
Vitamin A Promotes the Fusion of Autophagolysosomes and Prevents Excessive Inflammasome Activation in Dextran Sulfate Sodium-Induced Colitis

 , , , , and
, , , , and 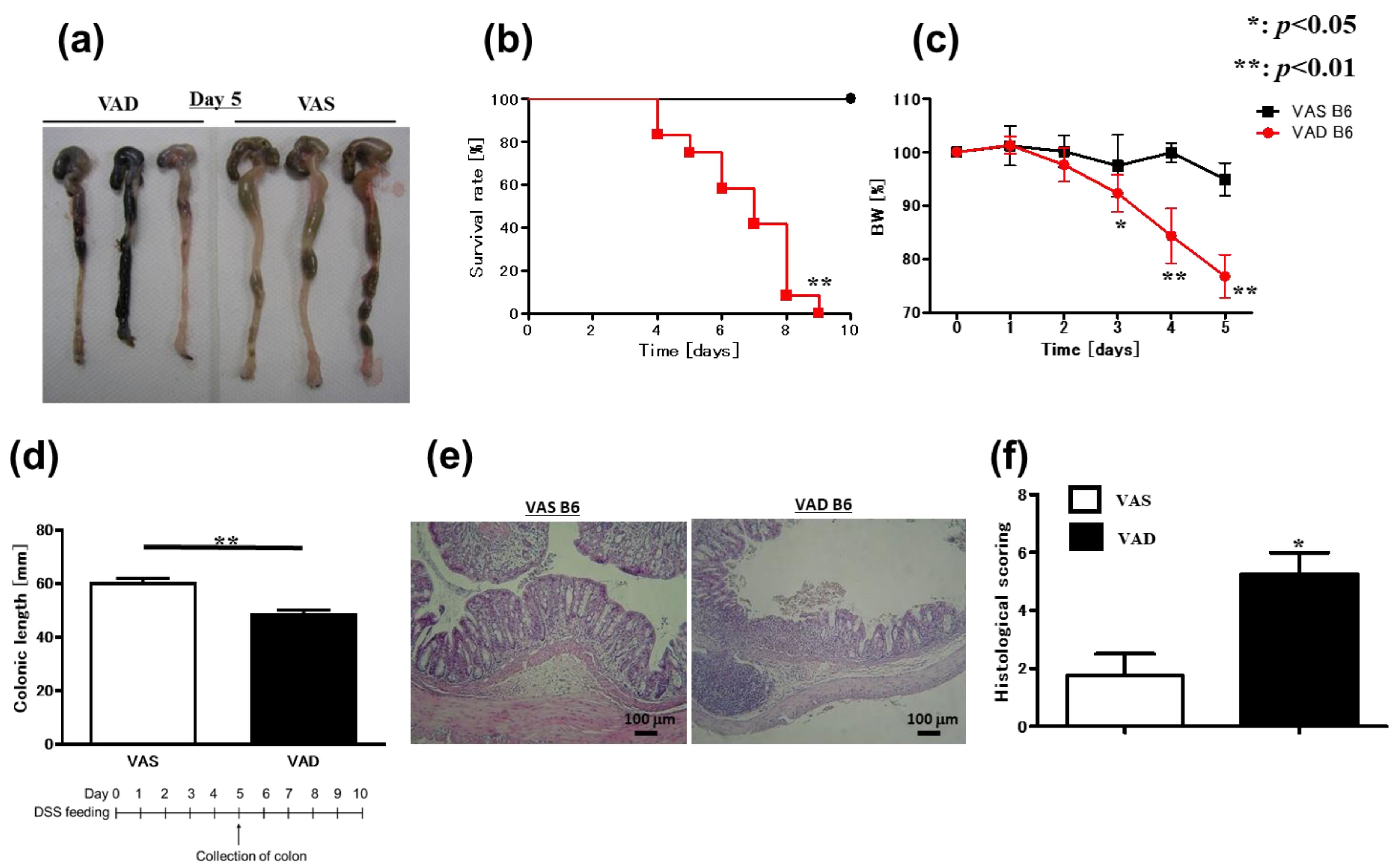{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
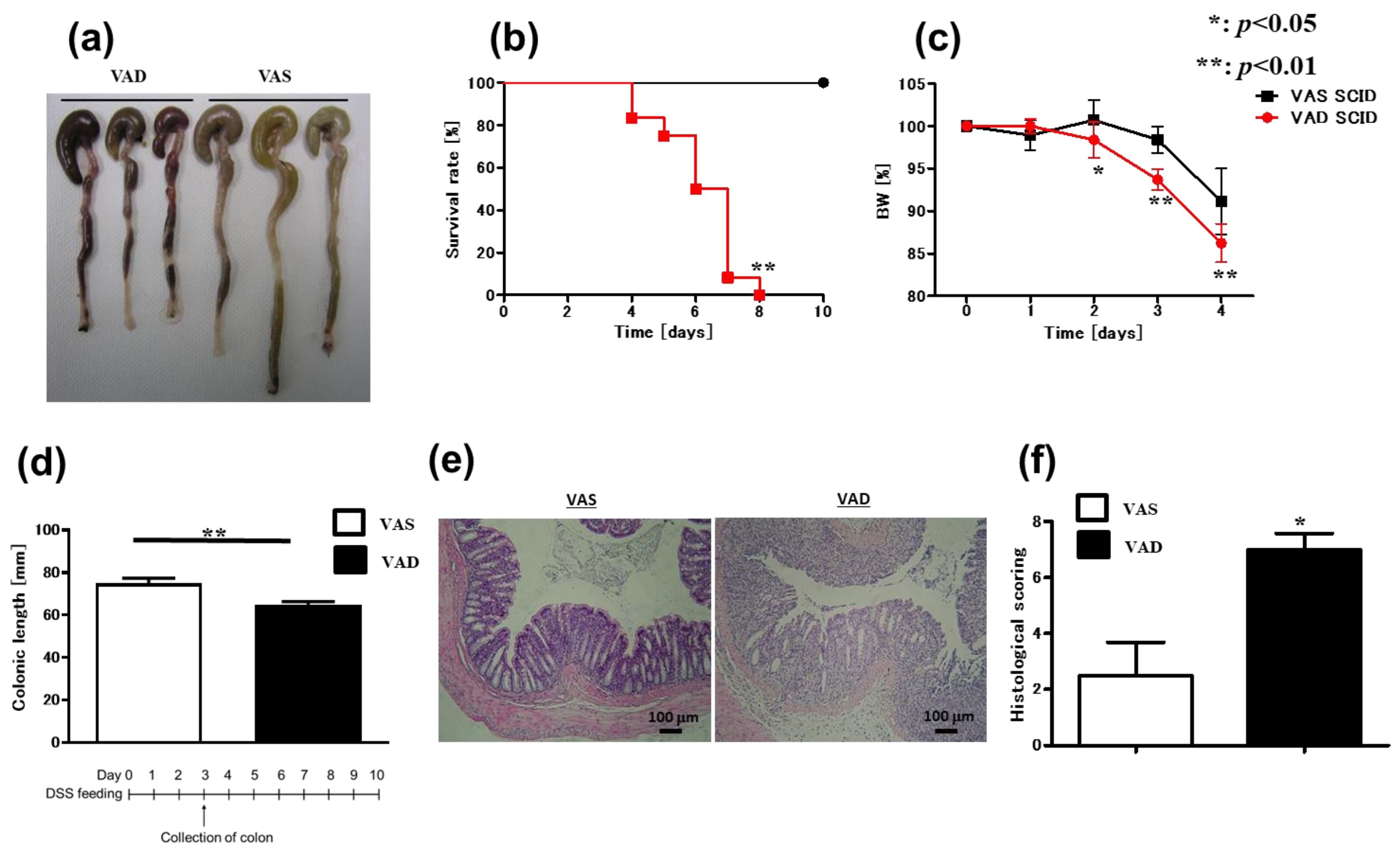
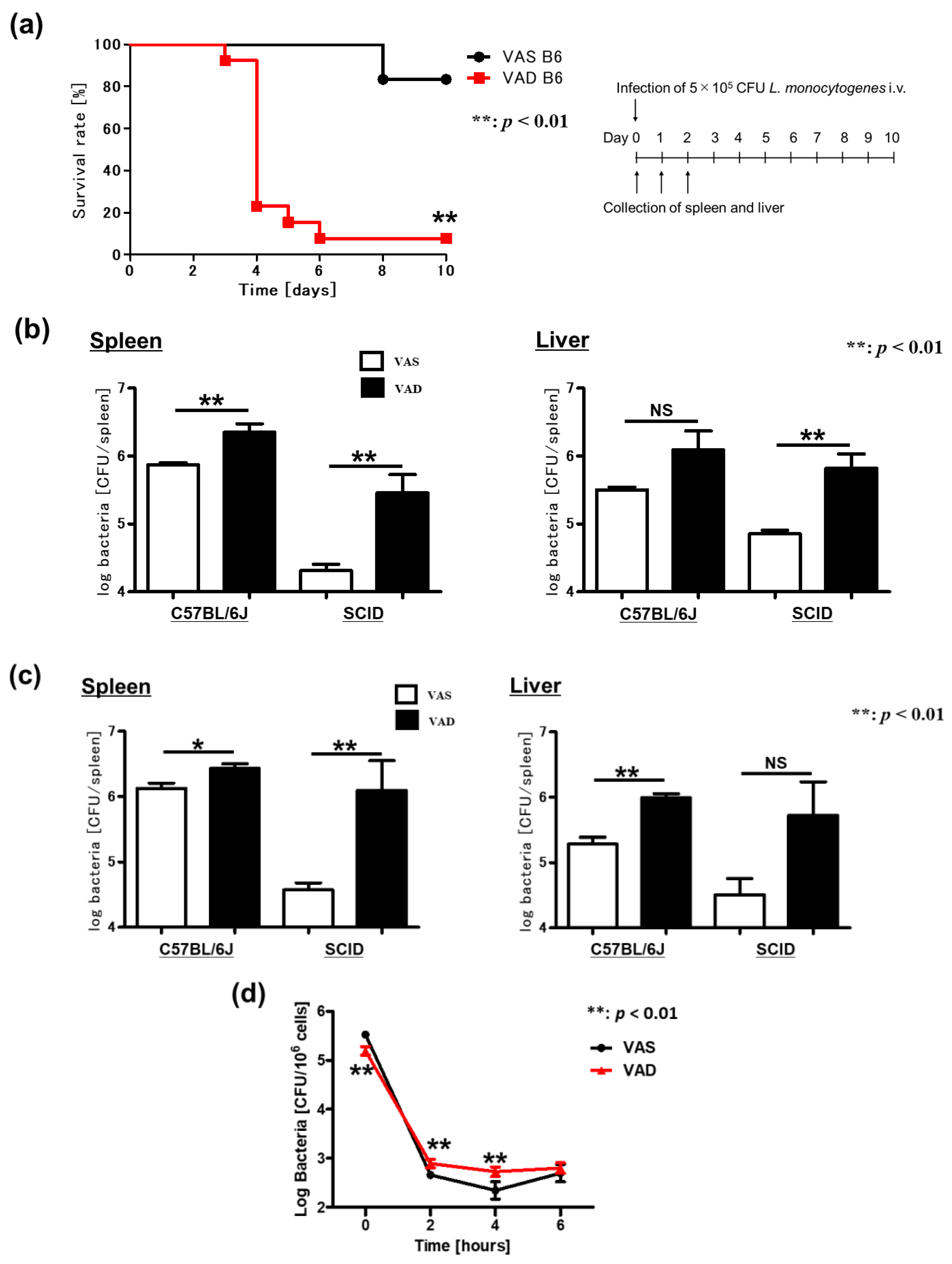
2.1. Vitamin A-Deficient (VAD) Mice Exhibit More Severe DSS-Induced Colitis Independent of T and B Cells
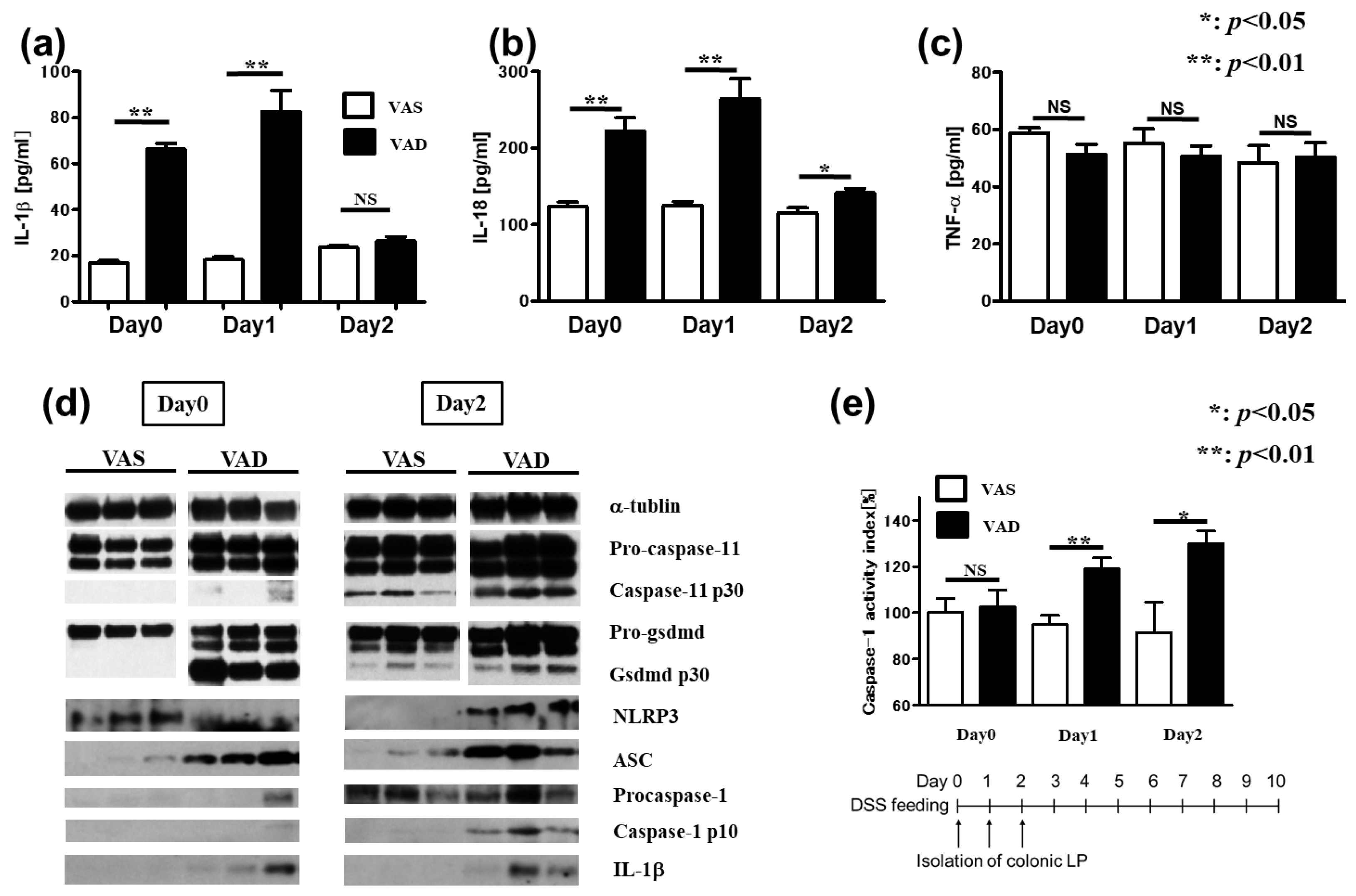
2.2. Inflammasome-Related Protein Levels Increase in the Colonic LP of VAD Mice
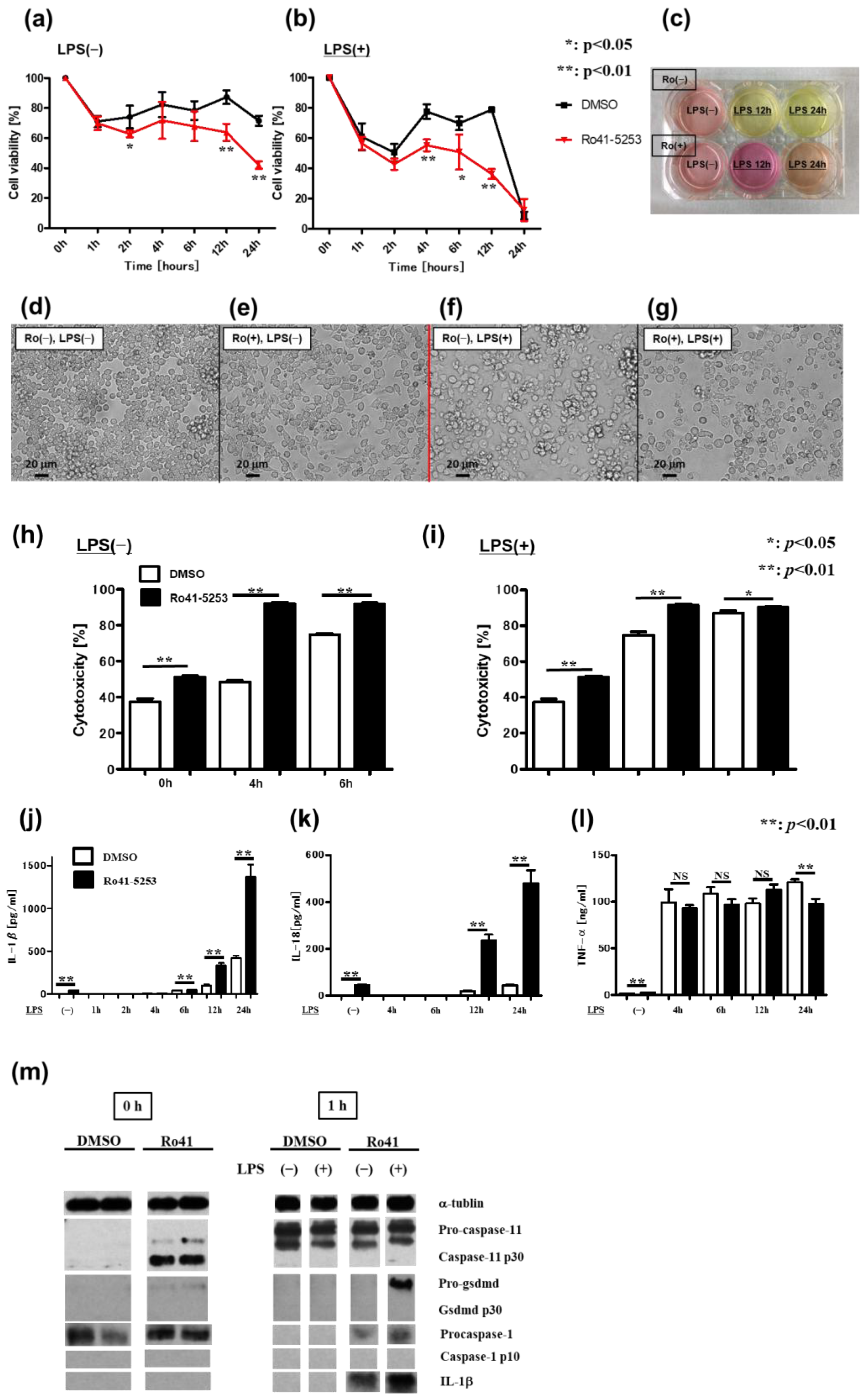
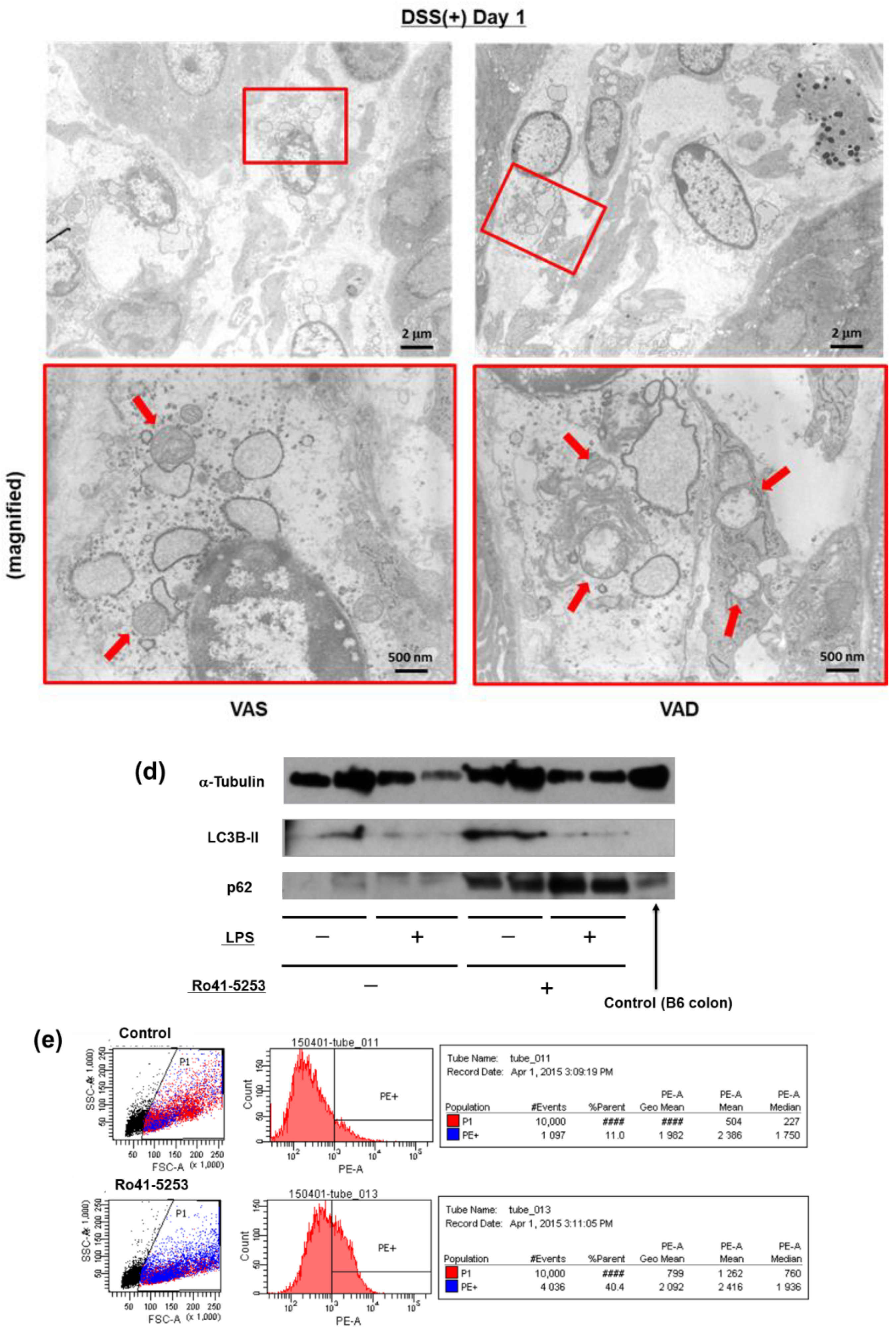
2.3. Pyroptosis via Non-Canonical Inflammasome Signaling Increases in Macrophages Pretreated with a RAR Antagonist
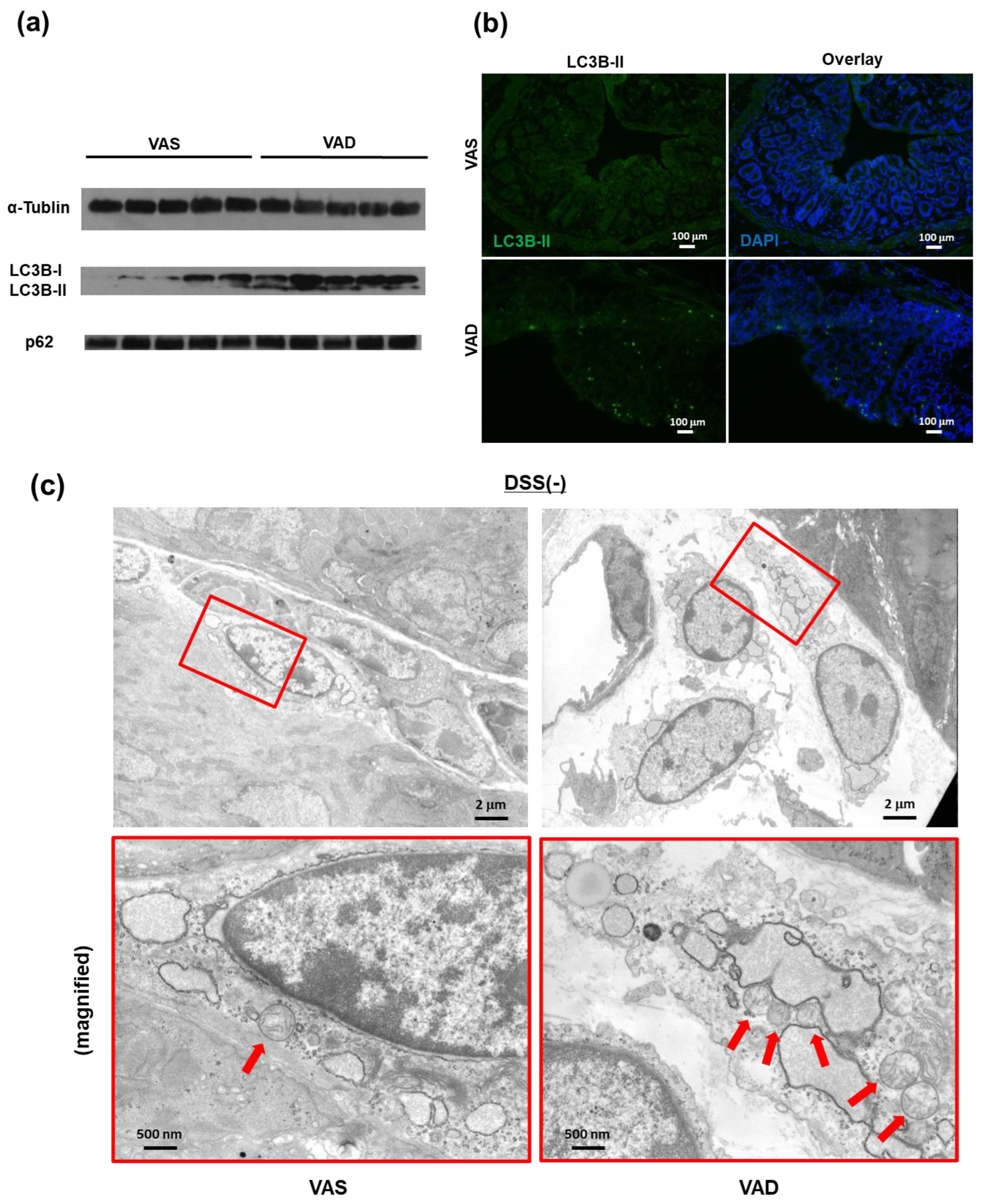
2.4. Vitamin A Deficiency Decreases Autophagic Flux via Reduced Fusion of Autophagosomes and Lysosomes
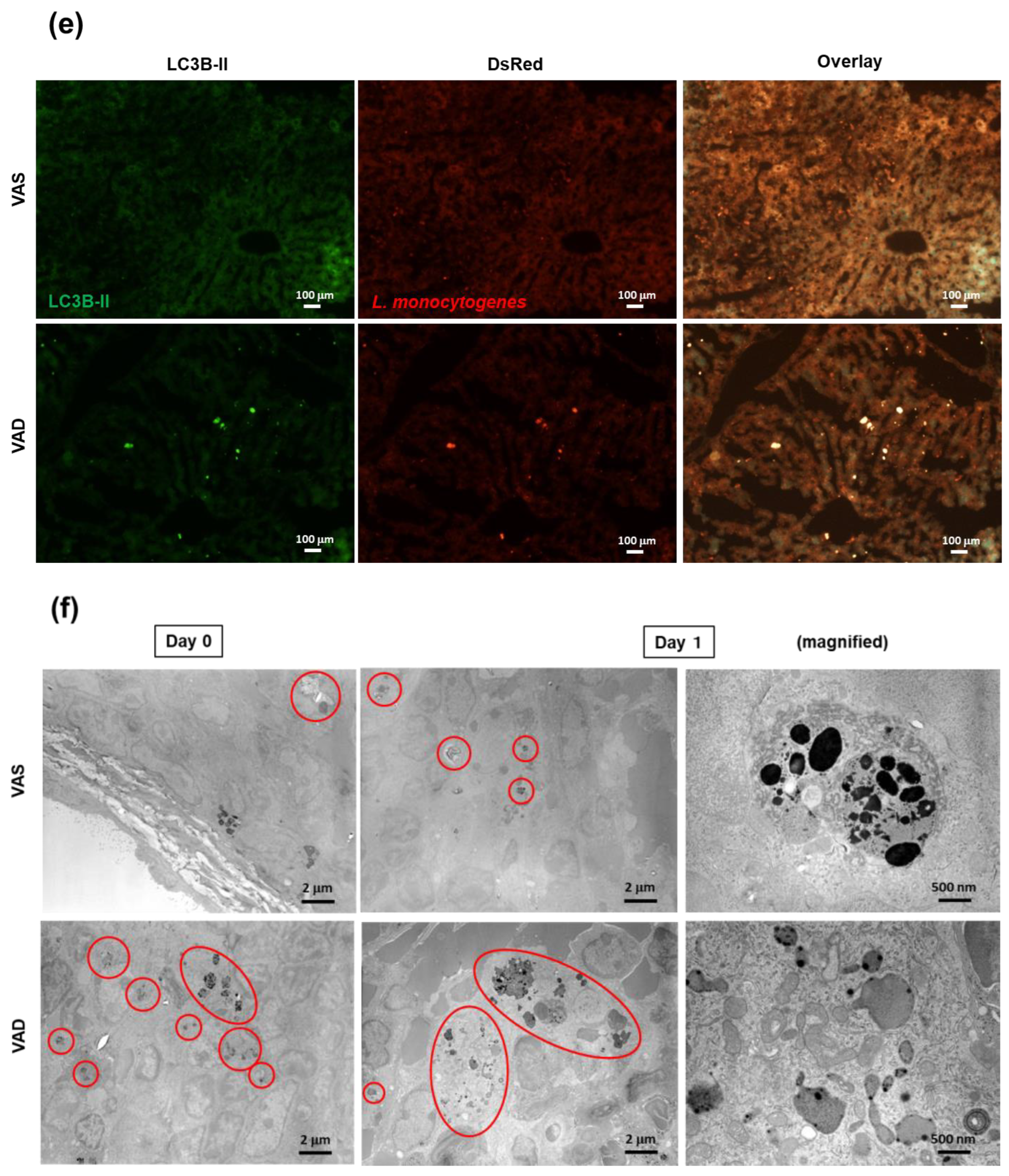
2.5. VAD Impairs the Autophagic and Listericidal Activities of Macrophages, Promoting Increased Susceptibility to Listeria Monocytogenes Infection
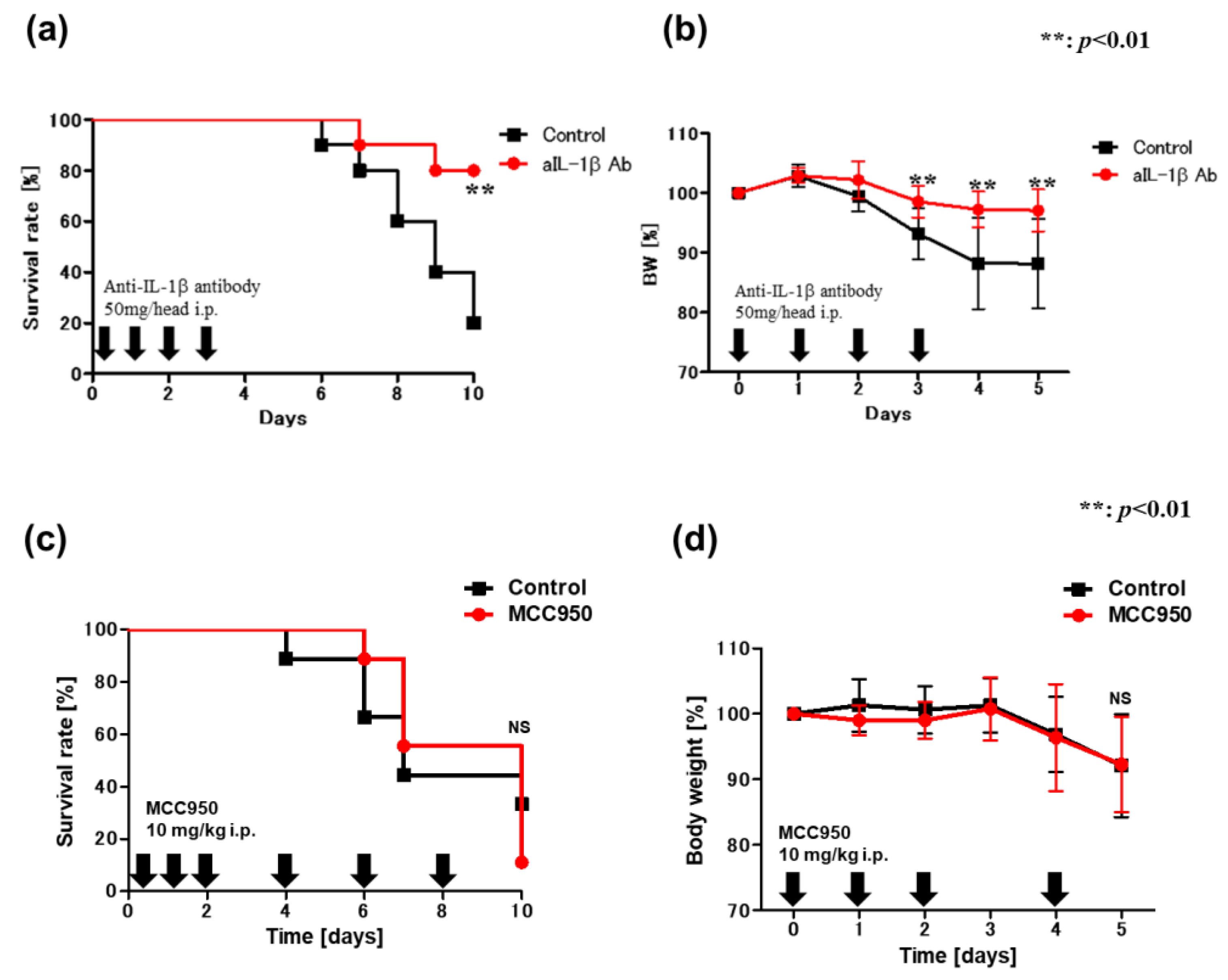
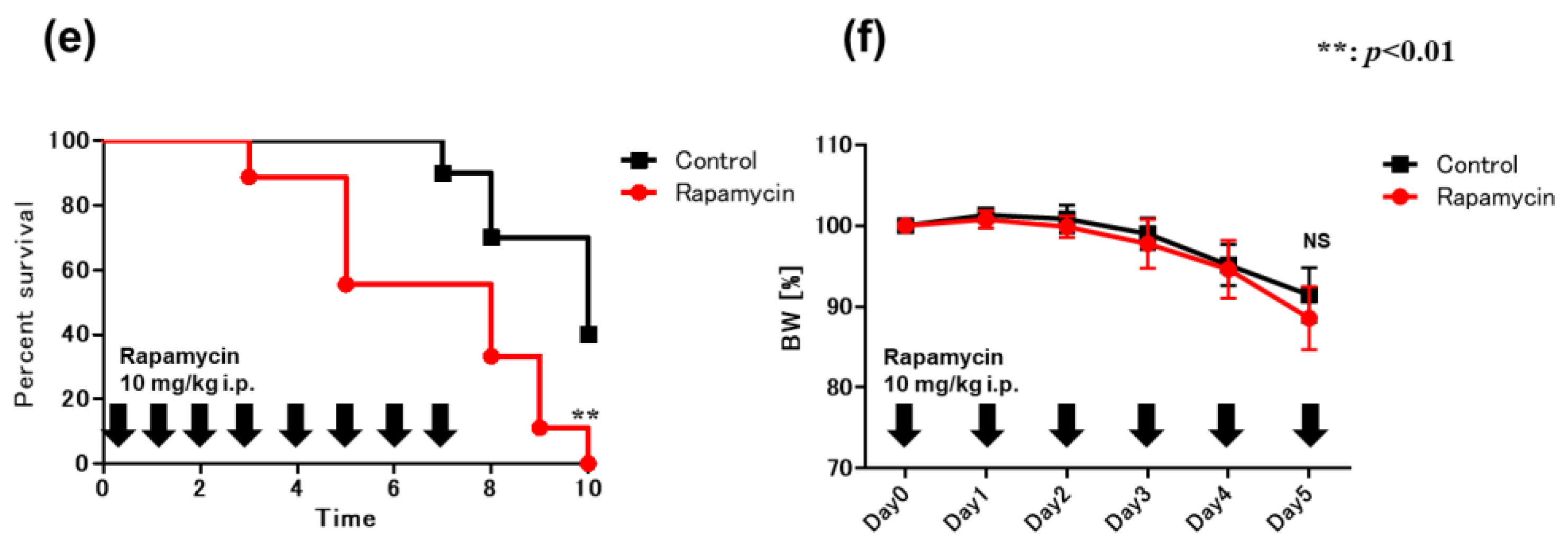
2.6. Blockade of IL-1β Ameliorates DSS-Induced Colitis in VAD Mice
3. Discussion
4. Materials and Methods
4.1. Preparation of VAD Mice
4.2. Induction of Colitis
4.3. Isolation of Colonic LP
4.4. Western Blotting and Caspase Activity
4.5. Bio-Plex
4.6. Histological Assessment of Colon Sections
4.7. Cell Culture
4.8. Cytotoxicity Assay
4.9. Flow Cytometric Analysis
4.10. Bacterial Strains, Plasmids, and Growth Conditions
4.11. Infection of Mice with L. Monocytogenes
4.12. Preparation of Splenic Macrophages and Bactericidal Activity
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kastner, T.; Walsh, K.K. The Optimal Evaluation and Treatment of Feeding Disorders in Children with Neurological Disorders. J. Am. Coll. Surg. 1995, 181, 383–386. [Google Scholar] [PubMed]
- Napoli, J.L. Retinoic Acid: Its Biosynthesis and Metabolism. Prog. Nucleic Acid Res. Mol. Biol. 1999, 63, 139–188. [Google Scholar] [CrossRef] [PubMed]
- Giguere, V.; Ong, E.S.; Segui, P.; Evans, R.M. Identification of a Receptor for the Morphogen Retinoic Acid. Nature 1987, 330, 624–629. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Ong, E.S.; Dyck, J.A.; Evans, R.M. Nuclear Receptor That Identifies a Novel Retinoic Acid Response Pathway. Nature 1990, 345, 224–229. [Google Scholar] [CrossRef]
- Petkovich, M.; Brand, N.J.; Krust, A.; Chambon, P. A Human Retinoic Acid Receptor Which Belongs to the Family of Nuclear Receptors. Nature 1987, 330, 444–450. [Google Scholar] [CrossRef] [PubMed]
- West, K.P., Jr.; Howard, G.R.; Sommer, A. Vitamin A and Infection: Public Health Implications. Annu. Rev. Nutr. 1989, 9, 63–86. [Google Scholar] [CrossRef]
- Iwata, M.; Hirakiyama, A.; Eshima, Y.; Kagechika, H.; Kato, C.; Song, S.Y. Retinoic Acid Imprints Gut-Homing Specificity on T Cells. Immunity 2004, 21, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.C.; Stephensen, C.B. Vitamin A and Retinoids in Antiviral Responses. FASEB J. 1996, 10, 979–985. [Google Scholar] [CrossRef]
- Wiedermann, U.; Hanson, L.A.; Bremell, T.; Kahu, H.; Dahlgren, U.I. Increased Translocation of Escherichia coli and Development of Arthritis in Vitamin A-Deficient Rats. Infect. Immun. 1995, 63, 3062–3068. [Google Scholar] [CrossRef]
- Twining, S.S.; Schulte, D.P.; Wilson, P.M.; Fish, B.L.; Moulder, J.E. Vitamin A Deficiency Alters Rat Neutrophil Function. J. Nutr. 1997, 127, 558–565. [Google Scholar] [CrossRef]
- Ongsakul, M.; Sirisinha, S.; Lamb, A.J. Impaired Blood Clearance of Bacteria and Phagocytic Activity in Vitamin A-Deficient Rats. Proc. Soc. Exp. Biol. Med. 1985, 178, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Dawson, H.D.; Ross, A.C. Chronic Marginal Vitamin A Status Affects the Distribution and Function of T Cells and Natural T Cells in Aging Lewis Rats. J. Nutr. 1999, 129, 1782–1790. [Google Scholar] [CrossRef] [PubMed]
- Stephensen, C.B. Vitamin A, Infection, and Immune Function. Annu. Rev. Nutr. 2001, 21, 167–192. [Google Scholar] [CrossRef]
- Kamada, N.; Hisamatsu, T.; Okamoto, S.; Sato, T.; Matsuoka, K.; Arai, K.; Nakai, T.; Hasegawa, A.; Inoue, N.; Watanabe, N.; et al. Abnormally Differentiated Subsets of Intestinal Macrophage Play a Key Role in Th1-Dominant Chronic Colitis through Excess Production of IL-12 and IL-23 in Response to Bacteria. J. Immunol. 2005, 175, 6900–6908. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.P.; Mee, A.S.; Parfitt, A.; Marks, I.N.; Burns, D.G.; Sherman, M.; Tigler-Wybrandi, N.; Isaacs, S. Vitamin A Therapy in Patients with Crohn’s Disease. Gastroenterology 1985, 88, 512–514. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nakahigashi, M.; Umegae, S.; Kitagawa, T.; Matsumoto, K. Impact of Elemental Diet on Mucosal Inflammation in Patients with Active Crohn’s Disease: Cytokine Production and Endoscopic and endoscopic and Histological Findings. Inflamm. Bowel Dis. 2005, 11, 580–588. [Google Scholar] [CrossRef]
- Aachoui, Y.; Leaf, I.A.; Hagar, J.A.; Fontana, M.F.; Campos, C.G.; Zak, D.E.; Tan, M.H.; Cotter, P.A.; Vance, R.E.; Aderem, A.; et al. Caspase-11 Protects Against Bacteria that Escape the Vacuole. Science 2013, 339, 975–978. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 Increases Susceptibility to Salmonella Infection in the Absence of Caspase-1. Nature 2012, 490, 288–291. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical Inflammasome Activation Targets Caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Vanaja, S.K.; Waggoner, L.; Sokolovska, A.; Becker, C.; Stuart, L.M.; Leong, J.M.; Fitzgerald, K.A. TRIF Licenses Caspase-11-Dependent NLRP3 Inflammasome Activation by Gram-Negative Bacteria. Cell 2012, 150, 606–619. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS Activates Caspase-11: Implications in TLR4-Independent Endotoxic Shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 Cleaves Gasdermin D for Non-canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential Activation of the Inflammasome by Caspase-1 Adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin Activates the Inflammasome in Response to Toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e6. [Google Scholar] [CrossRef]
- Ligumsky, M.; Simon, P.L.; Karmeli, F.; Rachmilewitz, D. Role of Interleukin 1 in Inflammatory Bowel Disease--Enhanced Production during Active Disease. Gut 1990, 31, 686–689. [Google Scholar] [CrossRef]
- Flood, B.; Oficjalska, K.; Laukens, D.; Fay, J.; O’Grady, A.; Caiazza, F.; Heetun, Z.; Mills, K.H.; Sheahan, K.; Ryan, E.J.; et al. Altered Expression of caspases-4 and −5 During Inflammatory Bowel Disease and Colorectal Cancer: Diagnostic and Therapeutic Potential. Clin. Exp. Immunol. 2015, 181, 39–50. [Google Scholar] [CrossRef]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A Genome-Wide Association Scan of Nonsynonymous SNPs Identifies a Susceptibility Variant for Crohn Disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1beta Production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Ozen, S.; Bilginer, Y. A Clinical Guide to Autoinflammatory Diseases: Familial Mediterranean Fever and Next-of-Kin. Nat. Rev. Rheumatol. 2014, 10, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.J.; Cho, Y.H.; Lee, G.S.; Cheng, J.; Liu, P.P.; Feigenbaum, L.; Katz, S.I.; Kastner, D.L. Gain-of-Function Pyrin Mutations Induce NLRP3 Protein-Independent Interleukin-1beta Activation and Severe Autoinflammation in Mice. Immunity 2011, 34, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Malik, A.; Guy, C.S.; Karki, R.; Vogel, P.; Kanneganti, T.D. Pyrin Inflammasome Regulates Tight Junction Integrity to Restrict Colitis and Tumorigenesis. Gastroenterology 2018, 154, 948–964.e8. [Google Scholar] [CrossRef]
- Arasawa, S.; Nakase, H.; Ozaki, Y.; Uza, N.; Matsuura, M.; Chiba, T. Mediterranean Mimicker. Lancet 2012, 380, 2052. [Google Scholar] [CrossRef]
- Dieleman, L.A.; Ridwan, B.U.; Tennyson, G.S.; Beagley, K.W.; Bucy, R.P.; Elson, C.O. Dextran Sulfate Sodium-Induced Colitis Occurs in Severe Combined Immunodeficient Mice. Gastroenterology 1994, 107, 1643–1652. [Google Scholar] [CrossRef]
- Mahida, Y.R.; Wu, K.; Jewell, D.P. Enhanced Production of Interleukin 1-Beta by Mononuclear Cells Isolated from Mucosa with Active Ulcerative Colitis of Crohn’s Disease. Gut 1989, 30, 835–838. [Google Scholar] [CrossRef]
- Pizarro, T.T.; Michie, M.H.; Bentz, M.; Woraratanadharm, J.; Smith, M.F.; Foley, E.; Moskaluk, C.A.; Bickston, S.J.; Cominelli, F. IL-18, a Novel Immunoregulatory Cytokine, Is Up-Regulated in Crohn’s Disease: Expression and Localization in Intestinal Mucosal Cells. J. Immunol. 1999, 162, 6829–6835. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Gründler, T.; Quednau, N.; Stump, C.; Orian-Rousseau, V.; Ishikawa, H.; Wolburg, H.; Schroten, H.; Tenenbaum, T.; Schwerk, C. The Surface Proteins InlA and InlB Are Interdependently Required for Polar Basolateral Invasion by Listeria monocytogenes in a Human Model of the Blood-Cerebrospinal Fluid Barrier. Microbes Infect. 2013, 15, 291–301. [Google Scholar] [CrossRef]
- Gluschko, A.; Herb, M.; Wiegmann, K.; Krut, O.; Neiss, W.F.; Utermöhlen, O.; Krönke, M.; Schramm, M. The beta2 Integrin Mac-1 Induces Protective LC3-Associated Phagocytosis of Listeria monocytogenes. Cell Host Microbe 2018, 23, 324–337.e5. [Google Scholar] [CrossRef] [PubMed]
- Phelps, C.C.; Vadia, S.; Arnett, E.; Tan, Y.; Zhang, X.; Pathak-Sharma, S.; Gavrilin, M.A.; Seveau, S. Relative Roles of Listeriolysin O, InlA, and InlB in Listeria monocytogenes Uptake by Host Cells. Infect. Immun. 2018, 86, e00555-18. [Google Scholar] [CrossRef] [PubMed]
- Wiedermann, U.; Tarkowski, A.; Bremell, T.; Hanson, L.A.; Kahu, H.; Dahlgren, U.I. Vitamin A Deficiency Predisposes to Staphylococcus aureus Infection. Infect. Immun. 1996, 64, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Fux, B.; Goodwin, M.; Dunay, I.R.; Strong, D.; Miller, B.C.; Cadwell, K.; Delgado, M.A.; Ponpuak, M.; Green, K.G.; et al. Autophagosome-Independent Essential Function for the Autophagy Protein Atg5 in Cellular Immunity to Intracellular Pathogens. Cell Host Microbe 2008, 4, 458–469. [Google Scholar] [CrossRef]
- Yano, T.; Mita, S.; Ohmori, H.; Oshima, Y.; Fujimoto, Y.; Ueda, R.; Takada, H.; Goldman, W.E.; Fukase, K.; Silverman, N.; et al. Autophagic Control of Listeria through Intracellular Innate Immune Recognition in Drosophila. Nat. Immunol. 2008, 9, 908–916. [Google Scholar] [CrossRef]
- Carter, J.D.; Valeriano, J.; Vasey, F.B. Crohn Disease Worsened by Anakinra Administration. J. Clin. Rheumatol. 2003, 9, 276–277. [Google Scholar] [CrossRef]
- Kaly, L.; Rozenbaum, M.; Rimar, D.; Slobodin, G.; Boulman, N.; Awisat, A.; Ginsberg, S.; Jiries, N.; Rosner, I. Ulcerative Colitis and Familial Mediterranean Fever: Can Anakinra Treat Both? ACG Case Rep. J. 2019, 6, e00143. [Google Scholar] [CrossRef]
- Okayasu, I.; Hana, K.; Nemoto, N.; Yoshida, T.; Saegusa, M..; Yokota-Nakatsuma, A.; Song, S.Y.; Iwata, M. Vitamin A Inhibits Development of Dextran Sulfate Sodium-Induced Colitis and Colon Cancer in a Mouse Model. Biomed. Res. Int. 2016, 2016, 4874809. [Google Scholar] [CrossRef]
- Reifen, R.; Nur, T.; Ghebermeskel, K.; Zaiger, G.; Urizky, R.; Pines, M. Vitamin A Deficiency Exacerbates Inflammation in a Rat Model of Colitis through Activation of Nuclear Factor-kappaB and Collagen Formation. J. Nutr. 2002, 132, 2743–2747. [Google Scholar] [CrossRef]
- Hong, K.; Zhang, Y.; Guo, Y.; Xie, J.; Wang, J.; He, X.; Lu, N.; Bai, A. All-Trans Retinoic Acid Attenuates Experimental Colitis through Inhibition of NF-kappaB Signaling. Immunol. Lett. 2014, 162, 34–40. [Google Scholar] [CrossRef]
- Bauer, C.; Duewell, P.; Mayer, C.; Lehr, H.A.; Fitzgerald, K.A.; Dauer, M.; Tschopp, J.; Endres, S.; Latz, E.; Schnurr, M. Colitis Induced in Mice with Dextran Sulfate Sodium (DSS) Is Mediated by the NLRP3 Inflammasome. Gut 2010, 59, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Chen, W.; Wang, Y.; Chen, C.; Guo, L.; Ju, R.; Li, J.; Zhang, D.; Zhu, L.; Ye, C. Therapeutic Efficacy of Carboxyamidotriazole on 2,4,6-Trinitrobenzene Sulfonic Acid-Induced Colitis Model Is Associated with the Inhibition of NLRP3 Inflammasome and NF-κB Activation. Int. Immunopharmacol. 2017, 45, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Liu, X.; Zhang, X.; Tang, J.; Li, Z.; Wang, Q.; Hu, R. Oroxylin A Inhibits Colitis by Inactivating NLRP3 Inflammasome. Oncotarget 2017, 8, 58903–58917. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 Inflammasome Protects Against Loss of Epithelial Integrity and Mortality during Experimental Colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, G.; Trapasso, F.; Parrello, T.; Biancone, L.; Stella, A.; Iuliano, R.; Luzza, F.; Fusco, A.; Pallone, F. Bioactive IL-18 Expression Is Up-Regulated in Crohn’s Disease. J. Immunol. 1999, 163, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Demon, D.; Kuchmiy, A.; Fossoul, A.; Zhu, Q.; Kanneganti, T.D.; Lamkanfi, M. Caspase-11 Is Expressed in the Colonic Mucosa and Protects Against Dextran Sodium Sulfate-Induced Colitis. Mucosal Immunol. 2014, 7, 1480–1491. [Google Scholar] [CrossRef]
- Oficjalska, K.; Raverdeau, M.; Aviello, G.; Wade, S.C.; Hickey, A.; Sheehan, K.M.; Corr, S.C.; Kay, E.W.; O’Neill, L.A.; Mills, K.H.; et al. Protective Role for Caspase-11 during Acute Experimental Murine Colitis. J. Immunol. 2015, 194, 1252–1260. [Google Scholar] [CrossRef]
- Bulek, K.; Zhao, J.; Liao, Y.; Rana, N.; Corridoni, D.; Antanaviciute, A.; Chen, X.; Wang, H.; Qian, W.; Miller-Little, W.A.; et al. Epithelial-Derived Gasdermin D Mediates Nonlytic IL-1beta Release during Experimental Colitis. J. Clin. Investig. 2020, 130, 4218–4234. [Google Scholar] [CrossRef]
- Ma, C.; Yang, D.; Wang, B.; Wu, C.; Wu, Y.; Li, S.; Liu, X.; Lassen, K.; Dai, L.; Yang, S. Gasdermin D in Macrophages Restrains Colitis by Controlling cGAS-Mediated Inflammation. Sci. Adv. 2020, 6, eaaz6717. [Google Scholar] [CrossRef]
- Takahama, M.; Akira, S.; Saitoh, T. Autophagy Limits Activation of the Inflammasomes. Immunol. Rev. 2018, 281, 62–73. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of proIL-Beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.; Caution, K.; Badr, A.; Hamilton, K.; Saleh, A.; Patel, K.; Seveau, S.; Hall-Stoodley, L.; Hegazi, R.; Zhang, X.; et al. CASP4/caspase-11 Promotes Autophagosome Formation in Response to Bacterial Infection. Autophagy 2018, 14, 1928–1942. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.J.; Mellouk, N.; Osborne, S.E.; Ammendolia, D.A.; Dyer, D.N.; Li, R.; Brunen, D.; van Rijn, J.M.; Huang, J.; Czuczman, M.A.; et al. An ATG16L1-Dependent Pathway Promotes Plasma Membrane Repair and Limits Listeria monocytogenes Cell-to-Cell Spread. Nat. Microbiol. 2018, 3, 1472–1485. [Google Scholar] [CrossRef] [PubMed]
- Lassen, K.G.; McKenzie, C.I.; Mari, M.; Murano, T.; Begun, J.; Baxt, L.A.; Goel, G.; Villablanca, E.J.; Kuo, S.Y.; Huang, H.; et al. Genetic Coding Variant in GPR65 Alters Lysosomal pH and Links Lysosomal Dysfunction with Colitis Risk. Immunity 2016, 44, 1392–1405. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical Inflammasome Signaling Elicits Gasdermin D-Dependent Neutrophil Extracellular Traps. Sci. Immunol. 2018, 3, eaar6676. [Google Scholar] [CrossRef]
- Zanoni, I.; Tan, Y.; Di Gioia, M.; Broggi, A.; Ruan, J.; Shi, J.; Donado, C.A.; Shao, F.; Wu, H.; Springstead, J.R.; et al. An Endogenous Caspase-11 Ligand Elicits Interleukin-1 Release from Living Dendritic Cells. Science 2016, 352, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Pascual, V.; Allantaz, F.; Arce, E.; Punaro, M.; Banchereau, J. Role of Interleukin-1 (IL-1) in the Pathogenesis of Systemic Onset Juvenile Idiopathic Arthritis and Clinical Response to IL-1 Blockade. J. Exp. Med. 2005, 201, 1479–1486. [Google Scholar] [CrossRef]
- Priori, R.; Colafrancesco, S.; Alessandri, C.; Minniti, A.; Perricone, C.; Iaiani, G.; Palazzo, D.; Valesini, G. Interleukin 18: A Biomarker for Differential Diagnosis Between Adult-Onset Still’s Disease and Sepsis. J. Rheumatol. 2014, 41, 1118–1123. [Google Scholar] [CrossRef]
- Gono, T.; Sato, S.; Kawaguchi, Y.; Kuwana, M.; Hanaoka, M.; Katsumata, Y.; Takagi, K.; Baba, S.; Okamoto, Y.; Ota, Y.; et al. Anti-MDA5 Antibody, Ferritin and IL-18 Are Useful for the Evaluation of Response to Treatment in Interstitial Lung Disease with Anti-MDA5 Antibody-Positive Dermatomyositis. Rheumatology 2012, 51, 1563–1570. [Google Scholar] [CrossRef]
- Gono, T.; Miyake, K.; Kawaguchi, Y.; Kaneko, H.; Shinozaki, M.; Yamanaka, H. Hyperferritinaemia and Macrophage Activation in a Patient with Interstitial Lung Disease with Clinically Amyopathic DM. Rheumatology 2012, 51, 1336–1338. [Google Scholar] [CrossRef]
- Gono, T.; Kawaguchi, Y.; Satoh, T.; Kuwana, M.; Katsumata, Y.; Takagi, K.; Masuda, I.; Tochimoto, A.; Baba, S.; Okamoto, Y.; et al. Clinical Manifestation and Prognostic Factor in Anti-melanoma Differentiation-Associated Gene 5 Antibody-Associated Interstitial Lung Disease as a Complication of Dermatomyositis. Rheumatology 2010, 49, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Iwamasa, K.; Hatta, N.; Fujita, S. Behçet’s Disease Associated with Myelodysplastic Syndrome with Elevated Levels of Inflammatory Cytokines. Mod. Rheumatol. 2003, 13, 350–355. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines Including Interleukin-6 in COVID-19 Induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Sakuraba, H.; Ishiguro, Y.; Yamagata, K.; Munakata, A.; Nakane, A. Blockade of TGF-Beta Accelerates Mucosal Destruction through Epithelial Cell Apoptosis. Biochem. Biophys. Res. Commun. 2007, 359, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.O.; Marinova-Mutafchieva, L.; Feldmann, M.; Maini, R.N. Evaluation of TNF-alpha and IL-1 blockade in collagen-induced arthritis and comparison with combined anti-TNF-alpha/anti-CD4 therapy. Immunol. 2000, 165, 7240–7245. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Munoz-Pilaillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Pupyshev, A.B.; Tikhonova, M.A.; Akopyan, A.A.; Tenditnik, M.V.; Dubrovina, N.I.; Korolenko, T.A. Therapeutic activation of autophagy by combined treatment with rapamycin and trehalose in a mouse MPTP-induced model of Parkinson’s disease. Pharmacol. Biochem. Behav. 2019, 177, 1–11. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Mizoguchi, A.; Takedatsu, H.; Cario, E.; de Jong, Y.P.; Ooi, C.J.; Xavier, R.J.; Terhorst, C.; Podolsky, D.K.; Bhan, A.K. Role of Tumor Necrosis Factor Receptor 2 (TNFR2) in Colonic Epithelial Hyperplasia and Chronic Intestinal Inflammation in Mice. Gastroenterology 2002, 122, 134–144. [Google Scholar] [CrossRef]
- Prabhakar, U.; Eirikis, E.; Davis, H.M. Simultaneous Quantification of Proinflammatory Cytokines in Human Plasma Using the LabMAP Assay. J. Immunol. Methods 2002, 260, 207–218. [Google Scholar] [CrossRef]
- Cooper, H.S.; Murthy, S.N.; Shah, R.S.; Sedergran, D.J. Clinicopathologic Study of Dextran Sulfate Sodium Experimental Murine Colitis. Lab. Investig. 1993, 69, 238–249. [Google Scholar]
- Tal, M.C.; Sasai, M.; Lee, H.K.; Yordy, B.; Shadel, G.S.; Iwasaki, A. Absence of Autophagy Results in Reactive Oxygen Species-Dependent Amplification of RLR Signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 2770–2775. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis Through Enhancing Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef]
- Nakane, A.; Minagawa, T.; Yasuda, I.; Yu, C.; Kato, K. Prevention by Gamma Interferon of Fatal Infection with Listeria monocytogenes in Mice Treated with Cyclosporin A. Infect. Immun. 1988, 56, 2011–2015. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.B.; Roldgaard, B.B.; Lindner, A.B.; Christensen, B.B.; Licht, T.R. Construction of a Multiple Fluorescence Labelling System for Use in Co-invasion Studies of Listeria monocytogenes. BMC Microbiol. 2006, 6, 86. [Google Scholar] [CrossRef] [PubMed]
- Nakane, A.; Nishikawa, S.; Sasaki, S.; Miura, T.; Asano, M.; Kohanawa, M.; Ishiwata, K.; Minagawa, T. Endogenous Interleukin-4, but Not Interleukin-10, Is Involved in Suppression of Host Resistance against Listeria monocytogenes Infection in Interferon-Depleted Mice. Infect. Immun. 1996, 64, 1252–1258. [Google Scholar] [CrossRef]
- Sashinami, H.; Nakane, A.; Iwakura, Y.; Sasaki, M. Effective Induction of Acquired Resistance to Listeria monocytogenes by Immunizing Mice with In Vivo-Infected Dendritic Cells. Infect. Immun. 2003, 71, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Sashinami, H.; Mori, F.; Matsumiya, T.; Yoshida, H.; Nakane, A.; Wakabayashi, K.; Oyama, C.; Satoh, K. Listeria monocytogenes Induces the Expression of Retinoic Acid-Inducible Gene-I. Microbiol. Immunol. 2006, 50, 811–815. [Google Scholar] [CrossRef]
- Nakane, A.; Minagawa, T.; Kato, K. Endogenous Tumor Necrosis Factor (Cachectin) Is Essential to Host Resistance Against Listeria monocytogenes Infection. Infect. Immun. 1988, 56, 2563–2569. [Google Scholar] [CrossRef]
- Ruedel, C.; Bachmann, M.F. CTL priming by CD8+ and CD8− dendritic cells in vivo. Eur. J. Immunol. 1999, 29, 3762–3767. [Google Scholar] [CrossRef]
- Toyonaga, T.; Matsuura, M.; Mori, K.; Honzawa, Y.; Minami, N.; Yamada, S.; Kobayashi, T.; Hibi, T.; Nakase, H. Lipocalin 2 prevents intestinal inflammation by enhancing phagocytic bacterial clearance in macrophages. Sci. Rep. 2016, 6, 35014. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiraga, H.; Chinda, D.; Maeda, T.; Murai, Y.; Ogasawara, K.; Muramoto, R.; Ota, S.; Hasui, K.; Sakuraba, H.; Ishiguro, Y.; et al. Vitamin A Promotes the Fusion of Autophagolysosomes and Prevents Excessive Inflammasome Activation in Dextran Sulfate Sodium-Induced Colitis. Int. J. Mol. Sci. 2023, 24, 8684. https://doi.org/10.3390/ijms24108684
Hiraga H, Chinda D, Maeda T, Murai Y, Ogasawara K, Muramoto R, Ota S, Hasui K, Sakuraba H, Ishiguro Y, et al. Vitamin A Promotes the Fusion of Autophagolysosomes and Prevents Excessive Inflammasome Activation in Dextran Sulfate Sodium-Induced Colitis. International Journal of Molecular Sciences. 2023; 24(10):8684. https://doi.org/10.3390/ijms24108684
Chicago/Turabian StyleHiraga, Hiroto, Daisuke Chinda, Takato Maeda, Yasuhisa Murai, Kohei Ogasawara, Ryutaro Muramoto, Shinji Ota, Keisuke Hasui, Hirotake Sakuraba, Yoh Ishiguro, and et al. 2023. "Vitamin A Promotes the Fusion of Autophagolysosomes and Prevents Excessive Inflammasome Activation in Dextran Sulfate Sodium-Induced Colitis" International Journal of Molecular Sciences 24, no. 10: 8684. https://doi.org/10.3390/ijms24108684