Paraquat Administration in Rats: An Animal Model of Preclinical Stages of Parkinson’s Disease? Eur. J. Neurosci. 2005, 22, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Peng, L.; Stevenson, F.F.; Doctrow, S.R.; Andersen, J.K. Iron and Paraquat as Synergistic Environmental Risk Factors in Sporadic Parkinson’s Disease Accelerate Age-Related Neurodegeneration. J. Neurosci. 2007, 27, 6914–6922. [Google Scholar] [CrossRef] [PubMed]
- Bastías-Candia, S.; Di Benedetto, M.; D’Addario, C.; Candeletti, S.; Romualdi, P. Combined Exposure to Agriculture Pesticides, Paraquat and Maneb, Induces Alterations in the N/OFQ-NOPr and PDYN/KOPr Systems in Rats: Relevance to Sporadic Parkinson’s Disease. Environ. Toxicol. 2015, 30, 656–663. [Google Scholar] [CrossRef]
- Caputi, F.F.; Carretta, D.; Lattanzio, F.; Palmisano, M.; Candeletti, S.; Romualdi, P. Proteasome Subunit and Opioid Receptor Gene Expression Down-Regulation Induced by Paraquat and Maneb in Human Neuroblastoma SH-SY5Y Cells. Environ. Toxicol. Pharmacol. 2015, 40, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Hisata, J.S. Lake and Stream Rehabilitation: Rotenone Use and Health Risks. In Final Supplemental Environmental Impact Statement; Washington Department of Fish and Wildlife: Olympia, WA, USA, 2002. [Google Scholar]
- Grealish, S.; Diguet, E.; Kirkeby, A.; Mattsson, B.; Heuer, A.; Bramoulle, Y.; Van Camp, N.; Perrier, A.L.; Hantraye, P.; Björklund, A.; et al. Human ESC-Derived Dopamine Neurons Show Similar Preclinical Efficacy and Potency to Fetal Neurons When Grafted in a Rat Model of Parkinson’s Disease. Cell Stem Cell 2014, 15, 653–665. [Google Scholar] [CrossRef]
- Chiueh, C.C.; Markey, S.P.; Burns, R.S.; Johannessen, J.N.; Pert, A.; Kopin, I.J. Neurochemical and Behavioral Effects of Systemic and Intranigral Administration of N-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine in the Rat. Eur. J. Pharmacol. 1984, 100, 189–194. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V.; Naini, A.B.; Jakowec, M.; Petzinger, G.; Miller, R.; Akram, M. The Parkinsonian Toxin 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP): A Technical Review of Its Utility and Safety. J. Neurochem. 2001, 76, 1265–1274. [Google Scholar] [CrossRef]
- Duty, S.; Jenner, P. Animal Models of Parkinson’s Disease: A Source of Novel Treatments and Clues to the Cause of the Disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic Systemic Pesticide Exposure Reproduces Features of Parkinson’s Disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A Highly Reproducible Rotenone Model of Parkinson’s Disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Javed, H.; Azimullah, S.; Abul Khair, S.B.; Ojha, S.; Haque, M.E. Neuroprotective Effect of Nerolidol against Neuroinflammation and Oxidative Stress Induced by Rotenone. BMC Neurosci. 2016, 17, 58. [Google Scholar] [CrossRef]
- Gan, X.; Ren, J.; Huang, T.; Wu, K.; Li, S.; Duan, Y.; Wang, Z.; Si, W.; Wei, J. Pathological α-Synuclein Accumulation, CSF Metabolites Changes and Brain Microstructures in Cynomolgus Monkeys Treated with 6-Hydroxydopamine. Neurotoxicology 2023, 94, 172–181. [Google Scholar] [CrossRef]
- Joers, V.; Dilley, K.; Rahman, S.; Jones, C.; Shultz, J.; Simmons, H.; Emborg, M.E. Cardiac Sympathetic Denervation in 6-OHDA-Treated Nonhuman Primates. PLoS ONE 2014, 9, e104850. [Google Scholar] [CrossRef] [PubMed]
- Thiele, S.L.; Warre, R.; Nash, J.E. Development of a Unilaterally-Lesioned 6-OHDA Mouse Model of Parkinson’s Disease. J. Vis. Exp. 2012, 3234. [Google Scholar] [CrossRef]
- Khan, M.M.; Raza, S.S.; Javed, H.; Ahmad, A.; Khan, A.; Islam, F.; Safhi, M.M.; Islam, F. Rutin Protects Dopaminergic Neurons from Oxidative Stress in an Animal Model of Parkinson’s Disease. Neurotox. Res. 2012, 22, 1–15. [Google Scholar] [CrossRef]
- Boix, J.; Padel, T.; Paul, G. A Partial Lesion Model of Parkinson’s Disease in Mice--Characterization of a 6-OHDA-Induced Medial Forebrain Bundle Lesion. Behav. Brain Res. 2015, 284, 196–206. [Google Scholar] [CrossRef]
- Stott, S.R.W.; Barker, R.A. Time Course of Dopamine Neuron Loss and Glial Response in the 6-OHDA Striatal Mouse Model of Parkinson’s Disease. Eur. J. Neurosci. 2014, 39, 1042–1056. [Google Scholar] [CrossRef]
- Bagga, V.; Dunnett, S.B.; Fricker, R.A. The 6-OHDA Mouse Model of Parkinson’s Disease—Terminal Striatal Lesions Provide a Superior Measure of Neuronal Loss and Replacement than Median Forebrain Bundle Lesions. Behav. Brain Res. 2015, 288, 107–117. [Google Scholar] [CrossRef]
- Vieira, J.C.F.; Bassani, T.B.; Santiago, R.M.; de O Guaita, G.; Zanoveli, J.M.; da Cunha, C.; Vital, M.A.B.F. Anxiety-like Behavior Induced by 6-OHDA Animal Model of Parkinson’s Disease May Be Related to a Dysregulation of Neurotransmitter Systems in Brain Areas Related to Anxiety. Behav. Brain Res. 2019, 371, 111981. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.K.; Bomhoff, G.L.; Stanford, J.A.; Geiger, P.C. Neurodegeneration in an Animal Model of Parkinson’s Disease Is Exacerbated by a High-Fat Diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R1082–R1090. [Google Scholar] [CrossRef] [PubMed]
- Goes, A.T.R.; Jesse, C.R.; Antunes, M.S.; Lobo Ladd, F.V.; Lobo Ladd, A.A.B.; Luchese, C.; Paroul, N.; Boeira, S.P. Protective Role of Chrysin on 6-Hydroxydopamine-Induced Neurodegeneration a Mouse Model of Parkinson’s Disease: Involvement of Neuroinflammation and Neurotrophins. Chem. Biol. Interact. 2018, 279, 111–120. [Google Scholar] [CrossRef]
- Hernandez-Baltazar, D.; Zavala-Flores, L.M.; Villanueva-Olivo, A. The 6-Hydroxydopamine Model and Parkinsonian Pathophysiology: Novel Findings in an Older Model. Neurología 2017, 32, 533–539. [Google Scholar] [CrossRef]
- Luchtman, D.W.; Shao, D.; Song, C. Behavior, Neurotransmitters and Inflammation in Three Regimens of the MPTP Mouse Model of Parkinson’s Disease. Physiol. Behav. 2009, 98, 130–138. [Google Scholar] [CrossRef]
- Martin, H.L.; Santoro, M.; Mustafa, S.; Riedel, G.; Forrester, J.V.; Teismann, P. Evidence for a Role of Adaptive Immune Response in the Disease Pathogenesis of the MPTP Mouse Model of Parkinson’s Disease. Glia 2016, 64, 386–395. [Google Scholar] [CrossRef]
- Chen, L.; Huang, Y.; Yu, X.; Lu, J.; Jia, W.; Song, J.; Liu, L.; Wang, Y.; Huang, Y.; Xie, J.; et al. Corynoxine Protects Dopaminergic Neurons Through Inducing Autophagy and Diminishing Neuroinflammation in Rotenone-Induced Animal Models of Parkinson’s Disease. Front. Pharmacol. 2021, 12, 642900. [Google Scholar] [CrossRef] [PubMed]
- Morais, L.H.; Hara, D.B.; Bicca, M.A.; Poli, A.; Takahashi, R.N. Early Signs of Colonic Inflammation, Intestinal Dysfunction, and Olfactory Impairments in the Rotenone-Induced Mouse Model of Parkinson’s Disease. Behav. Pharmacol. 2018, 29, 199–210. [Google Scholar] [CrossRef]
- Zhang, Z.-N.; Zhang, J.-S.; Xiang, J.; Yu, Z.-H.; Zhang, W.; Cai, M.; Li, X.-T.; Wu, T.; Li, W.-W.; Cai, D.-F. Subcutaneous Rotenone Rat Model of Parkinson’s Disease: Dose Exploration Study. Brain Res. 2017, 1655, 104–113. [Google Scholar] [CrossRef]
- von Wrangel, C.; Schwabe, K.; John, N.; Krauss, J.K.; Alam, M. The Rotenone-Induced Rat Model of Parkinson’s Disease: Behavioral and Electrophysiological Findings. Behav. Brain Res. 2015, 279, 52–61. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, J.-D.; Song, L.-K.; Li, J.; Chu, S.-F.; Yuan, Y.-H.; Chen, N.-H. Environment-Contact Administration of Rotenone: A New Rodent Model of Parkinson’s Disease. Behav. Brain Res. 2015, 294, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Khadrawy, Y.A.; Salem, A.M.; El-Shamy, K.A.; Ahmed, E.K.; Fadl, N.N.; Hosny, E.N. Neuroprotective and Therapeutic Effect of Caffeine on the Rat Model of Parkinson’s Disease Induced by Rotenone. J. Diet. Suppl. 2017, 14, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Z.-K.; Lang, J.; Xu, G.; Li, H.-Y.; Zhang, Y.; Wang, L.; Su, Y.; Sun, A.-J. Excessive Levels of Nitric Oxide in Rat Model of Parkinson’s Disease Induced by Rotenone. Exp. Ther. Med. 2015, 9, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Parkhe, A.; Parekh, P.; Nalla, L.V.; Sharma, N.; Sharma, M.; Gadepalli, A.; Kate, A.; Khairnar, A. Protective Effect of Alpha Mangostin on Rotenone Induced Toxicity in Rat Model of Parkinson’s Disease. Neurosci. Lett. 2020, 716, 134652. [Google Scholar] [CrossRef]
- Ablat, N.; Lv, D.; Ren, R.; Xiaokaiti, Y.; Ma, X.; Zhao, X.; Sun, Y.; Lei, H.; Xu, J.; Ma, Y.; et al. Neuroprotective Effects of a Standardized Flavonoid Extract from Safflower against a Rotenone-Induced Rat Model of Parkinson’s Disease. Molecules 2016, 21, 1107. [Google Scholar] [CrossRef]
- De Miranda, B.R.; Rocha, E.M.; Bai, Q.; El Ayadi, A.; Hinkle, D.; Burton, E.A.; Greenamyre, J.T. Astrocyte-Specific DJ-1 Overexpression Protects against Rotenone-Induced Neurotoxicity in a Rat Model of Parkinson’s Disease. Neurobiol. Dis. 2018, 115, 101–114. [Google Scholar] [CrossRef]
- Normando, E.M.; Davis, B.M.; De Groef, L.; Nizari, S.; Turner, L.A.; Ravindran, N.; Pahlitzsch, M.; Brenton, J.; Malaguarnera, G.; Guo, L.; et al. The Retina as an Early Biomarker of Neurodegeneration in a Rotenone-Induced Model of Parkinson’s Disease: Evidence for a Neuroprotective Effect of Rosiglitazone in the Eye and Brain. Acta Neuropathol. Commun. 2016, 4, 86. [Google Scholar] [CrossRef]
- Fathy, S.M.; El-Dash, H.A.; Said, N.I. Neuroprotective Effects of Pomegranate (Punica granatum L.) Juice and Seed Extract in Paraquat-Induced Mouse Model of Parkinson’s Disease. BMC Complement. Med. Ther. 2021, 21, 130. [Google Scholar] [CrossRef]
- Chen, Y.-B.; Wang, Y.-Q.; Wu, J.-R.; Cui, Y.-L. A Novel Idea for Establishing Parkinson’s Disease Mouse Model by Intranasal Administration of Paraquat. Neurol. Res. 2021, 43, 267–277. [Google Scholar] [CrossRef]
- Chao, C.-C.; Huang, C.-L.; Cheng, J.-J.; Chiou, C.-T.; Lee, I.-J.; Yang, Y.-C.; Hsu, T.-H.; Yei, C.-E.; Lin, P.-Y.; Chen, J.-J.; et al. SRT1720 as an SIRT1 Activator for Alleviating Paraquat-Induced Models of Parkinson’s Disease. Redox Biol. 2022, 58, 102534. [Google Scholar] [CrossRef]
- Cristóvão, A.C.; Campos, F.L.; Je, G.; Esteves, M.; Guhathakurta, S.; Yang, L.; Beal, M.F.; Fonseca, B.M.; Salgado, A.J.; Queiroz, J.; et al. Characterization of a Parkinson’s Disease Rat Model Using an Upgraded Paraquat Exposure Paradigm. Eur. J. Neurosci. 2020, 52, 3242–3255. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, B.; Tian, T.; Zhang, B.; Shi, G.; Zhang, C.; Li, G.; Huang, M. Taurine Protects Dopaminergic Neurons in Paraquat-Induced Parkinson’s Disease Mouse Model through PI3K/Akt Signaling Pathways. Amino Acids 2022, 54, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Siedlak, S.L.; Torres, S.L.; Xu, Q.; Tang, B.; Zhu, X. Conditional Haploinsufficiency of β-Catenin Aggravates Neuronal Damage in a Paraquat-Based Mouse Model of Parkinson Disease. Mol. Neurobiol. 2019, 56, 5157–5166. [Google Scholar] [CrossRef]
- Prakash, J.; Yadav, S.K.; Chouhan, S.; Singh, S.P. Neuroprotective Role of Withania Somnifera Root Extract in Maneb-Paraquat Induced Mouse Model of Parkinsonism. Neurochem. Res. 2013, 38, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Sun, F.; Sun, W.; Zhang, L.; Wang, Q. Lesion of the Locus Coeruleus Damages Learning and Memory Performance in Paraquat and Maneb-Induced Mouse Parkinson’s Disease Model. Neuroscience 2019, 419, 129–140. [Google Scholar] [CrossRef]
- Ahmad, M.H.; Fatima, M.; Ali, M.; Rizvi, M.A.; Mondal, A.C. Naringenin Alleviates Paraquat-Induced Dopaminergic Neuronal Loss in SH-SY5Y Cells and a Rat Model of Parkinson’s Disease. Neuropharmacology 2021, 201, 108831. [Google Scholar] [CrossRef] [PubMed]
- Vegh, C.; Wear, D.; Okaj, I.; Huggard, R.; Culmone, L.; Eren, S.; Cohen, J.; Rishi, A.K.; Pandey, S. Combined Ubisol-Q10 and Ashwagandha Root Extract Target Multiple Biochemical Mechanisms and Reduces Neurodegeneration in a Paraquat-Induced Rat Model of Parkinson’s Disease. Antioxidants 2021, 10, 563. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Song, B.; Yang, H.; Huang, B.; Chi, B.; Guo, Y.; Liu, H. Central Nervous System Damage Due to Acute Paraquat Poisoning: An Experimental Study with Rat Model. Neurotoxicology 2013, 35, 62–70. [Google Scholar] [CrossRef]
- Ellwanger, J.H.; Franke, S.I.R.; Bordin, D.L.; Prá, D.; Henriques, J.A. Biological Functions of Selenium and Its Potential Influence on Parkinson’s Disease. An. Acad. Bras. Ciênc. 2016, 88, 1655–1674. [Google Scholar] [CrossRef]
- Kumar, A.; Leinisch, F.; Kadiiska, M.B.; Corbett, J.; Mason, R.P. Formation and Implications of Alpha-Synuclein Radical in Maneb- and Paraquat-Induced Models of Parkinson’s Disease. Mol. Neurobiol. 2016, 53, 2983–2994. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Gasser, T. Molecular Pathogenesis of Parkinson Disease: Insights from Genetic Studies. Expert Rev. Mol. Med. 2009, 11, e22. [Google Scholar] [CrossRef]
- Sanchez, G.; Varaschin, R.K.; Büeler, H.; Marcogliese, P.C.; Park, D.S.; Trudeau, L.-E. Unaltered Striatal Dopamine Release Levels in Young Parkin Knockout, Pink1 Knockout, DJ-1 Knockout and LRRK2 R1441G Transgenic Mice. PLoS ONE 2014, 9, e94826. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Juri, C.; Collantes, M.; Peñuelas, I.; Prieto, E.; Iglesias, E.; Martí-Climent, J.; Arbizu, J.; Zubieta, J.L.; Rodríguez-Oroz, M.C.; et al. Progression of Dopaminergic Depletion in a Model of MPTP-Induced Parkinsonism in Non-Human Primates. An (18)F-DOPA and (11)C-DTBZ PET Study. Neurobiol. Dis. 2010, 38, 456–463. [Google Scholar] [CrossRef]
- Brucale, M.; Sandal, M.; Di Maio, S.; Rampioni, A.; Tessari, I.; Tosatto, L.; Bisaglia, M.; Bubacco, L.; Samorì, B. Pathogenic Mutations Shift the Equilibria of α-Synuclein Single Molecules towards Structured Conformers. ChemBioChem 2009, 10, 176–183. [Google Scholar] [CrossRef]
- Lautenschläger, J.; Stephens, A.D.; Fusco, G.; Ströhl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-Terminal Calcium Binding of α-Synuclein Modulates Synaptic Vesicle Interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef]
- Tóth, G.; Gardai, S.J.; Zago, W.; Bertoncini, C.W.; Cremades, N.; Roy, S.L.; Tambe, M.A.; Rochet, J.-C.; Galvagnion, C.; Skibinski, G.; et al. Targeting the Intrinsically Disordered Structural Ensemble of α-Synuclein by Small Molecules as a Potential Therapeutic Strategy for Parkinson’s Disease. PLoS ONE 2014, 9, e87133. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic Loss and Inclusion Body Formation in α-Synuclein Mice: Implications for Neurodegenerative Disorders. Science 2000, 287, 1265–1269. [Google Scholar] [CrossRef] [PubMed]
- Kilpeläinen, T.; Julku, U.H.; Svarcbahs, R.; Myöhänen, T.T. Behavioural and Dopaminergic Changes in Double Mutated Human A30P*A53T Alpha-Synuclein Transgenic Mouse Model of Parkinson’s Disease. Sci. Rep. 2019, 9, 17382. [Google Scholar] [CrossRef]
- Lesage, S.; Houot, M.; Mangone, G.; Tesson, C.; Bertrand, H.; Forlani, S.; Anheim, M.; Brefel-Courbon, C.; Broussolle, E.; Thobois, S.; et al. Genetic and Phenotypic Basis of Autosomal Dominant Parkinson’s Disease in a Large Multi-Center Cohort. Front. Neurol. 2020, 11, 682. [Google Scholar] [CrossRef]
- Lunati, A.; Lesage, S.; Brice, A. The Genetic Landscape of Parkinson’s Disease. Rev. Neurol. 2018, 174, 628–643. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s Disease Alpha-Synuclein Transgenic Mice Develop Neuronal Mitochondrial Degeneration and Cell Death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice Lacking Alpha-Synuclein Display Functional Deficits in the Nigrostriatal Dopamine System. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef]
- Thomas, B.; Mandir, A.S.; West, N.; Liu, Y.; Andrabi, S.A.; Stirling, W.; Dawson, V.L.; Dawson, T.M.; Lee, M.K. Resistance to MPTP-Neurotoxicity in α-Synuclein Knockout Mice Is Complemented by Human α-Synuclein and Associated with Increased β-Synuclein and Akt Activation. PLoS ONE 2011, 6, e16706. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M.-Y. Neuronal α-Synucleinopathy with Severe Movement Disorder in Mice Expressing A53T Human α-Synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tang, B.; Zhao, G.; Pan, Q.; Xia, K.; Bodmer, R.; Zhang, Z. Dispensable Role of Drosophila Ortholog of LRRK2 Kinase Activity in Survival of Dopaminergic Neurons. Mol. Neurodegener. 2008, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Marcogliese, P.C.; Abuaish, S.; Kabbach, G.; Abdel-Messih, E.; Seang, S.; Li, G.; Slack, R.S.; Haque, M.E.; Venderova, K.; Park, D.S. LRRK2(I2020T) Functional Genetic Interactors That Modify Eye Degeneration and Dopaminergic Cell Loss in Drosophila. Hum. Mol. Genet. 2017, 26, 1247–1257. [Google Scholar] [CrossRef]
- Li, A.; Song, N.-J.; Riesenberg, B.P.; Li, Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front. Immunol. 2020, 10, 3154. [Google Scholar] [CrossRef]
- Lee, B.D.; Shin, J.-H.; VanKampen, J.; Petrucelli, L.; West, A.B.; Ko, H.S.; Lee, Y.-I.; Maguire-Zeiss, K.A.; Bowers, W.J.; Federoff, H.J.; et al. Inhibitors of Leucine-Rich Repeat Kinase-2 Protect against Models of Parkinson’s Disease. Nat. Med. 2010, 16, 998–1000. [Google Scholar] [CrossRef]
- West, A.B. Achieving Neuroprotection with LRRK2 Kinase Inhibitors in Parkinson Disease. Exp. Neurol. 2017, 298, 236–245. [Google Scholar] [CrossRef]
- Song, P.; Li, S.; Wu, H.; Gao, R.; Rao, G.; Wang, D.; Chen, Z.; Ma, B.; Wang, H.; Sui, N.; et al. Parkin Promotes Proteasomal Degradation of P62: Implication of Selective Vulnerability of Neuronal Cells in the Pathogenesis of Parkinson’s Disease. Protein Cell 2016, 7, 114–129. [Google Scholar] [CrossRef]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial Pathology and Apoptotic Muscle Degeneration in Drosophila Parkin Mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef]
- Bian, M.; Liu, J.; Hong, X.; Yu, M.; Huang, Y.; Sheng, Z.; Fei, J.; Huang, F. Overexpression of Parkin Ameliorates Dopaminergic Neurodegeneration Induced by 1- Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine in Mice. PLoS ONE 2012, 7, e39953. [Google Scholar] [CrossRef]
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M.M. Neuroprotective Function of DJ-1 in Parkinson’s Disease. Oxid. Med. Cell. Longev. 2013, 2013, 683920. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cagniard, B.; Mathews, T.; Jones, S.; Koh, H.C.; Ding, Y.; Carvey, P.M.; Ling, Z.; Kang, U.J.; Zhuang, X. Age-Dependent Motor Deficits and Dopaminergic Dysfunction in DJ-1 Null Mice. J. Biol. Chem. 2005, 280, 21418–21426. [Google Scholar] [CrossRef] [PubMed]
- Aleyasin, H.; Rousseaux, M.W.C.; Marcogliese, P.C.; Hewitt, S.J.; Irrcher, I.; Joselin, A.P.; Parsanejad, M.; Kim, R.H.; Rizzu, P.; Callaghan, S.M.; et al. DJ-1 Protects the Nigrostriatal Axis from the Neurotoxin MPTP by Modulation of the AKT Pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 3186–3191. [Google Scholar] [CrossRef] [PubMed]
- Dagda, R.K.; Pien, I.; Wang, R.; Zhu, J.; Wang, K.Z.Q.; Callio, J.; Banerjee, T.D.; Dagda, R.Y.; Chu, C.T. Beyond the Mitochondrion: Cytosolic PINK1 Remodels Dendrites through Protein Kinase A. J. Neurochem. 2014, 128, 864–877. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.-M.; et al. Mitochondrial Dysfunction in Drosophila PINK1 Mutants Is Complemented by Parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila Pink1 Is Required for Mitochondrial Function and Interacts Genetically with Parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Fox, S.H.; Chuang, R.; Brotchie, J.M. Masliah. Mov. Disord. 2009, 24, 1255–1266. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, W.; Oo, T.F.; Wang, L.; Tang, Y.; Jackson-Lewis, V.; Zhou, C.; Geghman, K.; Bogdanov, M.; Przedborski, S.; et al. Mutant LRRK2(R1441G) BAC Transgenic Mice Recapitulate Cardinal Features of Parkinson’s Disease. Nat. Neurosci. 2009, 12, 826–828. [Google Scholar] [CrossRef]
- Chang, E.E.S.; Ho, P.W.-L.; Liu, H.-F.; Pang, S.Y.-Y.; Leung, C.-T.; Malki, Y.; Choi, Z.Y.-K.; Ramsden, D.B.; Ho, S.-L. LRRK2 Mutant Knock-in Mouse Models: Therapeutic Relevance in Parkinson’s Disease. Transl. Neurodegener. 2022, 11, 10. [Google Scholar] [CrossRef]
- Seegobin, S.P.; Heaton, G.R.; Liang, D.; Choi, I.; Blanca Ramirez, M.; Tang, B.; Yue, Z. Progress in LRRK2-Associated Parkinson’s Disease Animal Models. Front. Neurosci. 2020, 14, 674. [Google Scholar] [CrossRef]
- Kitada, T.; Tong, Y.; Gautier, C.A.; Shen, J. Absence of Nigral Degeneration in Aged Parkin/DJ-1/PINK1 Triple Knockout Mice. J. Neurochem. 2009, 111, 696. [Google Scholar] [CrossRef]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson Phenotype in Aged PINK1-Deficient Mice Is Accompanied by Progressive Mitochondrial Dysfunction in Absence of Neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef] [PubMed]
- Hauser, D.N.; Primiani, C.T.; Langston, R.G.; Kumaran, R.; Cookson, M.R. The Polg Mutator Phenotype Does Not Cause Dopaminergic Neurodegeneration in DJ-1-Deficient Mice. eNeuro 2015, 2, ENEURO.0075-14.2015. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gehrke, S.; Haque, M.E.; Imai, Y.; Kosek, J.; Yang, L.; Beal, M.F.; Nishimura, I.; Wakamatsu, K.; Ito, S.; et al. Inactivation of Drosophila DJ-1 Leads to Impairments of Oxidative Stress Response and Phosphatidylinositol 3-Kinase/Akt Signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 13670–13675. [Google Scholar] [CrossRef]
- Imai, Y.; Gehrke, S.; Wang, H.-Q.; Takahashi, R.; Hasegawa, K.; Oota, E.; Lu, B. Phosphorylation of 4E-BP by LRRK2 Affects the Maintenance of Dopaminergic Neurons in Drosophila. EMBO J. 2008, 27, 2432–2443. [Google Scholar] [CrossRef]
- Martella, G.; Madeo, G.; Maltese, M.; Vanni, V.; Puglisi, F.; Ferraro, E.; Schirinzi, T.; Valente, E.M.; Bonanni, L.; Shen, J.; et al. Exposure to Low-Dose Rotenone Precipitates Synaptic Plasticity Alterations in PINK1 Heterozygous Knockout Mice. Neurobiol. Dis. 2016, 91, 21–36. [Google Scholar] [CrossRef]
- Schirinzi, T.; Martella, G.; Pisani, A. Double Hit Mouse Model of Parkinson’s Disease. Oncotarget 2016, 7, 80109–80110. [Google Scholar] [CrossRef]
- Lopes, F.M.; Bristot, I.J.; da Motta, L.L.; Parsons, R.B.; Klamt, F. Mimicking Parkinson’s Disease in a Dish: Merits and Pitfalls of the Most Commonly Used Dopaminergic In Vitro Models. Neuromol. Med. 2017, 19, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Billings, J.L.; Gordon, S.L.; Rawling, T.; Doble, P.A.; Bush, A.I.; Adlard, P.A.; Finkelstein, D.I.; Hare, D.J. L-3,4-Dihydroxyphenylalanine (l-DOPA) Modulates Brain Iron, Dopaminergic Neurodegeneration and Motor Dysfunction in Iron Overload and Mutant Alpha-Synuclein Mouse Models of Parkinson’s Disease. J. Neurochem. 2019, 150, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR Method for MinION and Illumina Sequencing of Zika and Other Virus Genomes Directly from Clinical Samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- Galet, B.; Cheval, H.; Ravassard, P. Patient-Derived Midbrain Organoids to Explore the Molecular Basis of Parkinson’s Disease. Front. Neurol. 2020, 11, 1005. [Google Scholar] [CrossRef] [PubMed]
- Smits, L.M.; Schwamborn, J.C. Midbrain Organoids: A New Tool to Investigate Parkinson’s Disease. Front. Cell Dev. Biol. 2020, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Smits, L.M.; Reinhardt, L.; Reinhardt, P.; Glatza, M.; Monzel, A.S.; Stanslowsky, N.; Rosato-Siri, M.D.; Zanon, A.; Antony, P.M.; Bellmann, J.; et al. Modeling Parkinson’s Disease in Midbrain-like Organoids. NPJ Park. Dis. 2019, 5, 5. [Google Scholar] [CrossRef]
- Windrem, M.S.; Schanz, S.J.; Morrow, C.; Munir, J.; Chandler-Militello, D.; Wang, S.; Goldman, S.A. A Competitive Advantage by Neonatally Engrafted Human Glial Progenitors Yields Mice Whose Brains Are Chimeric for Human Glia. J. Neurosci. 2014, 34, 16153–16161. [Google Scholar] [CrossRef]


{kind=link}
| Toxin | Mode of Action | Host Species | Key features | Applications | Refs. | |||
|---|---|---|---|---|---|---|---|---|
| Nigro-Striatal Tract Damage/α-syn Spreading | DA Neuron Loss | Lewy Body-Like Structure | Phenotype/ Motor Symptoms (Behavior) | |||||
| 6-OHDA | auto-oxidation of 6-OHDA/formation of hydrogen peroxides due to the action of monoamine oxidase/ direct inhibition of mitochondrial respiratory chain complex I | Monkeys (rhesus/ cynomolgus) Mice (Tg/male and female/ C57BL6) Rats (male Wistar/ Fischer 344) | ✓ ✓ ✓ | ✓ ✓ ✓ | ✓ ✗ ✗ | ✓ ✓ (deficit in locomotor activity and decrease in motor coordination/ rotational bias) ✓ diminished locomotor activity observed/anxiety- like behavior portrayed. | In general, model systems have been used to investigate neuroprotective effects of different ‘disease modifying strategies’/drugs. They have been used to establish and characterize PD features and develop protocols for the same. Depending upon the objective, factors such as neuroinflammation, various moto/non-motor symptoms are investigated. | [96,97,98,99,100,101,102,103,104,105,106] |
| MPTP | Mitochondrial complex I inhibition | Mouse (male C57BL6_Tg/Wt) Male Wistar rats Monkey (Macaca fascicularis, Macaca mulatta) | ✓ ✓ ✓ | ✓ ✓ ✓ | ✓ ✗ ✓ | ✓ ✓ ✓ | Investigate and compare between various MPTP regimens found in the literature. To observe the role of adaptive immune response in PD pathogenesis (the role of the immune system). Observe neuroprotective effects of certain drugs/observe neuroinflammation, cytotoxic effects of microglia and astrocytes. Model system used to investigate chronobiological parameters, and cognitive and motor symptoms upon MPTP administration. Used to observe the relation between MPTP-induced inflammation and gut microbiota, along with any possible differences in PD progression between genders. | [95,107,108,109,110,111,112,113,114,115,116,117,118,119] |
| ROTENONE | Mitochondrial complex I inhibition | Mice (C57BL6/Swiss) Rats | ✓ | ✓ | ✗ | ✓ | Observation of the effect of ‘stress’ on disease progression. Studies observed dysfunction in gut –brain access. Use of a lower dose of this neurotoxin to develop a PD model system. Assess effect of social recognition system, GI functioning, and olfactory system. Development of model via environmental contact and investigate underlying pathological and molecular processes. Mostly rotenone rat model system used for looking at neuroprotective effects/ therapeutic interventions. | [59,120,121,122,123,124,125,126,127,128,129,130] |
| PARAQUAT | Alteration in the redox cycling of ‘glutathione and thioredoxin’ | Mice (C57BL6, albino/Tg) Rats (albino male Wistar/Sprague Dawley/male Wistar/long Evans hooded rats | ✓ | ✓ | ✗ | ✓ | Used to develop a model system to observe neuroprotective effects of pomegranate seed extract and pomegranate juice. One study found intra-nasal administration route showcasing better survivability along with observation of neuronal loss in the SNc and other essential PD-like signs. Generally, the rodents were used to develop a PD model system and identify an underlying molecular mechanism that aids in the disease’s progression. | [131,132,133,134,135,136,137,138,139,140,141,142,143] |
| Gene | Protein | Host System | Key Features | Applications | Refs. | |||
|---|---|---|---|---|---|---|---|---|
| DA Neuronal Loss | Lewy Body-Like Structure | Phenotype/ Behavior | Mitochondrial Defects | |||||
| SNCA | α-synuclein | Mice Rat Monkey Zebra fish C. elegans Drosophila | ✓ ✓ ✓ ✓ ✓ ✓ | ✓ ✗ ✓ ✓ - ✓ | ✓ ✓ ✓ ✓ ✓ ✓ (climbing defects) | ✓ | The pathological development of PD takes a long time in this model. However, in the case of Drosophila it takes less time for PD development. Drosophila model systems are useful for suppressor– enhancer screening. | [1,5,49,56,65,142,146,147,148,149,150,173] |
| LRRK2 | Leucine-rich repeat kinase 2 | Mice Rat Monkey C. elegans Zebra Fish Drosophila | ✓ ✗ decreased neuronal viability ✓ - ✓ | - - - - - - | ✓ ✓ - ✓ Increases in locomotion in adult stages ✓ (climbing defects) | ✓ - No mito defect seen, however, increased ROS observed due to increased kinase activity - - - | This model lacks α-synuclein inclusions and dopaminergic neuronal manifestation of PD. This is usually appropriate for LRRK2-specific drug testing. | [1,46,65,162,174,175,176] |
| PARKIN | Parkin | Mice and rat C. elegans Zebrafish Drosophila | ✗ ✓ ✓ ✓ | ✗ | ✓ No disturbances in swimming behavior ✓ (climbing defects) | ✓ ✓ ✓ | Drosophila model systems are useful for suppressor–enhancer screening and require less time for PD development. | [1,46,56,65,165] |
| PINK1 | PTEN-induced putative kinase protein 1 | Mice and rats C. elegans Zebra fish Drosophila | ✓ ✗ disturbed DA projection, no substantial loss of DA neurons. ✓ | ✗ - | - ✓ ✓ abnormal swimming ✓ (climbing defects) | - ✓ ✓ ✓ | Drosophila model systems are useful for suppressor–enhancer screening and require less time for PD development. | [1,46,56,65,171,172,177,178] |
| DJ-1 | DJ1 | Mice Rats Drosophila C. elegans | ✗ ✓ ✓ ✗ | ✗ | ✓ demonstrate age-dependent motor deficits of PD. ✓ (climbing defects) | ✓ | Drosophila model systems are useful for suppressor–enhancer screening and require less time for PD development. | [1,46,65,168,177,179,180,181] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, E.; Hasan, I.; Haque, M.E. Parkinson’s Disease: Exploring Different Animal Model Systems. Int. J. Mol. Sci. 2023, 24, 9088. https://doi.org/10.3390/ijms24109088
Khan E, Hasan I, Haque ME. Parkinson’s Disease: Exploring Different Animal Model Systems. International Journal of Molecular Sciences. 2023; 24(10):9088. https://doi.org/10.3390/ijms24109088
Chicago/Turabian StyleKhan, Engila, Ikramul Hasan, and M. Emdadul Haque. 2023. "Parkinson’s Disease: Exploring Different Animal Model Systems" International Journal of Molecular Sciences 24, no. 10: 9088. https://doi.org/10.3390/ijms24109088