
Neurosteroids Mediate Neuroprotection in an In Vitro Model of Hypoxic/Hypoglycaemic Excitotoxicity via δ-GABAA Receptors without Affecting Synaptic Plasticity
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Neuroprotective Properties against Ischaemic-Induced Excitotoxicity
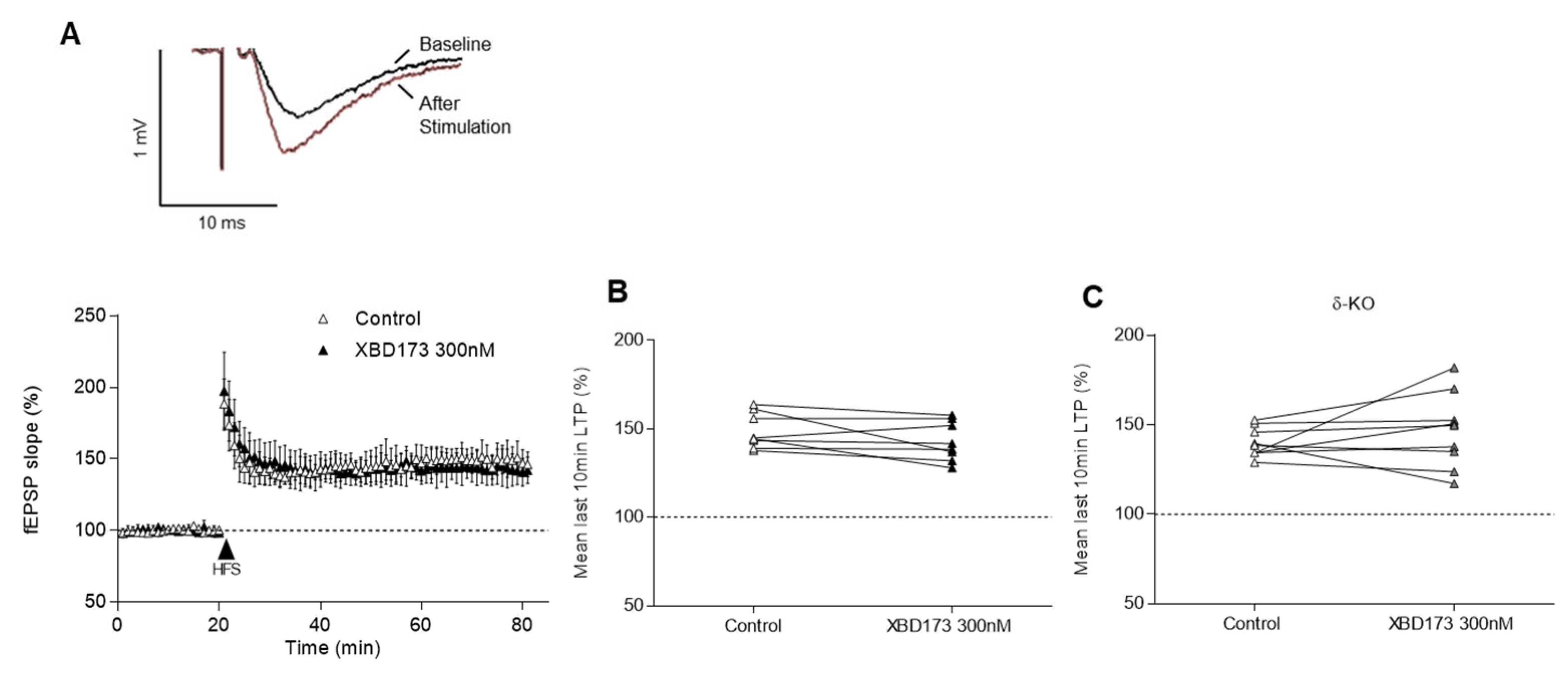
2.2. Effects of XBD173, THDOC and Allopregnanolone on LTP
2.3. Recoding of Spontaneous Inhibitory Postsynaptic Currents
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Acute Brain Slice Preparation
4.3. Oxygen–Glucose Deprivation Model: H/H Measurements
4.4. Long-Term Potentiation Recordings
4.5. Whole-Cell Patch-Clamp Recordings
4.6. Experimental Design and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, D.; Lee, S.; Pyeon, T.; Jeong, S. Use of triazolam and alprazolam as premedication for general anesthesia. Korean J. Anesthesiol. 2015, 68, 346–351. [Google Scholar] [CrossRef]
- Olkkola, K.T.; Ahonen, J. Midazolam and other benzodiazepines. Handb. Exp. Pharmacol. 2008, 182, 335–360. [Google Scholar]
- Young, C.C.; Prielipp, R.C. Benzodiazepines in the intensive care unit. Crit. Care Clin. 2001, 17, 843–862. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, B.A.; Suresh, S.; Hertz, S.; Evers, A.S.; Orser, B.A. Anesthetic Neurotoxicity—Clinical Implications of Animal Models. N. Engl. J. Med. 2015, 372, 796–797. [Google Scholar] [CrossRef] [PubMed]
- Buffett-Jerrott, S.; Stewart, S. Cognitive and Sedative Effects of Benzodiazepine Use. Curr. Pharm. Des. 2002, 8, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Luo, R.Y.; Chen, C.; Li, X.; Ao, J.S.; Liu, Y.; Yin, Y.Q. Effects of propofol, dexmedetomidine, and midazolam on postoperative cognitive dysfunction in elderly patients: A randomized controlled preliminary trial. Chin. Med. J. 2019, 132, 437–445. [Google Scholar] [CrossRef]
- Smith, H.A.; Gangopadhyay, M.; Goben, C.M.; Jacobowski, N.L.; Chestnut, M.H.; Thompson, J.L.; Chandrasekhar, R.; Williams, S.R.; Griffith, K.; Ely, E.; et al. Delirium and Benzodiazepines Associated with Prolonged ICU Stay in Critically Ill Infants and Young Children. Crit. Care Med. 2017, 45, 1427–1435. [Google Scholar] [CrossRef]
- Sneyd, J.R.; Gambus, P.L.; Rigby-Jones, A.E. Current status of perioperative hypnotics, role of benzodiazepines, and the case for remimazolam: A narrative review. Br. J. Anaesth. 2021, 127, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, A.R.; Levin, H.S. Cognitive Sequelae of Traumatic Brain Injury. Psychiatr. Clin. N. Am. 2014, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Larson, E.B.; Zollman, F.S. The effect of sleep medications on cognitive recovery from traumatic brain injury. J. Head Trauma Rehabil. 2010, 25, 61–67. [Google Scholar] [CrossRef]
- Kowark, A.; Rossaint, R.; Keszei, A.P.; Bischoff, P.; Czaplik, M.; Drexler, B.; Kienbaum, P.; Kretzschmar, M.; Rex, C.; Saller, T.; et al. Impact of PReOperative Midazolam on OuTcome of Elderly patients (I-PROMOTE): Study protocol for a multicentre randomised controlled trial. Trials 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.S.; Viola-McCabe, K.E. Midazolam inhibits long-term potentiation through modulation of GABAA receptors. Neuropharmacology 1996, 35, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Puig-Bosch, X.; Bieletzki, S.; Zeilhofer, H.U.; Rudolph, U.; Antkowiak, B.; Rammes, G. Midazolam at Low Nanomolar Concentrations Affects Long-term Potentiation and Synaptic Transmission Predominantly via the α1-γ-Aminobutyric Acid Type A Receptor Subunit in Mice. Anesthesiology 2022, 136, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Athanassoglou, V.; Cozowicz, C.; Zhong, H.; Illescas, A.; Poeran, J.; Liu, J.; Poultsides, L.; Memtsoudis, S.G. Association of perioperative midazolam use and complications: A population-based analysis. Reg. Anesth. Pain Med. 2022, 47, 228–233. [Google Scholar] [CrossRef]
- Rupprecht, R.; Holsboer, F. Neuroactive steroids: Mechanisms of action and neuropsychopharmacological perspectives. Trends Neurosci. 1999, 22, 410–416. [Google Scholar] [CrossRef]
- Reddy, D.S.; Rogawski, M.A. Neurosteroids—Endogenous Regulators of Seizure Susceptibility and Role in the Treatment of Epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies, 4th ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2012. [Google Scholar]
- Brown, N.; Kerby, J.; Bonnert, T.P.; Whiting, P.J.; Wafford, K.A. Pharmacological characterization of a novel cell line expressing human alpha(4)beta(3)delta GABA(A) receptors. Br. J. Pharmacol. 2002, 136, 965–974. [Google Scholar] [CrossRef]
- Carver, C.M.; Reddy, D.S. Neurosteroid structure-activity relationships for functional activation of extrasynaptic δGABAA receptors. J. Pharmacol. Exp. Ther. 2016, 357, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Kita, A.; Kinoshita, T.; Kohayakawa, H.; Furukawa, K.; Akaike, A. Lack of tolerance to anxiolysis and withdrawal symptoms in mice repeatedly treated with AC-5216, a selective TSPO ligand. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2009, 33, 1040–1045. [Google Scholar] [CrossRef]
- Rupprecht, R.; Rammes, G.; Eser, D.; Baghai, T.C.; Schüle, C.; Nothdurfter, C.; Troxler, T.; Gentsch, C.; Kalkman, H.O.; Chaperon, F.; et al. Translocator protein (18 kD) as target for anxiolytics without benzodiazepine-like side effects. Science 2009, 325, 490–493. [Google Scholar] [CrossRef]
- Schumacher, M.; Baulieu, E.E. Neurosteroids: Synthesis and Functions in the Central and Peripheral Nervous Systems. In Ciba Foundation Symposium 191—Non-Reproductive Actions of Sex Steroids; John Wiley & Sons: Hoboken, NJ, USA, 2007; pp. 90–121. [Google Scholar]
- Moralí, G.; Montes, P.; Hernández-Morales, L.; Monfil, T.; Espinosa-García, C.; Cervantes, M. Neuroprotective effects of progesterone and allopregnanolone on long-term cognitive outcome after global cerebral ischemia. Restor. Neurol. Neurosci. 2011, 29, 1–15. [Google Scholar] [CrossRef]
- Zauner, A.; Bullock, R. The role of excitatory amino acids in severe brain trauma: Opportunities for therapy: A review. J. Neurotrauma 1995, 12, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Rupprecht, R.; Papadopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010, 9, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.A. Long-term potentiation and memory. Physiol. Rev. 2004, 84, 87–136. [Google Scholar] [CrossRef] [PubMed]
- Seabrook, G.R.; Easter, A.; Dawson, G.R.; Bowery, B.J. Modulation of long-term potentiation in CA1 region of mouse hippocampal brain slices by GABA(a) receptor benzodiazepine site ligands. Neuropharmacology 1997, 36, 823–830. [Google Scholar] [CrossRef]
- Mihalek, R.M.; Banerjee, P.K.; Korpi, E.R.; Quinlan, J.J.; Firestone, L.L.; Mi, Z.P.; Lagenaur, C.; Tretter, V.; Sieghart, W.; Anagnostaras, S.G.; et al. Attenuated sensitivity to neuroactive steroids in gamma -aminobutyrate type A receptor delta subunit knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 12905–12910. [Google Scholar] [CrossRef]
- Vicini, S.; Losi, G.; Homanics, G.E. GABAAreceptor δ subunit deletion prevents neurosteroid modulation of inhibitory synaptic currents in cerebellar neurons. Neuropharmacology 2002, 43, 646–650. [Google Scholar] [CrossRef]
- Clarkson, A.N.; Boothman-Burrell, L.; Dósa, Z.; Nagaraja, R.Y.; Jin, L.; Parker, K.; Van Nieuwenhuijzen, P.S.; Neumann, S.; Gowing, E.K.; Gavande, N.; et al. The flavonoid, 2′-methoxy-6-methylflavone, affords neuroprotection following focal cerebral ischaemia. J. Cereb. Blood Flow Metab. 2019, 39, 1266–1282. [Google Scholar] [CrossRef]
- Brown, A.R.; Mitchell, S.J.; Peden, D.R.; Herd, M.B.; Seifi, M.; Swinny, J.D.; Belelli, D.; Lambert, J.J. During postnatal development endogenous neurosteroids influence GABA-ergic neurotransmission of mouse cortical neurons. Neuropharmacology 2016, 103, 163–173. [Google Scholar] [CrossRef]
- Bukanova, J.; Solntseva, E.; Kondratenko, R.; Kudova, E. Epipregnanolone as a positive modulator of gabaa receptor in rat cerebellar and hippocampus neurons. Biomolecules 2021, 11, 791. [Google Scholar] [CrossRef]
- Taylor, C.; Weber, M. Effect of temperature on synaptic function after reduced oxygen and glucose in hippocampal slices. Neuroscience 1993, 52, 555–562. [Google Scholar] [CrossRef]
- Mages, K.; Grassmann, F.; Jägle, H.; Rupprecht, R.; Weber, B.H.F.; Hauck, S.M.; Grosche, A. The agonistic TSPO ligand XBD173 attenuates the glial response thereby protecting inner retinal neurons in a murine model of retinal ischemia. J. Neuroinflamm. 2019, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; Boothman-Burrell, L.; Gowing, E.K.; Jacobsen, T.A.; Ahring, P.K.; Young, S.L.; Sandager-Nielsen, K.; Clarkson, A.N. The Delta-Subunit Selective GABAA Receptor Modulator, DS2, Improves Stroke Recovery via an Anti-inflammatory Mechanism. Front. Neurosci. 2019, 13, 1133. [Google Scholar] [CrossRef] [PubMed]
- Glykys, J.; Mody, I. The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J. Physiol. 2007, 582, 1163–1178. [Google Scholar] [CrossRef] [PubMed]
- Puia, G.; Santi, M.; Vicini, S.; Pritchett, D.B.; Purdy, R.H.; Paul, S.M.; Seeburg, P.H.; Costa, E. Neurosteroids Act on Recombinant GABAA receptors. Neuron 1990, 4, 759–765. [Google Scholar] [CrossRef]
- Wohlfarth, K.M.; Bianchi, M.T.; Macdonald, R.L. Enhanced neurosteroid potentiation of ternary GABAA receptors containing the delta subunit. J. Neurosci. 2002, 22, 1541–1549. [Google Scholar] [CrossRef]
- Izumi, Y.; Murayama, K.; Tokuda, K.; Krishnan, K.; Covey, D.F.; Zorumski, C.F. GABAergic neurosteroids mediate the effects of ethanol on long-term potentiation in rat hippocampal slices. Eur. J. Neurosci. 2007, 26, 1881–1888. [Google Scholar] [CrossRef]
- Simon-O’Brien, E.; Gauthier, D.; Riban, V.; Verleye, M. Etifoxine improves sensorimotor deficits and reduces glial activation, neuronal degeneration, and neuroinflammation in a rat model of traumatic brain injury. J. Neuroinflamm. 2016, 13, 1–15. [Google Scholar] [CrossRef]
- Kokate, T.G.; Yamaguchi, S.; Pannell, L.K.; Rajamani, U.; Carroll, D.M.; Grossman, A.B.; Rogawski, M. Lack of Anticonvulsant Tolerance to the Neuroactive Steroid Pregnanolone in Mice. J. Pharmacol. Exp. Ther. 1998, 287, 553–558. [Google Scholar]
- Reddy, D.S.; Estes, W.A. Clinical Potential of Neurosteroids for CNS Disorders. Trends Pharmacol. Sci. 2016, 37, 543–561. [Google Scholar] [CrossRef]
- Wieland, H.A.; Luddens, H.; Seeburg, P.H. A single histidine in GABA(A) receptors is essential for benzodiazepine agonist binding. J. Biol. Chem. 1992, 267, 1426–1429. [Google Scholar] [CrossRef]
- Boehm, S.L.; Homanics, G.E.; Blednov, Y.A.; Harris, R.A. δ-Subunit containing GABAA receptor knockout mice are less sensitive to the actions of 4,5,6,7-tetrahydroisoxazolo-[5,4-c]pyridin-3-ol. Eur. J. Pharmacol. 2006, 541, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Han, X.; Wang, J. Organotypic hippocampal slices as models for stroke and traumatic brain injury. Mol. Neurobiol. 2016, 53, 4226–4237. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puig-Bosch, X.; Ballmann, M.; Bieletzki, S.; Antkowiak, B.; Rudolph, U.; Zeilhofer, H.U.; Rammes, G. Neurosteroids Mediate Neuroprotection in an In Vitro Model of Hypoxic/Hypoglycaemic Excitotoxicity via δ-GABAA Receptors without Affecting Synaptic Plasticity. Int. J. Mol. Sci. 2023, 24, 9056. https://doi.org/10.3390/ijms24109056
Puig-Bosch X, Ballmann M, Bieletzki S, Antkowiak B, Rudolph U, Zeilhofer HU, Rammes G. Neurosteroids Mediate Neuroprotection in an In Vitro Model of Hypoxic/Hypoglycaemic Excitotoxicity via δ-GABAA Receptors without Affecting Synaptic Plasticity. International Journal of Molecular Sciences. 2023; 24(10):9056. https://doi.org/10.3390/ijms24109056
Chicago/Turabian StylePuig-Bosch, Xènia, Markus Ballmann, Stefan Bieletzki, Bernd Antkowiak, Uwe Rudolph, Hanns Ulrich Zeilhofer, and Gerhard Rammes. 2023. "Neurosteroids Mediate Neuroprotection in an In Vitro Model of Hypoxic/Hypoglycaemic Excitotoxicity via δ-GABAA Receptors without Affecting Synaptic Plasticity" International Journal of Molecular Sciences 24, no. 10: 9056. https://doi.org/10.3390/ijms24109056