
Evidence Synthesis of Gene Therapy and Gene Editing from Different Disorders—Implications for Individuals with Rett Syndrome: A Systematic Review

Abstract
:1. Introduction
2. Methods
2.1. Search Strategy
Primary Search Strategy
2.2. Search Terms
- Primary Search Terms
- Secondary Search Terms
2.3. Population Characteristics (Primary Search)
2.4. Intervention
2.5. Eligibility Criteria
- Inclusion Criteria
- Full-text records/articles in peer-reviewed academic/scientific journals available electronically from the last 10 years (2012 to present)
- All experiments are conducted on humans or human cells.
- Exclusion Criteria
- Articles that were not in English language.
- Studies using animal models.
- Information provided in reviews, preprints, letters to the editor, conference abstracts, book chapters and clinical trial protocols.
2.6. Extraction of Data and Thematic Analysis
3. Results
3.1. Article Characteristics
3.1.1. Leukodystrophies
3.1.2. Neuromuscular Disorders
3.1.3. Haemoglobinopathies
3.1.4. Lysosomal Storage Disorders
3.1.5. Rett Syndrome
3.1.6. Angelman Syndrome
3.1.7. Retinal Disorders
3.1.8. Immunodeficiencies
3.1.9. Familial Amyotrophic Lateral Sclerosis
3.1.10. Huntington’s Disease
3.2. Thematic Analysis of the Included Studies
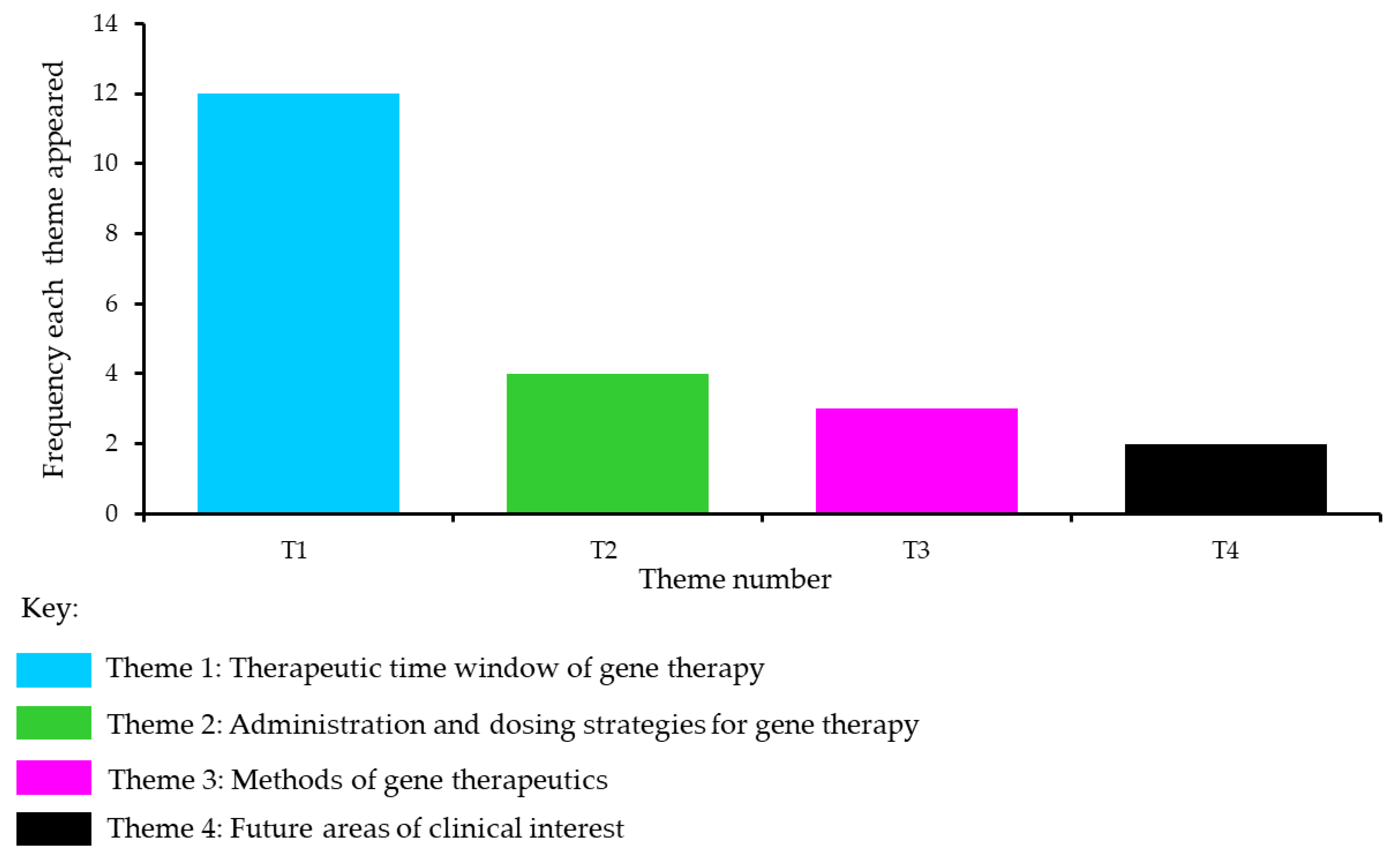
3.2.1. Theme 1: Therapeutic Time Window of Gene Therapy
3.2.2. Evidence against a Therapeutic Time Window
3.2.3. Theme 2: Administration and Dosing Strategies for Gene Therapy
3.2.4. Theme 3: Methods of Gene Therapeutics
3.2.5. Theme 4: Future Areas of Clinical Interest
3.2.6. Other Considerations
4. Discussion
- Is there a critical therapeutic time window for gene therapy or gene editing in individuals with Rett syndrome?
- What is the most appropriate route of administration and dosing strategy?
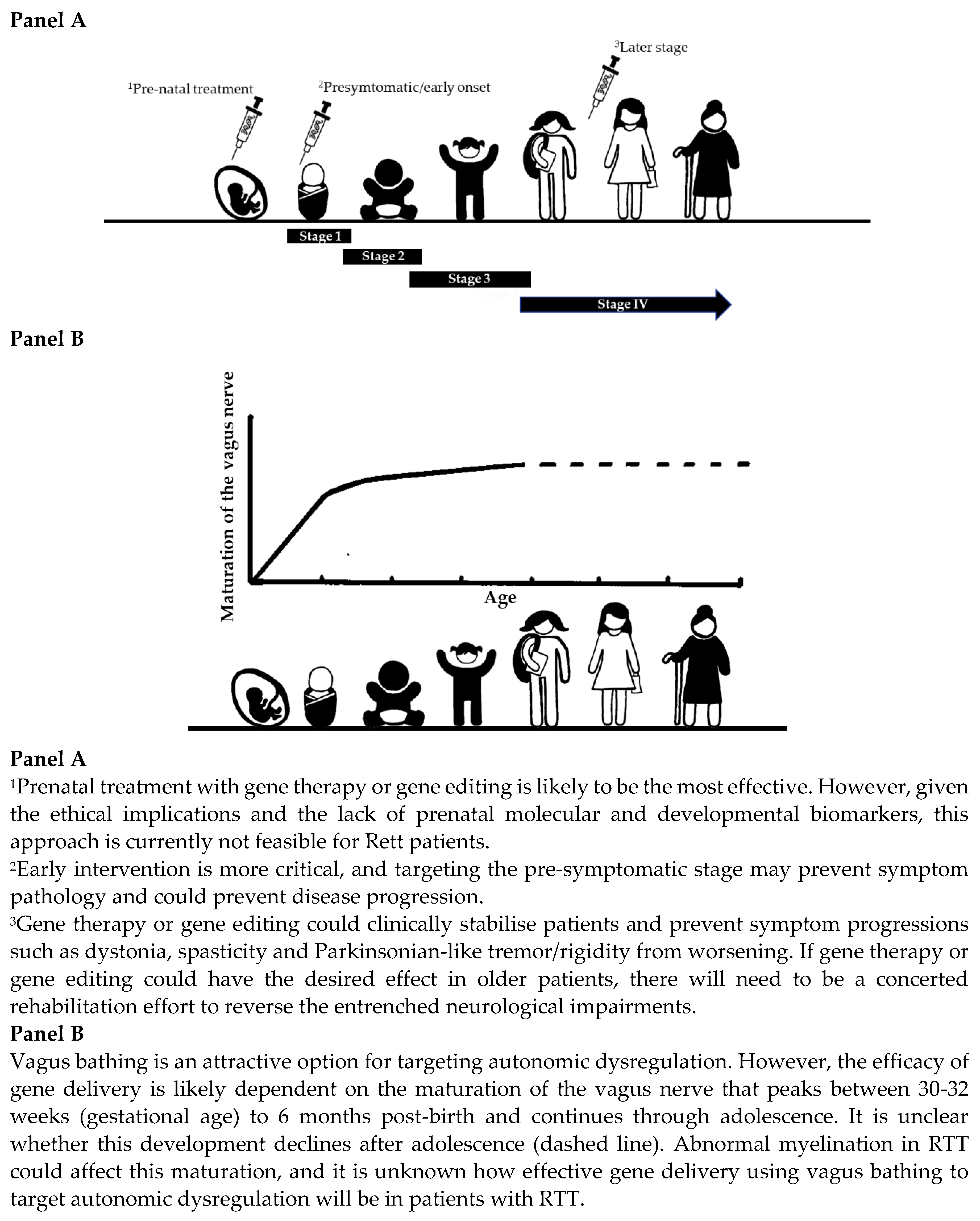
4.1. Is There a Critical Therapeutic Time Window for Gene Therapy or Gene Editing in Individuals with Rett Syndrome?
4.2. What Is the Most Appropriate Route of Administration and Dosing Strategy?
5. Conclusions
5.1. Future Directions
5.2. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tillotson, R.; Selfridge, J.; Koerner, M.V.; Gadalla, K.K.E.; Guy, J.; De Sousa, D.; Hector, R.D.; Cobb, S.R.; Bird, A. Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature 2017, 550, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Lyst, M.J.; Ekiert, R.; Ebert, D.H.; Merusi, C.; Nowak, J.; Selfridge, J.; Guy, J.; Kastan, N.R.; Robinson, N.D.; de Lima Alves, F.; et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013, 16, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Fu, X.; Zhang, Y.; Wang, S.-F.; Zhu, H.; Wang, W.-K.; Zhang, L.; Wu, P.; Wong, C.C.L.; Li, J.; et al. Rett syndrome linked to defects in forming the MeCP2/Rbfox/LASR complex in mouse models. Nat. Commun. 2021, 12, 5767. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Hamdan, H.; Yalamanchili, H.K.; Pang, K.; Pohodich, A.E.; Lopez, J.; Shao, Y.; Oses-Prieto, J.A.; Li, L.; Kim, W.; et al. Disruption of MeCP2-TCF20 complex underlies distinct neurodevelopmental disorders. Proc. Natl. Acad. Sci. USA 2022, 119, e2119078119. [Google Scholar] [CrossRef] [PubMed]
- Gabel, H.W.; Kinde, B.Z.; Stroud, H.; Gilbert, C.S.; Harmin, D.A.; Kastan, N.R.; Hemberg, M.; Ebert, D.H.; Greenberg, M.E. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 2015, 522, 89–93. [Google Scholar] [CrossRef]
- Haase, F.; Singh, R.; Gloss, B.; Tam, P.; Gold, W. Meta-Analysis Identifies BDNF and Novel Common Genes Differently Altered in Cross-Species Models of Rett Syndrome. Int. J. Mol. Sci. 2022, 23, 11125. [Google Scholar] [CrossRef]
- Cosentino, L.; Zidda, F.; Dukal, H.; Witt, S.H.; De Filippis, B.; Flor, H. Low levels of Methyl-CpG binding protein 2 are accompanied by an increased vulnerability to the negative outcomes of stress exposure during childhood in healthy women. Transl. Psychiatry 2022, 12, 506. [Google Scholar] [CrossRef]
- Singh, J.; Lanzarini, E.; Santosh, P. Autonomic dysfunction and sudden death in patients with Rett syndrome: A systematic review. J. Psychiatry Neurosci. 2020, 45, 150–181. [Google Scholar] [CrossRef]
- Singh, J.; Santosh, P. Key issues in Rett syndrome: Emotional, behavioural and autonomic dysregulation (EBAD)—A target for clinical trials. Orphanet. J. Rare Dis. 2018, 13, 128. [Google Scholar] [CrossRef]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef]
- McGraw, C.M.; Samaco, R.C.; Zoghbi, H.Y. Adult neural function requires MeCP2. Science 2011, 333, 186. [Google Scholar] [CrossRef] [PubMed]
- Ozlu, C.; Bailey, R.M.; Sinnett, S.; Goodspeed, K.D. Gene Transfer Therapy for Neurodevelopmental Disorders. Dev. Neurosci. 2021, 43, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Panayotis, N.; Ehinger, Y.; Felix, M.S.; Roux, J.C. State-of-the-art therapies for Rett syndrome. Dev. Med. Child Neurol. 2023, 65, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Coorey, B.; Haase, F.; Ellaway, C.; Clarke, A.; Lisowski, L.; Gold, W.A. Gene Editing and Rett Syndrome: Does It Make the Cut? CRISPR J. 2022, 5, 490–499. [Google Scholar] [CrossRef]
- Sandweiss, A.J.; Brandt, V.L.; Zoghbi, H.Y. Advances in understanding of Rett syndrome and MECP2 duplication syndrome: Prospects for future therapies. Lancet Neurol. 2020, 19, 689–698. [Google Scholar] [CrossRef]
- Clarke, A.J.; Abdala Sheikh, A.P. A perspective on “cure” for Rett syndrome. Orphanet. J. Rare Dis. 2018, 13, 44. [Google Scholar] [CrossRef]
- Gadalla, E.K.K.; Ross, D.P.; Hector, D.R.; Bahey, G.N.; Bailey, M.E.S.; Cobb, R.S. Gene therapy for Rett syndrome: Prospects and challenges. Future Neurol. 2015, 10, 467–483. [Google Scholar] [CrossRef]
- Ta, D.; Downs, J.; Baynam, G.; Wilson, A.; Richmond, P.; Leonard, H. A brief history of MECP2 duplication syndrome: 20-years of clinical understanding. Orphanet. J. Rare Dis. 2022, 17, 131. [Google Scholar] [CrossRef]
- Peters, S.U.; Fu, C.; Marsh, E.D.; Benke, T.A.; Suter, B.; Skinner, S.A.; Lieberman, D.N.; Standridge, S.; Jones, M.; Beisang, A.; et al. Phenotypic features in MECP2 duplication syndrome: Effects of age. Am. J. Med. Genet. Part A 2021, 185, 362–369. [Google Scholar] [CrossRef]
- Sinnett, S.E.; Boyle, E.; Lyons, C.; Gray, S.J. Engineered microRNA-based regulatory element permits safe high-dose miniMECP2 gene therapy in Rett mice. Brain 2021, 144, 3005–3019. [Google Scholar] [CrossRef]
- Singh, J.; Ameenpur, S.; Ahmed, R.; Basheer, S.; Chishti, S.; Lawrence, R.; Fiori, F.; Santosh, P. An Observational Study of Heart Rate Variability Using Wearable Sensors Provides a Target for Therapeutic Monitoring of Autonomic Dysregulation in Patients with Rett Syndrome. Biomedicines 2022, 10, 1684. [Google Scholar] [CrossRef] [PubMed]
- Gualniera, L.; Singh, J.; Fiori, F.; Santosh, P. Emotional Behavioural and Autonomic Dysregulation (EBAD) in Rett Syndrome-EDA and HRV monitoring using wearable sensor technology. J. Psychiatr. Res. 2021, 138, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Lanzarini, E.; Santosh, P. Organic features of autonomic dysregulation in paediatric brain injury-Clinical and research implications for the management of patients with Rett syndrome. Neurosci. Biobehav. Rev. 2020, 118, 809–827. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Turner, T.J.; Zourray, C.; Schorge, S.; Lignani, G. Recent advances in gene therapy for neurodevelopmental disorders with epilepsy. J. Neurochem. 2021, 157, 229–262. [Google Scholar] [CrossRef]
- Singh, J.; Lanzarini, E.; Nardocci, N.; Santosh, P. Movement disorders in patients with Rett syndrome: A systematic review of evidence and associated clinical considerations. Psychiatry Clin. Neurosci. 2021, 75, 369–393. [Google Scholar] [CrossRef]
- Fumagalli, F.; Calbi, V.; Sora, M.G.N.; Sessa, M.; Baldoli, C.; Rancoita, P.M.V.; Ciotti, F.; Sarzana, M.; Fraschini, M.; Zambon, A.A.; et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: Long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 2022, 399, 372–383. [Google Scholar] [CrossRef]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef]
- Bougnères, P.; Hacein-Bey-Abina, S.; Labik, I.; Adamsbaum, C.; Castaignède, C.; Bellesme, C.; Schmidt, M. Long-Term Follow-Up of Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. Hum. Gene Ther. 2021, 32, 1260–1269. [Google Scholar] [CrossRef]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; De Oliveira, S.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Lehman, K.; Nease, C.; Lowes, L.P.; Miller, N.F.; Iammarino, M.A.; Alfano, L.N.; Nicholl, A.; Al-Zaidy, S.; et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020, 77, 1122–1131. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Chicoine, L.G.; Al-Zaidy, S.A.; Sahenk, Z.; Lehman, K.; Lowes, L.; Miller, N.; Alfano, L.; Galliers, B.; Lewis, S.; et al. Gene Delivery for Limb-Girdle Muscular Dystrophy Type 2D by Isolated Limb Infusion. Hum. Gene Ther. 2019, 30, 794–801. [Google Scholar] [CrossRef]
- Mendell, J.R.; Sahenk, Z.; Malik, V.; Gomez, A.M.; Flanigan, K.M.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Meadows, E.; Lewis, S.; et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol. Ther. 2015, 23, 192–201. [Google Scholar] [CrossRef]
- Al-Zaidy, S.A.; Kolb, S.J.; Lowes, L.; Alfano, L.N.; Shell, R.; Church, K.R.; Nagendran, S.; Sproule, D.M.; Feltner, D.E.; Wells, C.; et al. AVXS-101 (Onasemnogene Abeparvovec) for SMA1: Comparative Study with a Prospective Natural History Cohort. J. Neuromuscul. Dis. 2019, 6, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Al-Zaidy, S.; Shell, R.; Arnold, W.D.; Rodino-Klapac, L.R.; Prior, T.W.; Lowes, L.; Alfano, L.; Berry, K.; Church, K.; et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1713–1722. [Google Scholar] [CrossRef]
- Pasi, K.J.; Rangarajan, S.; Mitchell, N.; Lester, W.; Symington, E.; Madan, B.; Laffan, M.; Russell, C.B.; Li, M.; Pierce, G.F.; et al. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia, A. N. Engl. J. Med. 2020, 382, 29–40. [Google Scholar] [CrossRef]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.; Ribeil, J.-A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef]
- Ribeil, J.-A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, J.; Roscoe, B.P.; Liu, P.; Yao, Q.; Lazzarotto, C.R.; Clement, K.; Cole, M.A.; Luk, K.; Baricordi, C.; et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019, 25, 776–783. [Google Scholar] [CrossRef]
- Flotte, T.R.; Cataltepe, O.; Puri, A.; Batista, A.R.; Moser, R.; McKenna-Yasek, D.; Douthwright, C.; Gernoux, G.; Blackwood, M.; Mueller, C.; et al. AAV gene therapy for Tay-Sachs disease. Nat. Med. 2022, 28, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Tardieu, M.; Zérah, M.; Husson, B.; de Bournonville, S.; Deiva, K.; Adamsbaum, C.; Vincent, F.; Hocquemiller, M.; Broissand, C.; Furlan, V.; et al. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: Results of a phase I/II trial. Hum. Gene Ther. 2014, 25, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hu, C.; Moufawad El Achkar, C.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Barber, D.L.; Huang, J.; Rupar, C.A.; Rip, J.W.; Auray-Blais, C.; Boutin, M.; O’Hoski, P.; Gargulak, K.; McKillop, W.M.; et al. Lentivirus-mediated gene therapy for Fabry disease. Nat. Commun. 2021, 12, 1178. [Google Scholar] [CrossRef]
- Croci, S.; ML, C.; Capitani, K.; Daga, S.; Donati, F.; Frullanti, E.; Lamacchia, V.; Tita, R.; Giliberti, A.; Valentino, F.; et al. High rate of HDR in gene editing of p.(Thr158Met) MECP2 mutational hotspot. Eur. J. Hum. Genet. 2020, 28, 1231–1242. [Google Scholar] [CrossRef]
- Croci, S.; Carriero, M.L.; Capitani, K.; Daga, S.; Donati, F.; Papa, F.T.; Frullanti, E.; Lopergolo, D.; Lamacchia, V.; Tita, R.; et al. AAV-mediated FOXG1 gene editing in human Rett primary cells. Eur. J. Hum. Genet. 2020, 28, 1446–1458. [Google Scholar] [CrossRef]
- Andoh-Noda, T.; Akamatsu, W.; Miyake, K.; Matsumoto, T.; Yamaguchi, R.; Sanosaka, T.; Okada, Y.; Kobayashi, T.; Ohyama, M.; Nakashima, K.; et al. Differentiation of multipotent neural stem cells derived from Rett syndrome patients is biased toward the astrocytic lineage. Mol. Brain. 2015, 8, 31. [Google Scholar] [CrossRef]
- Adhikari, A.; A Copping, N.; Beegle, J.; Cameron, D.L.; Deng, P.; O’geen, H.; Segal, D.J.; Fink, K.D.; Silverman, J.L.; Anderson, J.S. Functional rescue in an Angelman syndrome model following treatment with lentivector transduced hematopoietic stem cells. Hum Mol Genet. 2021, 30, 1067–1083. [Google Scholar] [CrossRef]
- Wolter, J.M.; Mao, H.; Fragola, G.; Simon, J.M.; Krantz, J.L.; Bazick, H.O.; Oztemiz, B.; Stein, J.L.; Zylka, M.J. Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature 2020, 587, 281–284. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Bainbridge, J.W.; Mehat, M.S.; Sundaram, V.; Robbie, S.J.; Barker, S.E.; Ripamonti, C.; Georgiadis, A.; Mowat, F.M.; Beattie, S.G.; Gardner, P.J.; et al. Long-term effect of gene therapy on Leber′s congenital amaurosis. N. Engl. J. Med. 2015, 372, 1887–1897. [Google Scholar] [CrossRef] [PubMed]
- Ferrua, F.; Cicalese, M.P.; Galimberti, S.; Giannelli, S.; Dionisio, F.; Barzaghi, F.; Migliavacca, M.; Bernardo, M.E.; Calbi, V.; Assanelli, A.A.; et al. Lentiviral haemopoietic stem/progenitor cell gene therapy for treatment of Wiskott-Aldrich syndrome: Interim results of a non-randomised, open-label, phase 1/2 clinical study. Lancet Haematol. 2019, 6, e239–e253. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey Abina, S.; Gaspar, H.B.; Blondeau, J.; Caccavelli, L.; Charrier, S.; Buckland, K.; Picard, C.; Six, E.; Himoudi, N.; Gilmour, K.; et al. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA 2015, 313, 1550–1563. [Google Scholar] [CrossRef] [PubMed]
- Mueller, C.; Berry, J.D.; McKenna-Yasek, D.M.; Gernoux, G.; Owegi, M.A.; Pothier, L.M.; Douthwright, C.L.; Gelevski, D.; Luppino, S.D.; Blackwood, M.; et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020, 383, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H.; et al. Phase 1–2a IONIS-HTTRx Study Site Teams. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N. Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Bird, L.M.; Ochoa-Lubinoff, C.; Tan, W.-H.; Heimer, G.; Melmed, R.D.; Rakhit, A.; Visootsak, J.; During, M.J.; Holcroft, C.; Burdine, R.D.; et al. The STARS Phase 2 Study: A Randomized Controlled Trial of Gaboxadol in Angelman Syndrome. Neurology 2021, 96, e1024–e1035. [Google Scholar] [CrossRef] [PubMed]
- Copping, N.A.; McTighe, S.M.; Fink, K.D.; Silverman, J.L. Emerging Gene and Small Molecule Therapies for the Neurodevelopmental Disorder Angelman Syndrome. Neurotherapeutics 2021, 18, 1535–1547. [Google Scholar] [CrossRef]
- A Study of the Safety and Tolerability of GTX-102 in Children with Angelman Syndrome. Available online: https://clinicaltrials.gov/ct2/show/NCT04259281?term=antisense&cond=Angelman+Syndrome&draw=2&rank=1ClinicalTrials.govIdentifier:NCT05127226 (accessed on 21 February 2023).
- A Study to Investigate The Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of RO7248824 in Participants with Angelman Syndrome. Identifier: NCT04428281. Available online: https://clinicaltrials.gov/ct2/show/NCT04428281?cond=Angelman+Syndrome&draw=4&rank=5ClinicalTrials.gov (accessed on 21 February 2023).
- HALOS: A Safety, Tolerability, Pharmacokinetics and Pharmacodynamics Study of Multiple Ascending Doses of ION582 in Participants with Angelman Syndrome. Identifier: NCT05127226. Available online: https://clinicaltrials.gov/ct2/show/NCT05127226?cond=Angelman+Syndrome&draw=3&rank=16ClinicalTrials.gov (accessed on 21 February 2023).
- Collins, B.E.; Neul, J.L. Rett Syndrome and MECP2 Duplication Syndrome: Disorders of MeCP2 Dosage. Neuropsychiatr. Dis. Treat. 2022, 18, 2813–2835. [Google Scholar] [CrossRef]
- Collins, A.L.; Levenson, J.M.; Vilaythong, A.P.; Richman, R.; Armstrong, D.L.; Noebels, J.L.; Sweatt, J.D.; Zoghbi, H.Y. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum. Mol. Genet. 2004, 13, 2679–2689. [Google Scholar] [CrossRef]
- Sztainberg, Y.; Chen, H.-M.; Swann, J.W.; Hao, S.; Tang, B.; Wu, Z.; Tang, J.; Wan, Y.-W.; Liu, Z.; Rigo, F.; et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 2015, 528, 123–126. [Google Scholar] [CrossRef]
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P.; Specchio, N.; Aronica, E. Advances in the genetics and neuropathology of tuberous sclerosis complex: Edging closer to targeted therapy. Lancet Neurol. 2022, 21, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Cheah, P.-S.; Prabhakar, S.; Yellen, D.; Beauchamp, R.L.; Zhang, X.; Kasamatsu, S.; Bronson, R.T.; Thiele, E.A.; Kwiatkowski, D.J.; Stemmer-Rachamimov, A.; et al. Gene therapy for tuberous sclerosis complex type 2 in a mouse model by delivery of AAV9 encoding a condensed form of tuberin. Sci. Adv. 2021, 7, eabb1703. [Google Scholar] [CrossRef]
- Eichmüller, O.L.; Corsini, N.S.; Vértesy, Á.; Morassut, I.; Scholl, T.; Gruber, V.-E.; Peer, A.M.; Chu, J.; Novatchkova, M.; Hainfellner, J.A.; et al. Amplification of human interneuron progenitors promotes brain tumors and neurological defects. Science 2022, 375, eabf5546. [Google Scholar] [CrossRef] [PubMed]
- Novartis Terminates Their Rett Syndrome Gene Replacement Program. Available online: https://reverserett.org/news/articles/novartis-terminates-their-rett-syndrome-gene-replacement-program/ (accessed on 21 February 2023).
- Safety and Efficacy of TSHA-102 in Adult Females with Rett Syndrome (REVEAL Adult Study). Available online: https://clinicaltrials.gov/ct2/show/NCT05606614?term=gene+therapy&cond=Rett+Syndrome&draw=2&rank=1ClinicalTrials.govIdentifier:NCT05606614 (accessed on 21 February 2023).
- Neurogene Announces FDA Clearance of IND for NGN-401 Gene Therapy for Children with Rett Syndrome. Available online: https://www.neurogene.com/press-releases/neurogene-announces-fda-clearance-of-ind-for-ngn-401-gene-therapy-for-children-with-rett-syndrome/ (accessed on 21 February 2023).
- Glascock, J.; Sampson, J.; Haidet-Phillips, A.; Connolly, A.; Darras, B.; Day, J.; Finkel, R.; Howell, R.R.; Klinger, K.; Kuntz, N.; et al. Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening. J. Neuromuscul. Dis. 2018, 5, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; A Chiriboga, C.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Bishop, K.M.; Foster, R.; Liu, Y.; Ramirez-Schrempp, D.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: Final report of a phase 2, open-label, multicentre, dose-escalation study. Lancet Child Adolesc. Health 2021, 5, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. ENDEAR Study Group. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. P.381-Nusinersen demonstrates greater efficacy in infants with shorter disease duration: End of study results from the ENDEAR study in infants with spinal muscular atrophy (SMA). Neuromuscul. Disord. 2017, 27 (Suppl 2), S211. [Google Scholar]
- Darras, B.T.; Chiriboga, C.A.; Iannaccone, S.T.; Swoboda, K.J.; Montes, J.; Mignon, L.; Xia, S.; Bennett, C.F.; Bishop, K.M.; Shefner, J.M.; et al. Nusinersen in later-onset spinal muscular atrophy: Long-term results from the phase 1/2 studies. Neurology 2019, 92, e2492–e2506. [Google Scholar] [CrossRef]
- Hagenacker, T.; Wurster, C.D.; Günther, R.; Schreiber-Katz, O.; Osmanovic, A.; Petri, S.; Weiler, M.; Ziegler, A.; Kuttler, J.; Koch, J.C.; et al. Nusinersen in adults with 5q spinal muscular atrophy: A non-interventional, multicentre, observational cohort study. Lancet Neurol. 2020, 19, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.C.; Wenninger, S.; Thiele, S.; Stauber, J.; Hiebeler, M.; Greckl, E.; Stahl, K.; Pechmann, A.; Lochmüller, H.; Kirschner, J.; et al. Safety and Treatment Effects of Nusinersen in Longstanding Adult 5q-SMA Type 3-A Prospective Observational Study. J. Neuromuscul. Dis. 2019, 6, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Han, S.T.; Hirt, A.; Nicoli, E.; Kono, M.; Toro, C.; Proia, R.L.; Tifft, C.J. Gene expression changes in Tay-Sachs disease begin early in fetal brain development. J. Inherit. Metab. Dis. 2023. [Google Scholar] [CrossRef] [PubMed]
- Sonzogni, M.; Zhai, P.; Mientjes, E.J.; Van Woerden, G.M.; Elgersma, Y. Assessing the requirements of prenatal UBE3A expression for rescue of behavioral phenotypes in a mouse model for Angelman syndrome. Mol. Autism. 2020, 11, 70. [Google Scholar] [CrossRef] [PubMed]
- Braz, B.Y.; Wennagel, D.; Ratié, L.; de Souza, D.A.R.; Deloulme, J.C.; Barbier, E.L.; Buisson, A.; Lanté, F.; Humbert, S. Treating early postnatal circuit defect delays Huntington′s disease onset and pathology in mice. Science 2022, 377, eabq5011. [Google Scholar] [CrossRef]
- Achilly, N.P.; Wang, W.; Zoghbi, H.Y. Presymptomatic training mitigates functional deficits in a mouse model of Rett syndrome. Nature 2021, 592, 596–600. [Google Scholar] [CrossRef]
- Newborn Blood Spot Test. Available online: https://www.nhs.uk/conditions/baby/newborn-screening/blood-spot-test/ (accessed on 25 February 2023).
- Morton, G.; Thomas, S.; Roberts, P.; Clark, V.; Imrie, J.; Morrison, A. The importance of early diagnosis and views on newborn screening in metachromatic leukodystrophy: Results of a Caregiver Survey in the UK and Republic of Ireland. Orphanet. J. Rare Dis. 2022, 17, 403. [Google Scholar] [CrossRef]
- England Rare Diseases Action Plan 2022, Department of Health and Social Care, February 2022. Available online: https://www.gov.uk/government/publications/england-rare-diseases-action-plan-2022 (accessed on 25 February 2023).
- Hopkins, H.; Kinsella, S.; Evans, G. A Findings Report, Hopkins Van Mil, July Implications of Whole Genome Sequencing for Newborn Screening. A Public Dialogue. Available online: https://www.gov.uk/government/publications/implications-of-whole-genome-sequencing-for-newborn-screening (accessed on 25 February 2023).
- Watson, P.; Black, G.; Ramsden, S.; Barrow, M.; Super, M.; Kerr, B.; Clayton-Smith, J. Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J. Med. Genet. 2001, 38, 224–228. [Google Scholar] [CrossRef]
- Curie, A.; Lesca, G.; Bussy, G.; Manificat, S.; Arnaud, V.; Gonzalez, S.; Revol, O.; Calender, A.; Gérard, D.; Portes, V.D. Asperger syndrome and early-onset schizophrenia associated with a novel MECP2 deleterious missense variant. Psychiatr. Genet. 2017, 27, 105–109. [Google Scholar] [CrossRef]
- Suter, B.; Treadwell-Deering, D.; Zoghbi, H.Y.; Glaze, D.G.; Neul, J.L. Brief report: MECP2 mutations in people without Rett syndrome. J. Autism Dev. Disord. 2014, 44, 703–711. [Google Scholar] [CrossRef]
- Maortua, H.; Martínez-Bouzas, C.; García-Ribes, A.; Martínez, M.-J.; Guillen, E.; Domingo, M.-R.; Calvo, M.-T.; Guitart, M.; Gabau, E.; Botella, M.-P.; et al. MECP2 gene study in a large cohort: Testing of 240 female patients and 861 healthy controls (519 females and 342 males). J. Mol. Diagn. 2013, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Ylisaukko-Oja, T.; Rehnström, K.; Vanhala, R.; Kempas, E.; von Koskull, H.; Tengström, C.; Mustonen, A.; Ounap, K.; Lähdetie, J.; Järvelä, I. MECP2 mutation analysis in patients with mental retardation. Am. J. Med. Genet. Part A 2005, 132A, 121–124. [Google Scholar] [CrossRef]
- Dragoumi, P.; O′Callaghan, F.; Zafeiriou, D.I. Diagnosis of tuberous sclerosis complex in the fetus. Eur. J. Paediatr. Neurol. 2018, 22, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, B.; Prohl, A.K.; Taquet, M.; Kapur, K.; Peters, J.M.; Tomas-Fernandez, X.; E Davis, P.; Bebin, E.M.; A Krueger, D.; Northrup, H.; et al. The Connectivity Fingerprint of the Fusiform Gyrus Captures the Risk of Developing Autism in Infants with Tuberous Sclerosis Complex. Cereb. Cortex. 2020, 30, 2199–2214. [Google Scholar] [CrossRef]
- Prohl, A.K.; Scherrer, B.; Tomas-Fernandez, X.; Davis, P.E.; Filip-Dhima, R.; Prabhu, S.P.; Peters, J.M.; Bebin, E.M.; Krueger, D.A.; Northrup, H. TACERN Study Group. Early white matter development is abnormal in tuberous sclerosis complex patients who develop autism spectrum disorder. J. Neurodev. Disord. 2019, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Hulshof, H.M.; Slot, E.M.; Lequin, M.; Breuillard, D.; Boddaert, N.; Jozwiak, S.; Kotulska, K.; Riney, K.; Feucht, M.; Samueli, S.; et al. EPISTOP consortium. Fetal Brain Magnetic Resonance Imaging Findings Predict Neurodevelopment in Children with Tuberous Sclerosis Complex. J. Pediatr. 2021, 233, 156–162.e2. [Google Scholar] [CrossRef]
- Pierpont, E.I.; Eisengart, J.B.; Shanley, R.; Nascene, D.; Raymond, G.V.; Shapiro, E.G.; Ziegler, R.S.; Orchard, P.J.; Miller, W.P. Neurocognitive Trajectory of Boys Who Received a Hematopoietic Stem Cell Transplant at an Early Stage of Childhood Cerebral Adrenoleukodystrophy. JAMA Neurol. 2017, 74, 710–717. [Google Scholar] [CrossRef]
- Li, D.; Mei, L.; Li, H.; Hu, C.; Zhou, B.; Zhang, K.; Qiao, Z.; Xu, X.; Xu, Q. Brain structural alterations in young girls with Rett syndrome: A voxel-based morphometry and tract-based spatial statistics study. Front. Neuroinform. 2022, 16, 962197. [Google Scholar] [CrossRef]
- Kong, Y.; Li, Q.-B.; Yuan, Z.-H.; Jiang, X.-F.; Zhang, G.-Q.; Cheng, N.; Dang, N. Multimodal Neuroimaging in Rett Syndrome With MECP2 Mutation. Front. Neurol. 2022, 13, 838206. [Google Scholar] [CrossRef]
- Sharifi, O.; Yasui, D.H. The Molecular Functions of MeCP2 in Rett Syndrome Pathology. Front. Genet. 2021, 12, 624290. [Google Scholar] [CrossRef]
- Marín, O. Developmental timing and critical windows for the treatment of psychiatric disorders. Nat. Med. 2016, 22, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Bosch, F.; Miksa, M.; Bittigau, P.; Vockler, J.; Dikranian, K.; Tenkova, T.I.; Stefovska, V.; Turski, L.; Olney, J.W. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 1999, 283, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front. Aging. Neurosci. 2020, 11, 373. [Google Scholar] [CrossRef]
- Jensen, T.L.; Gøtzsche, C.R.; Woldbye, D.P.D. Current and Future Prospects for Gene Therapy for Rare Genetic Diseases Affecting the Brain and Spinal Cord. Front. Mol. Neurosci. 2021, 14, 695937. [Google Scholar] [CrossRef] [PubMed]
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood-brain barrier opening in Alzheimer′s disease using MR-guided focused ultrasound. Nat. Commun. 2018, 9, 2336. [Google Scholar] [CrossRef]
- Gasca-Salas, C.; Fernández-Rodríguez, B.; Pineda-Pardo, J.A.; Rodríguez-Rojas, R.; Obeso, I.; Hernández-Fernández, F.; del Álamo, M.; Mata, D.; Guida, P.; Ordás-Bandera, C.; et al. Blood-brain barrier opening with focused ultrasound in Parkinson′s disease dementia. Nat. Commun. 2021, 12, 779. [Google Scholar] [CrossRef]
- Porges, S.W.; Furman, S.A. The Early Development of the Autonomic Nervous System Provides a Neural Platform for Social Behavior: A Polyvagal Perspective. Infant Child Dev. 2011, 20, 106–118. [Google Scholar] [CrossRef]
- Sachis, P.N.; Armstrong, D.L.; Becker, L.E.; Bryan, A.C. Myelination of the human vagus nerve from 24 weeks postconceptional age to adolescence. J. Neuropathol. Exp. Neurol. 1982, 41, 466–472. [Google Scholar] [CrossRef]
- Nguyen, M.V.; Felice, C.A.; Du, F.; Covey, M.V.; Robinson, J.K.; Mandel, G.; Ballas, N. Oligodendrocyte lineage cells contribute unique features to Rett syndrome neuropathology. J. Neurosci. 2013, 33, 18764–18774. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; Zhang, H.; Feng, S.; Lu, Y.; Wang, S.; Wang, H.; Sun, Y.E.; Chen, Y. White Matter Structural and Network Topological Changes Underlying the Behavioral Phenotype of MECP2 Mutant Monkeys. Cereb. Cortex. 2021, 31, 5396–5410. [Google Scholar] [CrossRef]
- Pejhan, S.; Siu, V.M.; Ang, L.C.; Del Bigio, M.R.; Rastegar, M. Differential brain region-specific expression of MeCP2 and BDNF in Rett Syndrome patients: A distinct grey-white matter variation. Neuropathol. Appl. Neurobiol. 2020, 46, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Petazzi, P.; Jorge-Torres, O.C.; Gomez, A.; Scognamiglio, I.; Serra-Musach, J.; Merkel, A.; Grases, D.; Xiol, C.; O’callaghan, M.; Armstrong, J.; et al. Global Impairment of Immediate-Early Genes Expression in Rett Syndrome Models and Patients Linked to Myelination Defects. Int. J. Mol. Sci. 2023, 24, 1453. [Google Scholar] [CrossRef] [PubMed]
- Phan, B.N.; Bohlen, J.F.; Davis, B.A.; Ye, Z.; Chen, H.Y.; Mayfield, B.; Sripathy, S.R.; Cerceo Page, S.; Campbell, M.N.; Smith, H.L. A myelin-related transcriptomic profile is shared by Pitt-Hopkins syndrome models and human autism spectrum disorder. Nat. Neurosci. 2020, 23, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.S.; Markov, V.; Wojtas, I.D.; Gray, S.; McCown, T.; Samulski, R.J.; Figueroa, M.; Leone, P. Preclinical biodistribution, tropism, and efficacy of oligotropic AAV/Olig001 in a mouse model of congenital white matter disease. Mol. Ther. Methods Clin. Dev. 2021, 20, 520–534. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.-L.; Sanchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; E Deverman, B.; et al. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Luoni, M.; Giannelli, S.; Indrigo, M.T.; Niro, A.; Massimino, L.; Iannielli, A.; Passeri, L.; Russo, F.; Morabito, G.; Calamita, P.; et al. Whole brain delivery of an instability-prone Mecp2 transgene improves behavioral and molecular pathological defects in mouse models of Rett syndrome. eLife 2020, 9, e52629. [Google Scholar] [CrossRef] [PubMed]
- Le, T.T.H.; Tran, N.T.; Dao, T.M.L.; Nguyen, D.D.; Do, H.D.; Ha, T.L.; Kühn, R.; Nguyen, T.L.; Rajewsky, K.; Chu, V.T. Efficient and Precise CRISPR/Cas9-Mediated MECP2 Modifications in Human-Induced Pluripotent Stem Cells. Front. Genet. 2019, 10, 625. [Google Scholar] [CrossRef]
- Bak, R.O.; Porteus, M.H. CRISPR-Mediated Integration of Large Gene Cassettes Using AAV Donor Vectors. Cell Rep. 2017, 20, 750–756. [Google Scholar] [CrossRef]
- Javed, S.; Selliah, T.; Lee, Y.J.; Huang, W.H. Dosage-sensitive genes in autism spectrum disorders: From neurobiology to therapy. Neurosci. Biobehav. Rev. 2020, 118, 538–567. [Google Scholar] [CrossRef]
- Bajikar, S.S.; Anderson, A.G.; Zhou, J.; Durham, M.A.; Trostle, A.J.; Wan, Y.W.; Liu, Z.; Zoghbi, H.Y. MeCP2 regulates Gdf11, a dosage-sensitive gene critical for neurological function. eLife 2023, 12, e83806. [Google Scholar] [CrossRef]
- Koerner, M.V.; FitzPatrick, L.; Selfridge, J.; Guy, J.; De Sousa, D.; Tillotson, R.; Kerr, A.; Sun, Z.; Lazar, M.A.; Lyst, M.J.; et al. Toxicity of overexpressed MeCP2 is independent of HDAC3 activity. Genes Dev. 2018, 32, 1514–1524. [Google Scholar] [CrossRef] [PubMed]
- Jugloff, D.G.; Vandamme, K.; Logan, R.; Visanji, N.P.; Brotchie, J.M.; Eubanks, J.H. Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2-deficient mice. Hum. Mol. Genet. 2008, 17, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Matagne, V.; Borloz, E.; Ehinger, Y.; Saidi, L.; Villard, L.; Roux, J.C. Severe off target effects following intravenous delivery of AAV9-MECP2 in a female mouse model of Rett syndrome. Neurobiol. Dis. 2021, 149, 105235. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.E.; Merritt, J.K.; Erickson, K.R.; Neul, J.L. Safety and efficacy of genetic MECP2 supplementation in the R294X mouse model of Rett syndrome. Genes Brain Behav. 2022, 21, e12739. [Google Scholar] [CrossRef]
- Chen, X.; Lim, D.A.; Lawlor, M.W.; Dimmock, D.; Vite, C.H.; Lester, T.; Tavakkoli, F.; Sadhu, C.; Prasad, S.; Gray, S.J. Biodistribution of Adeno-Associated Virus Gene Therapy Following Cerebrospinal Fluid-Directed Administration. Hum. Gene Ther. 2023, 34, 94–111. [Google Scholar] [CrossRef]
- Benatti, H.R.; Gray-Edwards, H.L. Adeno-Associated Virus Delivery Limitations for Neurological Indications. Hum. Gene Ther. 2022, 33, 1–7. [Google Scholar] [CrossRef]
- Petriti, U.; Dudman, D.C.; Scosyrev, E.; Lopez-Leon, S. Global prevalence of Rett syndrome: Systematic review and meta-analysis. Syst. Rev. 2023, 12, 5. [Google Scholar] [CrossRef]
- Grimm, N.B.; Lee, J.T. Selective Xi reactivation and alternative methods to restore MECP2 function in Rett syndrome. Trends Genet. 2022, 38, 920–943. [Google Scholar] [CrossRef]
- Sinnamon, J.R.; Jacobson, M.E.; Yung, J.F.; Fisk, J.R.; Jeng, S.; McWeeney, S.K.; Parmelee, L.K.; Chan, C.N.; Yee, S.-P.; Mandel, G. Targeted RNA editing in brainstem alleviates respiratory dysfunction in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. USA 2022, 119, e2206053119. [Google Scholar] [CrossRef]
- Qian, J.; Guan, X.; Xie, B.; Xu, C.; Niu, J.; Tang, X.; Li, C.H.; Colecraft, H.M.; Jaenisch, R.; Liu, X.S. Multiplex epigenome editing of MECP2 to rescue Rett syndrome neurons. Sci. Transl. Med. 2023, 15, eadd4666. [Google Scholar] [CrossRef]
- Biogen and Alcyone Therapeutics Announce License and Collaboration Agreement to Evaluate a Novel Device to Improve Patient Experience and Access to Neurological ASO Therapies. Available online: https://alcyonetx.com/pipeline-programs/actx-101/ (accessed on 20 March 2023).
- Neul, J.L.; Percy, A.K.; Benke, T.A.; Berry-Kravis, E.M.; Glaze, D.G.; Peters, S.U.; Jones, N.E.; Youakim, J.M. Design and outcome measures of LAVENDER, a phase 3 study of trofinetide for Rett syndrome. Contemp. Clin. Trials 2022, 114, 106704. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.E.; Sprouse, J.; Rebowe, N.; Hanania, T.; Klamer, D.; Missling, C.U. ANAVEX®2-73 (blarcamesine), a Sigma-1 receptor agonist, ameliorates neurologic impairments in a mouse model of Rett syndrome. Pharmacol. Biochem. Behav. 2019, 187, 172796. [Google Scholar] [CrossRef] [PubMed]
- Onasemnogene Abeparvovec for Treating Spinal Muscular Atrophy. Available online: https://www.nice.org.uk/guidance/hst15 (accessed on 10 March 2023).
- Atidarsagene Autotemcel for Treating Metachromatic Leukodystrophy. Available online: https://www.nice.org.uk/guidance/hst18 (accessed on 10 March 2023).
- Eladocagene Exuparvovec for Treating Aromatic L-Amino Acid Decarboxylase Deficiency. Available online: https://www.nice.org.uk/guidance/hst26 (accessed on 29 March 2023).
- Nusinersen for Treating Spinal Muscular Atrophy. Available online: https://www.nice.org.uk/guidance/ta588 (accessed on 10 March 2023).
- Qiu, Y.; O’neill, N.; Maffei, B.; Zourray, C.; Almacellas-Barbanoj, A.; Carpenter, J.C.; Jones, S.P.; Leite, M.; Turner, T.J.; Moreira, F.C.; et al. On-demand cell-autonomous gene therapy for brain circuit disorders. Science 2022, 378, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Swen, J.J.; van der Wouden, C.H.; Manson, L.E.; Abdullah-Koolmees, H.; Blagec, K.; Blagus, T.; Böhringer, S.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.-C.; et al. Ubiquitous Pharmacogenomics Consortium. A 12-gene pharmacogenetic panel to prevent adverse drug reactions: An open-label, multicentre, controlled, cluster-randomised crossover implementation study. Lancet 2023, 401, 347–356. [Google Scholar] [CrossRef]
- Chu, W.S.; Ng, J. Immunomodulation in Administration of rAAV: Preclinical and Clinical Adjuvant Pharmacotherapies. Front. Immunol. 2021, 12, 658038. [Google Scholar] [CrossRef] [PubMed]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Suresha, P.B.; O′Leary, H.; Tarquinio, D.C.; Von Hehn, J.; Clifford, G.D. Rett syndrome severity estimation with the BioStamp nPoint using interactions between heart rate variability and body movement. PLoS ONE 2023, 18, e0266351. [Google Scholar] [CrossRef]
- Singh, J.; Fiori, F.; Law, M.L.; Ahmed, R.; Ameenpur, S.; Basheer, S.; Chishti, S.; Lawrence, R.; Mastroianni, M.; Mosaddegh, A.; et al. Development and Psychometric Properties of the Multi-System Profile of Symptoms Scale in Patients with Rett Syndrome. J. Clin. Med. 2022, 11, 5094. [Google Scholar] [CrossRef]
- Arabi, F.; Mansouri, V.; Ahmadbeigi, N. Gene therapy clinical trials, where do we go? An overview. Biomed. Pharmacother. 2022, 153, 113324. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Source | Characteristics/Demographics | Clinical Characteristics | Assessment Methods | Relevant Information |
|---|---|---|---|---|
| [27] Fumagalli et al. (2022) | Long-term follow up of lentivirus haematopoietic stem cell gene therapy in patients with metachromatic leukodystrophy Intention to treat set:
Natural history cohort (untreated control group):
|
| Primary endpoints included: Improvements (>10%) in (i) total scores of the gross motor function measure (GMFM-88) at 2 years after treatment and (ii) change in baseline of total peripheral blood mononuclear cell ARSA activity after 2 years |
|
| [28] Sessa et al. (2016) |
|
|
|
|
| [29] Biffi et al. (2013) |
|
|
|
|
| [30] Bougnères et al. (2021) |
|
|
|
|
| [31] Eichler et al. (2017) |
|
|
|
|
| [32] Mendell et al. (2020) | Randomized controlled trial of 4 patients (boys) aged 6, 5 and 4 years |
|
|
|
| [33] Mendell et al. (2019) | Isolated limb infusion (ILI) delivery at ascending doses of an AAV (scAAVrh74.tMCK.hSGCA) in six patients aged between 8 and 49 years with limb-girdle muscular dystrophy type 2D (LGMD2D) |
|
|
|
| [34] Mendell et al. (2015) |
|
|
|
|
| [35] Al-Zaidy et al. (2019) |
| Patients received a therapeutic dose of AVXS-101 and compared to age and gender matched cohorts of a natural history SMA 1 cohort and healthy controls | Outcome measures included event free survival, CHOP-INTEND scores, motor milestones muscle action potentials (CMAP) and adverse events |
|
| [36] Mendell et al. (2017) |
| All patients had a confirmed diagnosis of SMA1 |
|
|
| [37] Pasi et al. (2020) | AAV (AAV5-hFVIII-SQ) mediated gene therapy follow-up in adults with haemophilia A | Patients were males (n = 15) with severe haemophilia A and received a single infusion of AAV5-hFVIII-SQ at different dose levels | Parameters such as factor VIII levels, rate of bleeding events, safety and tolerability were followed for up to 3 years |
|
| [38] Thompson et al. (2018) |
| Patients with any genotype for β-thalassemia and had at least eight transfusions or 100 mL/kg of body weight packed red cells in the last 24 months were eligible for study enrolment | Assessment included monitoring of adverse events, lentivirus integration, levels of total haemoglobin and lentivirus marked β-globin |
|
| [39] Ribeil et al. (2017) |
|
| Outcomes were safety including MRI, changes in sickle cell disease related measures, engraftment and gene expression levels |
|
| [40] Wu et al. (2019) | Study assessing gene editing in haemopoietic stem cells | Hematopoietic stem cells were isolated from a patient with Sickle cell disease and a patient with β-thalassemia |
|
|
| [41] Flotte et al. (2022) |
|
|
|
|
| [42] Tardieu et al. (2014) |
|
|
|
|
| [43] Kim et al. (2019) | Proof of concept (n = 1) study in a 6-year girl with neuronal ceroid lipofuscinosis 7 (CLN7) |
| Neurological and neuropsychological assessments including the Vineland Adaptive Behaviour Scales, assessments for global motor function and seizure frequency |
|
| [44] Khan et al. (2021) |
|
|
|
|
| [45] Croci et al. (2020) | Characterisation of human neuronal model from Rett Syndrome (RTT) derived fibroblasts and iPSC-derived neurons |
| Gene editing CRISPR/Cas9 toolkit to correct the mutation in patient derived primary cells |
|
| [46] Croci et al. (2020) | Gene editing in human RTT primary cells |
| AAV mediated CRISPR/Cas9 targeting of FOXG1 variants in patient derived fibroblasts, iPSCs and iPSC-derived neurons |
|
| [47] Andoh-Noda et al. (2015) |
|
|
|
|
| [48] Adhikari et al. (2021) | Study evaluating an Angelman syndrome model using Ube3a expressing lentivector in haemopoietic stem cells to deliver functional UBE3A | Human CD34+ haemopoietic stem cells were obtained from umbilical cord blood |
|
|
| [49] Wolter et al. (2020) | Study exploring Cas9-mediated gene therapy for Angelman syndrome | Primary human neural progenitor-derived (phNPC) neurons from fetal brain tissue | To evaluate UBE3A expression and whether Cas9-mediated targeting of SNORD115 genes can unsilence paternal Ube3a in differentiated phNPC neurons |
|
| [50] Russell, et al. (2017) |
|
|
|
|
| [51] Bainbridge et al. (2015) |
|
|
|
|
| [52] Ferrua et al. (2019) | Lentivirus haemopoietic stem cell gene therapy in eight children with Wiskott–Aldrich syndrome (WAS) |
|
|
|
| [53] Hacein-Bey Abina et al. (2015) |
| All patients had a single infusion of lentivirus modified CD34+ cells |
|
|
| [54] Mueller, et al. (2020) | Case study of two patients (patient 1: 22-year-old and patient 2: 56-year-old) with familial amyotrophic lateral sclerosis (ALS) |
|
|
|
| [55] Tabrizi et al. (2019) |
|
|
|
|
| Disorder | Key Findings | Gene Therapy or Gene Editing Considerations for Individuals with Rett Syndrome (RTT) |
|---|---|---|
| Metachromatic Leukodystrophy (MLD) |
|
|
| Cerebral Adrenoleukodystrophy |
|
|
| Mucopolysaccharidosis Type IIIA (MPS Type IIIA) |
|
|
| Huntington’s disease |
|
|
| Tay–Sachs disease (TSD) |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, J.; Goodman-Vincent, E.; Santosh, P. Evidence Synthesis of Gene Therapy and Gene Editing from Different Disorders—Implications for Individuals with Rett Syndrome: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 9023. https://doi.org/10.3390/ijms24109023
Singh J, Goodman-Vincent E, Santosh P. Evidence Synthesis of Gene Therapy and Gene Editing from Different Disorders—Implications for Individuals with Rett Syndrome: A Systematic Review. International Journal of Molecular Sciences. 2023; 24(10):9023. https://doi.org/10.3390/ijms24109023
Chicago/Turabian StyleSingh, Jatinder, Ella Goodman-Vincent, and Paramala Santosh. 2023. "Evidence Synthesis of Gene Therapy and Gene Editing from Different Disorders—Implications for Individuals with Rett Syndrome: A Systematic Review" International Journal of Molecular Sciences 24, no. 10: 9023. https://doi.org/10.3390/ijms24109023