
Computational and Experimental Evaluation of the Immune Response of Neoantigens for Personalized Vaccine Design
 , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
2. Results
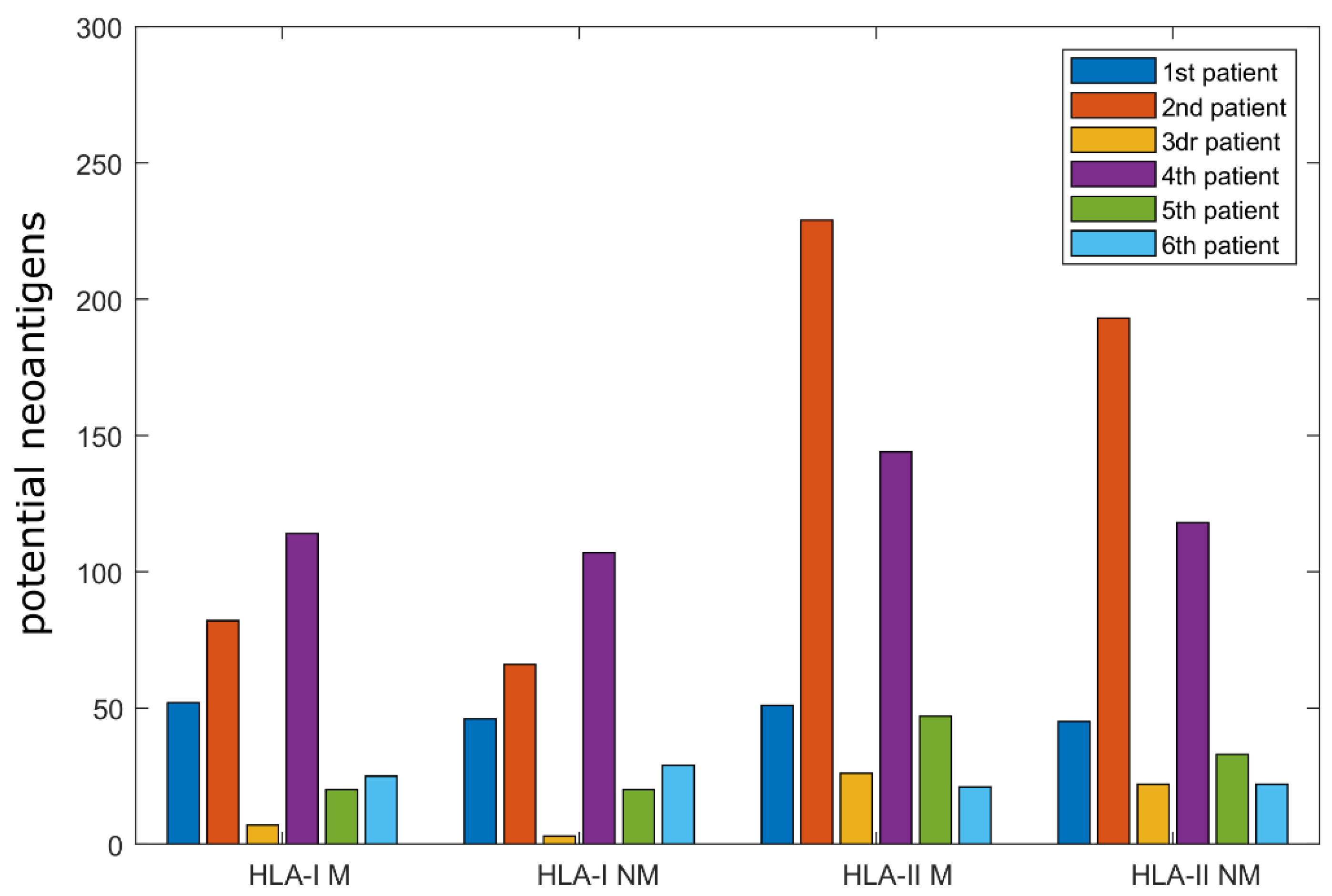
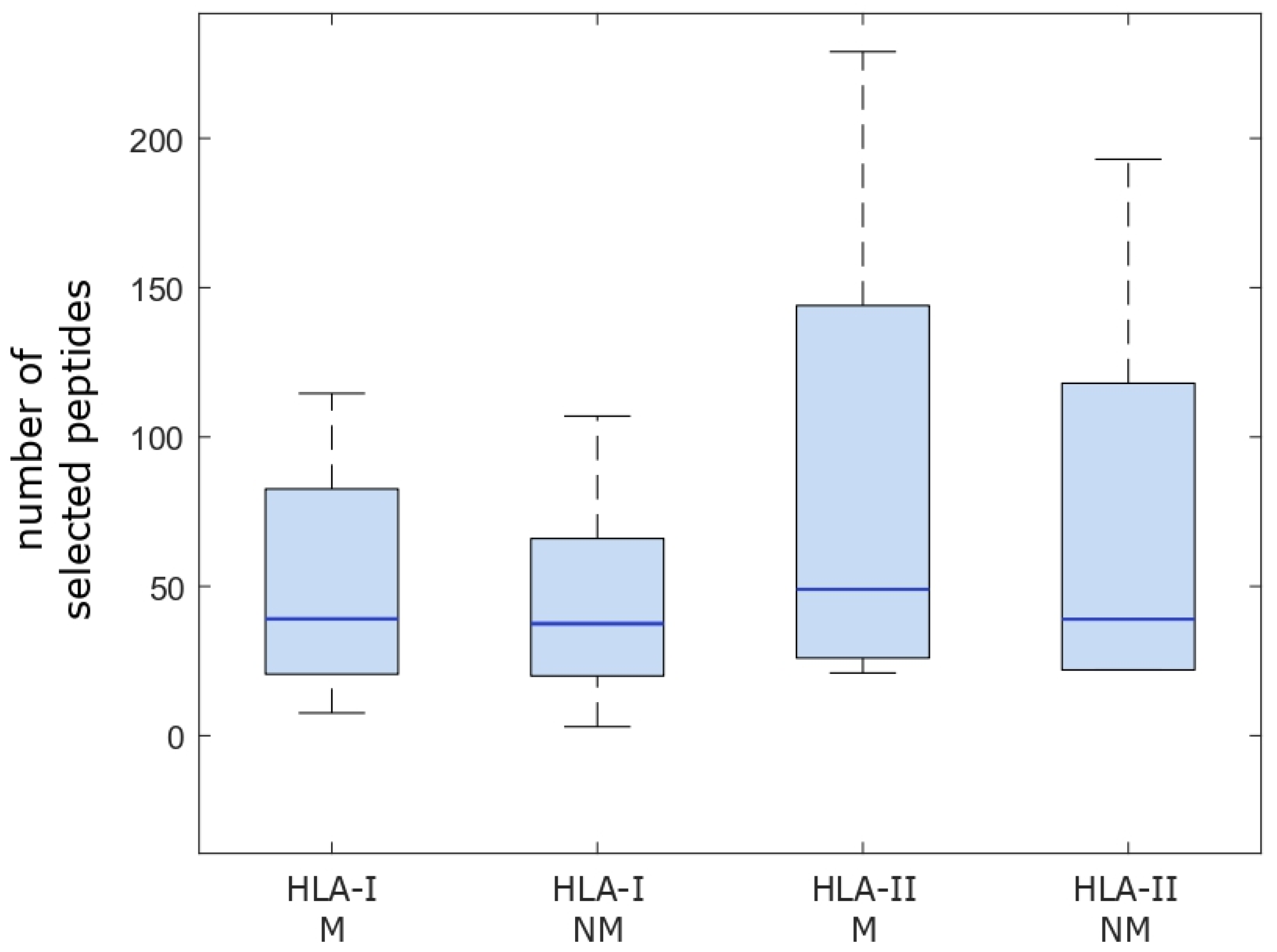
2.1. Computational Analysis of Immunological Characteristics
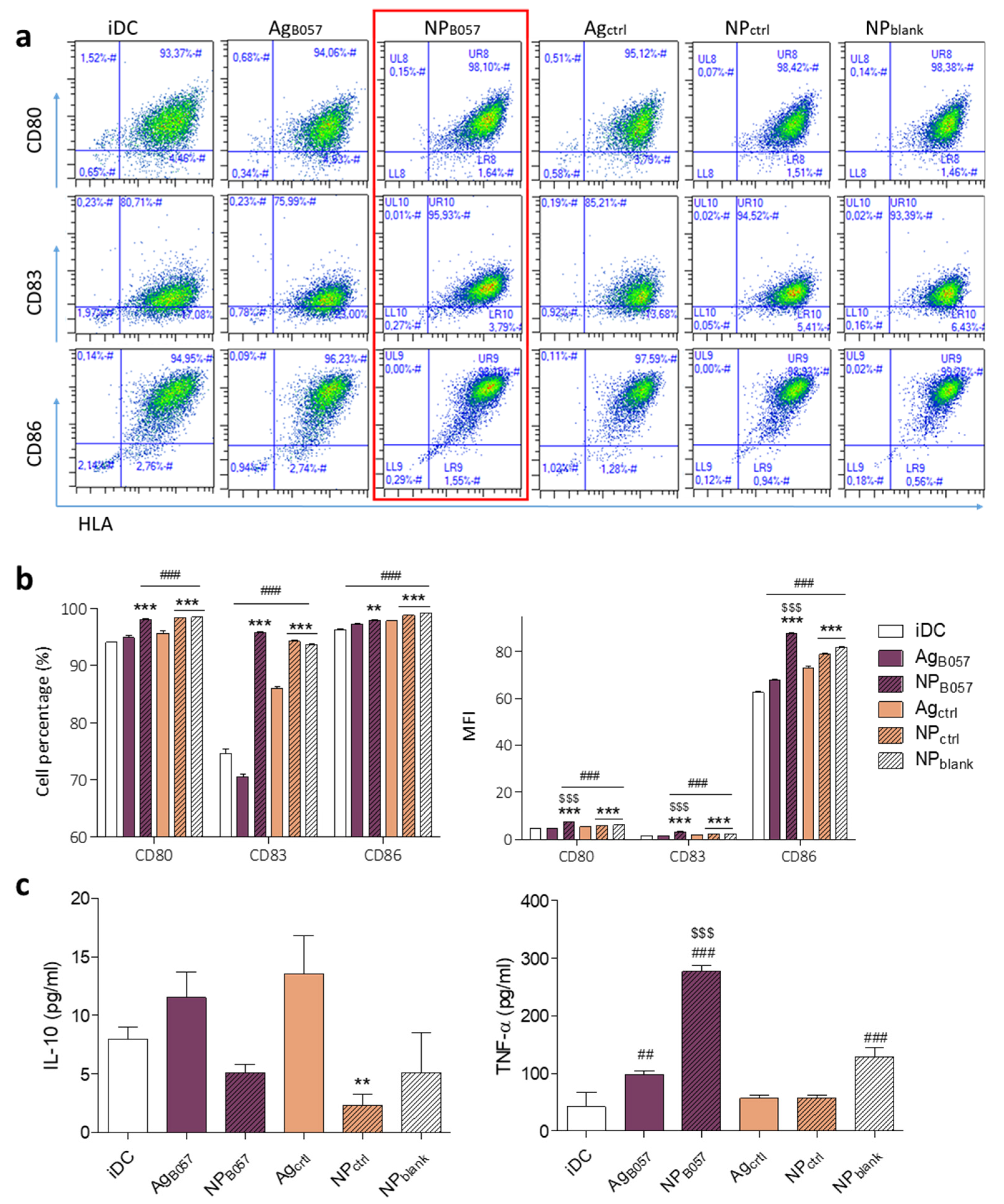
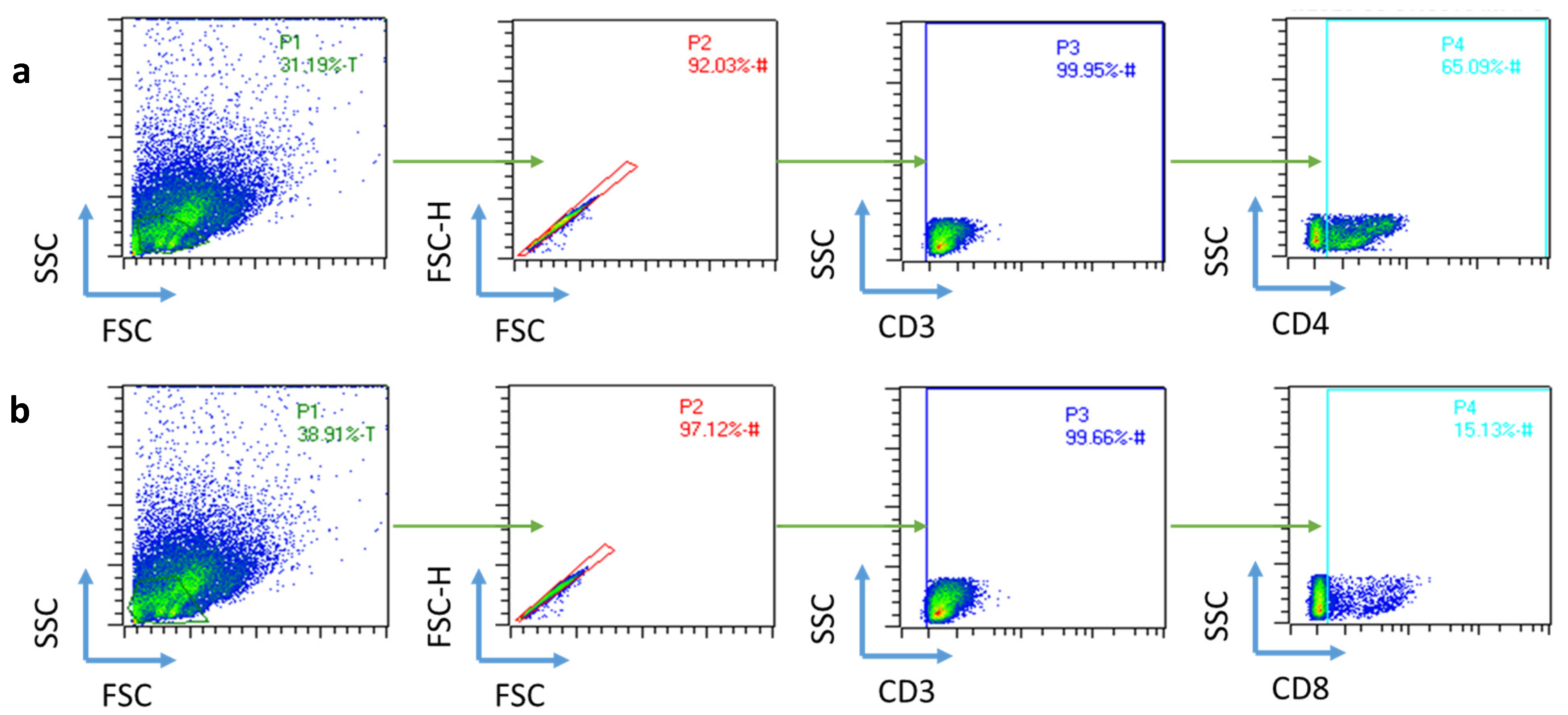
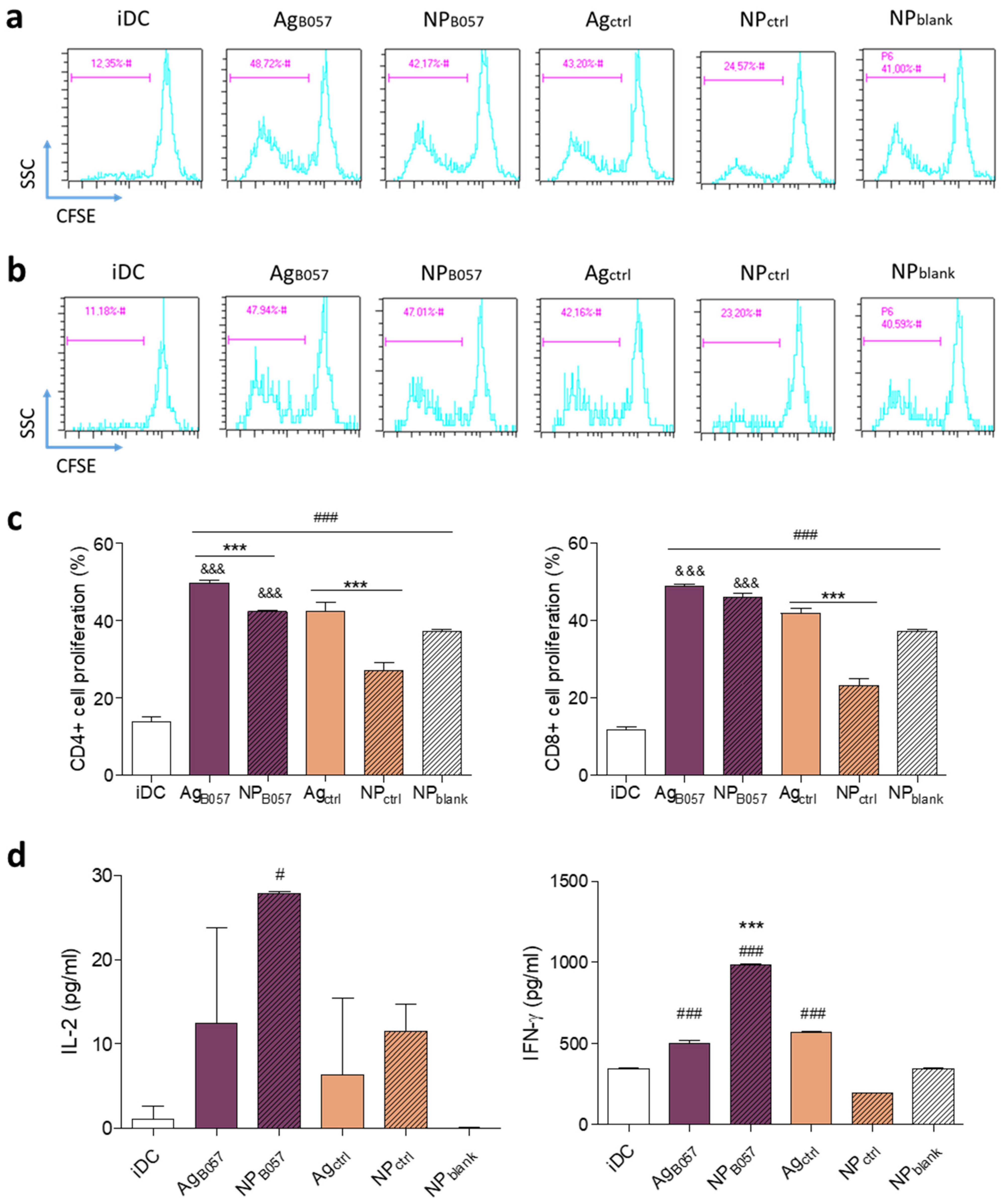
2.2. Ex Vivo Validation of the Immune Response
2.2.1. Optimization of Neoantigen Selection for Vaccine Design
- Peptide 1: DWLEWLRQLSLELLKFRDQSLSYHHTMVVQIGRFANYFRNLLPSN
- Peptide 2: MRHSFFSEVNWQDVYRLFMHHVFLEPITCVCSRRFYQFTKLLDSV
2.2.2. Optimization of Neoantigen Selection for Vaccine Design
3. Discussion
4. Materials and Methods
4.1. Computational Testing
4.1.1. Sample Acquisition for in Silico Assays
4.1.2. Detection of Genome Mutations
4.1.3. Determination of Potential Neoantigens and the Main Characteristics
4.1.4. Statistics
4.2. Experimental Testing
4.2.1. Sample Acquisition for Ex Vivo Assays
4.2.2. Estimation of Neoantigens’ Characteristics with Bioinformatic Tools
4.2.3. Optimization and Selection of Neoantigens
4.2.4. Peptide Synthesis
- DWLEWLRQLSLELLKFRDQSLSYHHTMVVQIGRFANYFRNLLPSN
- MRHSFFSEVNWQDVYRLFMHHVFLEPITCVCSRRFYQFTKLLDSV
- PSLQVITFKQRPRKLSHIRPYMNEIVTLMRFLPQVMPMFLNVIRV
- LKCVQFLSQVMPTFLIHCFENVISIMFLVAAGATLERAKTLSPGK
4.2.5. Preparation of PEI-Coated PLGA Nanoparticles (NPs)
4.2.6. Characterization of PEI-Coated PLGA NPs
4.2.7. Generation of Monocyte-Derived Dendritic Cells (DCs)
4.2.8. Dendritic Cell Maturation
4.2.9. DC/T Cell Co-Cultures
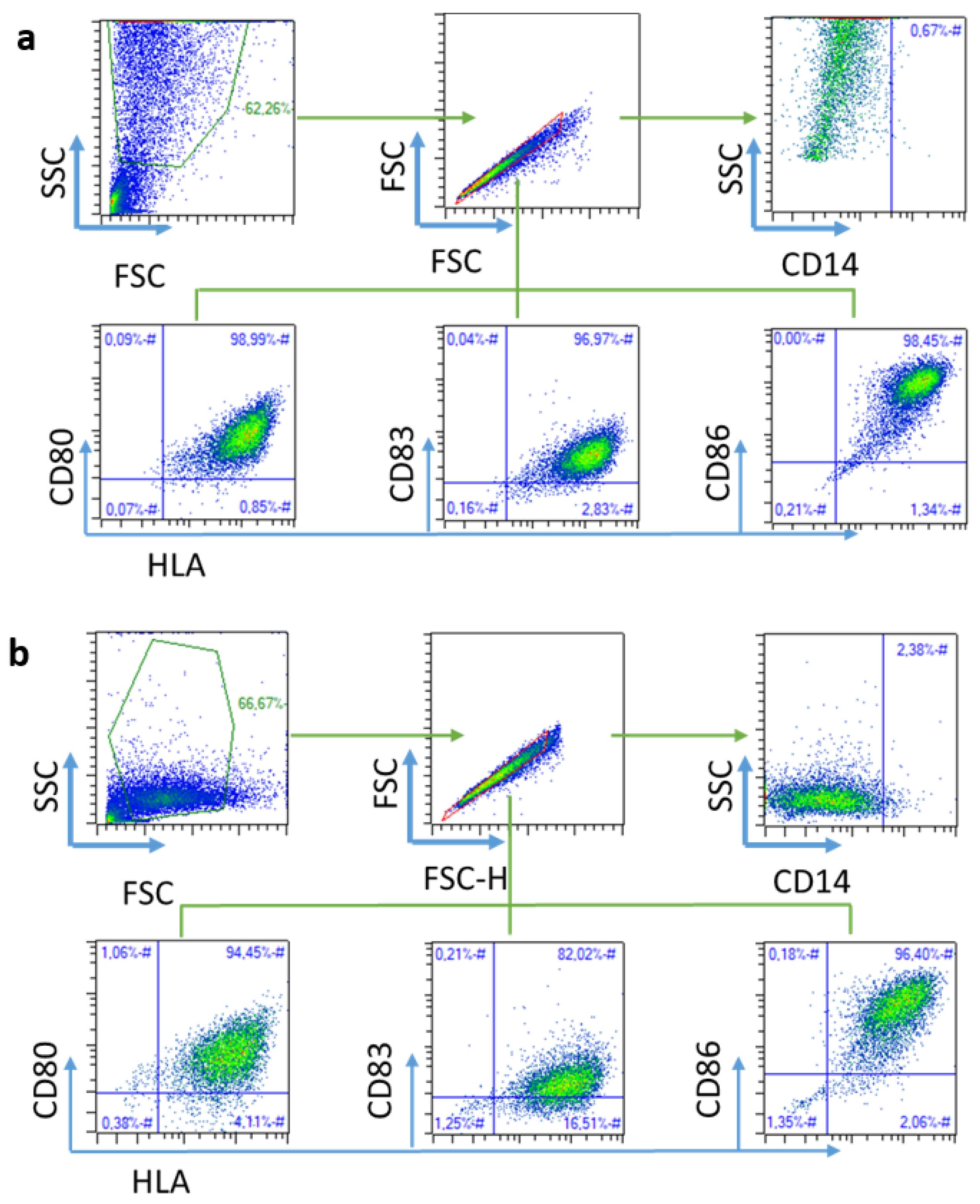
4.2.10. Flow Cytometry Analysis
4.2.11. ELISA from Supernatants
4.2.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACN | Acetonitrile |
| Ag | Antigen |
| APC | Antigen-Presenting Cell |
| CI | Confidence interval |
| DC | Dendritic cell |
| DCM | Dichloromethane |
| EE | Encapsulation efficacy |
| GRAVY | Grand average of hydropathicity index |
| HAS | Human serum |
| HLA | Human Leucocytic Antigen |
| IEDB | Immune epitope database |
| IFN | Interferon |
| IL | Interleukin |
| MHC | Major Histocompatibility Complex |
| NP | Nanoparticle |
| PBL | Peripheral blood lymphocyte |
| PBMC | Peripheral blood mononuclear cells |
| PdI | Polydispersity index |
| PEI | Polyethylenimine |
| PLGA | Polyethylenimine-coated L-lactic-co-glycolic acid |
| SAP | Surface Absorbed Protein |
| SD | Standard deviation |
| TAP | Transporter associated with antigen processing |
References
- Vormehr, M.; Diken, M.; Türeci, Ö.; Sahin, U.; Kreiter, S. Personalized Neo-Epitope Vaccines for Cancer Treatment. In Current Immunotherapeutic Strategies in Cancer; Springer: Cham, Switzerland, 2020; pp. 153–167. [Google Scholar]
- Vermaelen, K. Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Front. Immunol. 2019, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 7662. [Google Scholar] [CrossRef] [PubMed]
- Kakimi, K.; Karasaki, T.; Matsushita, H.; Sugie, T. Advances in personalized cancer immunotherapy. Breast Cancer 2017, 24, 16–24. [Google Scholar] [CrossRef]
- Stratton, M.R. Exploring the genomes of cancer cells: Progress and promise. Science 2011, 331, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Castle, J.C.; Türeci, Ö.; Sahin, U. Targeting the tumor mutanome for personalized vaccination therapy. Oncoimmunology 2012, 1, 768–769. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef]
- Vormehr, M.; Türeci, Ö.; Sahin, U. Harnessing Tumor Mutations for Truly Individualized Cancer Vaccines. Annu. Rev. Med. 2019, 70, 395–407. [Google Scholar] [CrossRef]
- Tanyi, J.L.; Bobisse, S.; Ophir, E.; Tuyaerts, S.; Roberti, A.; Genolet, R. Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 2018, 10, eaao5931. [Google Scholar] [CrossRef]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized; tumour-specific; therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168. [Google Scholar] [CrossRef]
- Fritsch, E.F.; Rajasagi, M.; Ott, P.A.; Brusic, V.; Hacohen, N.; Wu, C.J. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol. Res. 2014, 2, 522–529. [Google Scholar] [CrossRef]
- Lundegaard, C.; Lund, O.; Nielsen, M. Prediction of epitopes using neural network-based methods. J. Immunol. Methods 2011, 374, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, P.; Kim, Y.; Haste-Andersen, P.; Beaver, J.; Bourne, P.E.; Bui, H.-H.; Buus, S.; Frankild, S.; Greenbaum, J.; et al. Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 2008, 36, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Soria-Guerra, R.E.; Nieto-Gomez, R.; Govea-Alonso, D.O.; Rosales-Mendoza, S. An overview of bioinformatics tools for epitope prediction: Implications on vaccine development. J. Biomed. Inform. 2015, 53, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Martínez, L.; Milanič, M.; Malaina, I.; Álvarez, C.; Pérez, M.B.; De la Fuente, I.M. Weighted lambda superstrings applied to vaccine design. PLoS ONE 2019, 14, e0211714. [Google Scholar] [CrossRef] [PubMed]
- Malaina, I.; Legarreta, L.; Boyano, M.D.; Gardeazabal, J.; Bringas, C.; Martinez, L.; de la Fuente, I.M. Metastasis of Cutaneous Melanoma: Risk Factors; Detection and Forecasting. In Proceedings of the 6th International Work-Conference, Bioinformatics and Biomedical Engineering, IWBBIO 2018, Lecture Notes in Computer Science, Granada, Spain, 25–27 April 2018; p. 10813. [Google Scholar]
- Miller, A.J.; Mihm, M.C. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef]
- Yang, X.; Fan, J.; Wu, Y.; Ma, Z.; Huang, J.; Zhang, Y.; Zhou, Z.; Mo, F.; Liu, X.; Yuan, H.; et al. Synthetic multiepitope neoantigen DNA vaccine for personalized cancer immunotherapy. Nanomed. Nanotechnol. Biol. Med. 2021, 37, 102443. [Google Scholar] [CrossRef]
- Nam, J.; Son, S.; Park, K.S.; Moon, J.J. Modularly Programmable Nanoparticle Vaccine Based on Polyethyleneimine for Personalized Cancer Immunotherapy. Adv. Sci. 2021, 8, 2002577. [Google Scholar] [CrossRef]
- Arbelaez, C.A.; Estrada, J.; Gessner, M.A.; Glaus, C.; Morales, A.B.; Mohn, D.; Phee, H.; Lipford, J.R.; Johnston, J.A. A nanoparticle vaccine that targets neoantigen peptides to lymphoid tissues elicits robust antitumor T cell responses. NPJ Vaccines 2020, 5, 106. [Google Scholar] [CrossRef]
- Ni, Q.; Zhang, F.; Liu, Y.; Wang, Z.; Yu, G.; Liang, B.; Niu, G.; Su, T.; Zhu, G.; Lu, G.; et al. A bi-adjuvant nanovaccine that potentiates immunogenicity of neoantigen for combination immunotherapy of colorectal cancer. Sci. Adv. 2020, 6, eaaw6071. [Google Scholar] [CrossRef]
- Kohnepoushi, C.; Nejati, V.; Delirezh, N.; Biparva, P. Poly Lactic-co-Glycolic Acid Nanoparticles Containing Human Gastric Tumor Lysates as Antigen Delivery Vehicles for Dendritic Cell-Based Antitumor Immunotherapy. Immunol. Investig. 2019, 48, 794–808. [Google Scholar] [CrossRef]
- Moutaftsi, M.; Peters, B.; Pasquetto, V.; Tscharke, D.C.; Sidney, J.; Bui, H.H.; Grey, H.; Sette, A. A consensus epitope prediction approach identifies the breadth of murine T CD8+-cell responses to vaccinia virus. Nat. Biotechnol. 2006, 24, 817. [Google Scholar] [CrossRef] [PubMed]
- Kotturi, M.F.; Peters, B.; Buendia-Laysa, F.; Sidney, J.; Oseroff, C.; Botten, J.; Grey, H.; Buchmeier, M.J.; Sette, A. The CD8+ T-cell response to lymphocytic choriomeningitis virus involves the L antigen: Uncovering new tricks for an old virus. J. Virol. 2007, 81, 4928–4940. [Google Scholar] [CrossRef] [PubMed]
- Hopp, T.P.; Woods, K.R. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl. Acad. Sci. USA 1981, 78, 3824–3828. [Google Scholar] [CrossRef]
- Hopp, T.P.; Woods, K.R. A computer program for predicting protein antigenic determinants. Mol. Immunol. 1983, 20, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Adekiya, T.A.; Taiwo, R.; Khanyile, S.; Masamba, P.; Oyinloye, B.E.; Kappo, A.P. Structural Analysis and Epitope Prediction of MHC Class-1-Chain Related Protein-A for Cancer Vaccine Development. Vaccines 2018, 6, 1. [Google Scholar]
- Alcazer, V.; Bonaventura, P.; Tonon, L.; Wittmann, S.; Caux, C.; Depil, S. Neoepitopes-based vaccines: Challenges and perspectives. Eur. J. Cancer 2019, 108, 55–60. [Google Scholar] [CrossRef]
- Doytchinova, I.A.; Flower, D.R. A server for prediction of protective antigens; tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef]
- Bae, J.; Parayath, N.; Ma, W.; Amiji, M.; Munshi, N.; Anderson, K.C. BCMA peptide-engineered nanoparticles enhance induction and function of antigen-specific CD8+ cytotoxic T lymphocytes against multiple myeloma: Clinical applications. Leukemia 2020, 34, 210–223. [Google Scholar] [CrossRef]
- Iranpour, S.; Nejati, V.; Delirezh, N.; Biparva, P.; Shirian, S. Enhanced stimulation of anti-breast cancer T cells responses by dendritic cells loaded with poly lactic-co-glycolic acid (PLGA) nanoparticle encapsulated tumor antigens. J. Exp. Clin. Cancer Res. 2016, 35, 168. [Google Scholar] [CrossRef]
- Aerts-Toegaert, C.; Heirman, C.; Tuyaerts, S.; Corthals, J.; Aerts, J.L.; Bonehill, A.; Thielemans, K.; Breckpot, K. CD83 expression on dendritic cells and T cells: Correlation with effective immune responses. Eur. J. Immunol. 2007, 37, 686–695. [Google Scholar] [CrossRef]
- Lu, P.; Wang, Y.L.; Linsley, P.S. Regulation of self-tolerance by CD80/CD86 interactions. Curr. Opin. Immunol. 1997, 9, 858–862. [Google Scholar] [CrossRef]
- Suzuki, H.; Toyooka, T.; Ibuki, Y. Simple and easy method to evaluate uptake potential of nanoparticles in mammalian cells using a flow cytometric light scatter analysis. Environ. Sci. Technol. 2007, 41, 3018–3024. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Noh, Y.; Lim, Y.T. Polymer nanoparticles for cross-presentation of exogenous antigens and enhanced cytotoxic T-lymphocyte immune response. Int. J. Nanomed. 2016, 11, 3753–3764. [Google Scholar]
- Wajant, H. The role of TNF in cancer. In Death Receptors and Cognate Ligands in Cancer; Results and Problems in Cell Differentiation; Springer: Berlin/Heidelberg, Germany, 2009; Volume 49, pp. 1–15. [Google Scholar]
- Tough, D.F. Cytokines produced by dendritic cells. In Handbook of Dendritic Cells; John Wiley & Sons; Ltd.: Hoboken, NJ, USA, 2008; pp. 355–383. [Google Scholar]
- Schülke, S. Induction of interleukin-10 producing dendritic cells as a tool to suppress allergen-specific T helper 2 responses. Front. Immunol. 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Passarelli, A.; Mannavola, F.; Felici, C.; Stucci, L.S.; Cives, M.; Silvestris, F. Immune system evasion as hallmark of melanoma progression: The role of dendritic cells. Front. Oncol. 2019, 9, 1148. [Google Scholar] [CrossRef]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef]
- Flores, I.; Hevia, D.; Tittarelli, A.; Soto, D.; Rojas-Sepúlveda, D.; Pereda, C.; Tempio, F.; Fuentes, C.; Falcón-Beas, C.; Gatica, J.; et al. Dendritic cells loaded with heat shock-conditioned ovarian epithelial carcinoma cell lysates elicit T cell-dependent antitumor immune responses in vitro. J. Immunol. Res. 2019, 2019, 9631515. [Google Scholar] [CrossRef]
- Silva, A.L.; Soema, P.C.; Slütter, B.; Ossendorp, F.; Jiskoot, W. PLGA particulate delivery systems for subunit vaccines: Linking particle properties to immunogenicity. Hum. Vaccines Immunother. 2016, 12, 1056–1069. [Google Scholar] [CrossRef]
- Kim, H.; Cantor, H. CD4 T-cell subsets and tumor immunity: The helpful and the not-so-helpful. Cancer Immunol. Res. 2014, 2, 91–98. [Google Scholar] [CrossRef]
- Boyman, O.; Cho, J.; Sprent, J. The role of interleukin-2 in memory CD8 cell differentiation. Adv. Exp. Med. Biol. 2010, 684, 28–41. [Google Scholar]
- Bae, J.; Smith, R.; Daley, J.; Mimura, N.; Tai, Y.-T.; Anderson, K.C.; Munshi, N.C. Myeloma-specific multiple peptides able to generate cytotoxic T lymphocytes: A potential therapeutic application in multiple myeloma and other plasma cell disorders. Clin. Cancer Res. 2012, 18, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, M.; Bobisse, S.; Costa-Nunes, C.; Cesson, V.; Jichlinski, P.; Speiser, D.; Harari, A.; Coukos, G.; Romero, P.; Nardelli-Haefliger, D.; et al. High-throughput monitoring of human tumor-specific T-cell responses with large peptide pools. OncoImmunology 2015, 4, e1029702. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.-N.; Zhang, C.-N.; Xu, R.; Niu, J.-F.; Song, H.-J.; Zhang, X.-Y.; Wang, W.-W.; Wang, Y.-M.; Li, C.; Wei, X.-Q.; et al. Enhanced antitumor immunity by targeting dendritic cells with tumor cell lysate-loaded chitosan nanoparticles vaccine. Biomaterials 2017, 113, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Sun, Z.; Liang, J.; Wang, H.; Zhu, D.; Leng, X.; Wang, C.; Kong, D.; Lv, F. A visible fluorescent nanovaccine based on functional genipin crosslinked ovalbumin protein nanoparticles. Nanomedicine 2018, 14, 1087–1098. [Google Scholar] [CrossRef]
- Taams, L.S.; Van Eden, W.; Wauben, M.H. Antigen presentation by T cells versus professional antigen-presenting cells (APC): Differential consequences for T cell activation and subsequent T cell-APC interactions. Eur. J. Immunol. 1999, 29, 1543–1550. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbala, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Vormehr, M.; Schrörs, B.; Boegel, S.; Löwer, M.; Türeci, Ö.; Sahin, U. Mutanome engineered RNA immunotherapy: Towards patient-centered tumor vaccination. J. Immunol. Res. 2015, 2015, 595363. [Google Scholar] [CrossRef]
- Kardani, K.; Bolhassani, A.; Namvar, A. An overview of in silico vaccine design against different pathogens and cancer. Expert Rev. Vaccines 2020, 19, 699–726. [Google Scholar] [CrossRef]
- Jamari, J.; Ammarullah, M.I.; Santoso, G.; Sugiharto, S.; Supriyono, T.; Permana, M.S.; Winarni, T.I.; van der Heide, E. Adopted Walking Condition for Computational Simulation Approach on Bearing of Hip Joint Prosthesis: Review over the Past 30 Years. Heliyon 2022, 8, e12050. [Google Scholar] [CrossRef]
- Ammarullah, M.I.; Hartono, R.; Supriyono, T.; Santoso, G.; Sugiharto, S.; Permana, M.S. Polycrystalline Diamond as a Potential Material for the Hard-on-Hard Bearing of Total Hip Prosthesis: Von Mises Stress Analysis. Biomedicines 2023, 11, 951. [Google Scholar] [CrossRef] [PubMed]
- Tauviqirrahman, M.; Ammarullah, M.I.; Jamari, J.; Saputra, E.; Winarni, T.I.; Kurniawan, F.D.; Shiddiq, S.A.; van der Heide, E. Analysis of Contact Pressure in a 3D Model of Dual-Mobility Hip Joint Prosthesis under a Gait Cycle. Sci. Rep. 2023, 13, 3564. [Google Scholar] [CrossRef]
- Ammarullah, M.I.; Santoso, G.; Sugiharto, S.; Supriyono, T.; Wibowo, D.B.; Kurdi, O.; Tauviqirrahman, M.; Jamari, J. Minimizing Risk of Failure from Ceramic-on-Ceramic Total Hip Prosthesis by Selecting Ceramic Materials Based on Tresca Stress. Sustainability 2022, 14, 13413. [Google Scholar] [CrossRef]
- Ammarullah, M.I.; Afif, I.Y.; Maula, M.I.; Winarni, T.I.; Tauviqirrahman, M.; Akbar, I.; Basri, H.; van der Heide, E.; Jamari, J. Tresca Stress Simulation of Metal-on-Metal Total Hip Arthroplasty during Normal Walking Activity. Materials 2021, 14, 7554. [Google Scholar] [CrossRef] [PubMed]
- Salaha, Z.F.M.; Ammarullah, M.I.; Abdullah, N.N.A.A.; Aziz, A.U.A.; Gan, H.-S.; Abdullah, A.H.; Abdul Kadir, M.R.; Ramlee, M.H. Biomechanical Effects of the Porous Structure of Gyroid and Voronoi Hip Implants: A Finite Element Analysis Using an Experimentally Validated Model. Materials 2023, 16, 3298. [Google Scholar] [CrossRef]
- Raghava, G.P.; Han, J.H. Correlation and Prediction of Gene Expression Level from Amino Acid and Dipeptide Composition of Its Protein. BMC Bioinformatics 2005, 6, 59. [Google Scholar] [CrossRef]
- Beltrán, J. Computational prediction of linear B-cell epitopes in the E5 oncoprotein of the human papillomavirus type 16 using several bioinformatics tools. Vacunas: Investig. Y Práctica 2016, 17, 18–26. [Google Scholar] [CrossRef]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; Roemer, N.V.D.; Graaf, J.D.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. AACR (Am. Assoc. Cancer Res.) 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Thompson, J.A. The revised American Joint Committee on Cancer staging system for melanoma. Semin. Oncol. 2002, 29, 361–369. [Google Scholar] [CrossRef]
- The Basque BioBank. Available online: https://www.biobancovasco.org/ (accessed on 21 December 2021).
- Edlundh-Rose, E.; Egyha, S.; Omholt, K.; Mansson-Brahme, E.; Platz, A.; Hansson, J.; Lundeberg, J. NRAS and BRAF mutations in melanoma tumours in relation to clinical characteristics: A study based on mutation screening by pyrosequencing. Melanoma Res. 2006, 16, 471–478. [Google Scholar] [CrossRef]
- Nikolaev, S.I.; Rimoldi, D.; Iseli, C.; Valsesia, A.; Robyr, D.; Gehrig, C.; Harshman, K.; Guipponi, M.; Bukach, O.; Zoete, V.; et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat. Genet. 2012, 44, 133. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://genome.ucsc.edu/ (accessed on 10 February 2022).
- Available online: https://pypi.org/project/pyliftover/ (accessed on 10 February 2022).
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217. [Google Scholar] [CrossRef] [PubMed]
- Trolle, T.; McMurtrey, C.P.; Sidney, J.; Bardet, W.; Osborn, S.C.; Kaever, T.; Sette, A.; Hildebrand, W.H.; Nielsen, M.; Peters, B. The length distribution of class I–restricted T cell epitopes is determined by both peptide supply and MHC allele–specific binding preference. J. Immunol. 2016, 196, 1480–1487. [Google Scholar] [CrossRef]
- Mann, E.R.; Li, X. Intestinal antigen-presenting cells in mucosal immune homeostasis: Crosstalk between dendritic cells; macrophages and B-cells. World J. Gastroenterol. WJG 2014, 20, 9653. [Google Scholar] [CrossRef]
- López-Martínez, A.; Chávez-Muñoz, C.; Granados, J. Función biológica del complejo principal de histocompatibilidad. Rev. De Investig. Clínica 2005, 57, 132–141. [Google Scholar]
- IEDB Class-I Immunogenicity. Available online: http://tools.iedb.org/immunogenicity/ (accessed on 14 March 2022).
- IEDB MHC-I Binding. Available online: http://tools.iedb.org/mhci/ (accessed on 14 March 2022).
- IEDB MHC-II Binding. Available online: http://tools.iedb.org/mhcii/ (accessed on 14 March 2022).
- VaxiJen Database. Available online: http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html (accessed on 14 March 2022).
- ExPASy Database. Available online: https://web.expasy.org/protparam/protparam-doc.html (accessed on 14 March 2022).
- IEDB TAP/Transport. Available online: http://tools.iedb.org/processing/ (accessed on 14 March 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HLA-I | 1st Patient | 2nd Patient | 3rd Patient | 4th Patient | 5th Patient | 6th Patient | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1st | 2nd | 1st | 2nd | 1st | 2nd | 1st | 2nd | 1st | 2nd | 1st | 2nd | |
| A | 01:01:01 | 02:01:01 | 02:01:01 | 32:01:01 | 24:02:01 | 32:01:01 | 02:01:01 | 11:01:01 | 24:02:01 | 24:02:01 | 03:01:01 | 26:01:01 |
| B | 08:01:01 | 44:27:01 | 35:11:01 | 51:01:01 | 07:02:01 | 51:01:01 | 27:05:02 | 40:01:02 | 35:01:01 | 40:01:03 | 07:02:01 | 18:01:01 |
| C | 07:01:01 | 07:04:01 | 02:02:02 | 04:01:01 | 07:02:01 | 15:02:01 | 01:02:01 | 03:04:01 | 03:04:01 | 04:01:01 | 02:02:02 | 07:02:01 |
| HLA-II | 1st Patient | 2nd Patient | 3rd Patient | 4th Patient | 5th Patient | 6th Patient | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1st | 2nd | 1st | 2nd | 1st | 2nd | 1st | 2nd | 1st | 2nd | 1st | 2nd | |
| DPA1 | 01:03:01 | 02:01:01 | 01:03:01 | 01:03:01 | 01:03:01 | 01:03:01 | 01:03:01 | 01:03:01 | 01:03:01 | 01:03:01 | 01:03:01 | 02:01:01 |
| DPB1 | 02:01:02 | 14:01:01 | 02:01:02 | 04:01:01 | --:01:-- | --:01:-- | 04:01:01 | 06:01:-- | 04:01:01 | 04:01:01 | 02:01:02 | 11:01:01 |
| DQA1 | 01:02:02 | 01:04:01 | 01:02:01 | 05:05:01 | 01:02:01 | 01:03:01 | 03:01:01 | 04:01:01 | 01:01:01 | 04:01:01 | 01:03:01 | 05:05:01 |
| DQB1 | 05:02:01 | 05:03:01 | 03:01:01 | 06:02:01 | 06:02:01 | 06:03:01 | 03:02:01 | 04:02:01 | 04:02:01 | 05:01:01 | 03:01:01 | 06:03:01 |
| DRB1 | 16:01:01 | 14:54:01 | 15:01:01 | 11:04:01 | 15:01:01 | 13:01:01 | 08:01:-- | 04:04:01 | 01:01:01 | 08:02:01 | --:--:-- | --:--:-- |
| DRB3 | 02:02:01 | --:--:-- | 02:02:01 | --:--:-- | 01:01:02 | --:--:-- | --:--:-- | --:--:-- | --:--:-- | --:--:-- | 01:01:02 | 01:01:02 |
| DRB4 | --:--:-- | --:--:-- | --:--:-- | --:--:-- | --:--:-- | --:--:-- | 01:03:01 | 01:03:01 | --:--:-- | --:--:-- | --:--:-- | --:--:-- |
| DRB5 | 02:02:-- | --:--:-- | 01:01:01 | --:--:-- | 01:01:01 | --:--:-- | --:--:-- | --:--:-- | --:--:-- | --:--:- | --:--:-- | --:--:-- |
| Neoantigen | Immunogenicity | HLA-I | HLA-II | Hydrophilicity | TAP, Proteosome | VaxiJen | Variant Frequency |
|---|---|---|---|---|---|---|---|
| DWLEWLRQLSLELLK | 0.556 | 0.875 | 1 | 0.366 | 0.722 | 0.516 | 0.199 |
| FRDQSLSYHHTMVVQ | 0.335 | 1 | 0 | 0.501 | 0.558 | 0.453 | 0.304 |
| IGRFANYFRNLLPSN | 0.851 | 0.665 | 0.529 | 0.41 | 0.567 | 0.555 | 0.333 |
| MRHSFFSEVNWQDVY | 0.878 | 0 | 0.452 | 0.535 | 0.72 | 0.427 | 0.339 |
| RLFMHHVFLEPITCV | 1 | 0.366 | 0.432 | 0 | 0.603 | 0 | 0.762 |
| CSRRFYQFTKLLDSV | 0.518 | 0.562 | 0.788 | 0.398 | 0.608 | 0.379 | 0.466 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malaina, I.; Gonzalez-Melero, L.; Martínez, L.; Salvador, A.; Sanchez-Diez, A.; Asumendi, A.; Margareto, J.; Carrasco-Pujante, J.; Legarreta, L.; García, M.A.; et al. Computational and Experimental Evaluation of the Immune Response of Neoantigens for Personalized Vaccine Design. Int. J. Mol. Sci. 2023, 24, 9024. https://doi.org/10.3390/ijms24109024
Malaina I, Gonzalez-Melero L, Martínez L, Salvador A, Sanchez-Diez A, Asumendi A, Margareto J, Carrasco-Pujante J, Legarreta L, García MA, et al. Computational and Experimental Evaluation of the Immune Response of Neoantigens for Personalized Vaccine Design. International Journal of Molecular Sciences. 2023; 24(10):9024. https://doi.org/10.3390/ijms24109024
Chicago/Turabian StyleMalaina, Iker, Lorena Gonzalez-Melero, Luis Martínez, Aiala Salvador, Ana Sanchez-Diez, Aintzane Asumendi, Javier Margareto, Jose Carrasco-Pujante, Leire Legarreta, María Asunción García, and et al. 2023. "Computational and Experimental Evaluation of the Immune Response of Neoantigens for Personalized Vaccine Design" International Journal of Molecular Sciences 24, no. 10: 9024. https://doi.org/10.3390/ijms24109024