
Mix24X, a Lab-Assembled Reference to Evaluate Interpretation Procedures for Tandem Mass Spectrometry Proteotyping of Complex Samples

Abstract
:1. Introduction
2. Results
2.1. Assembly of 24 Bacterial Peptide Digests According to a Predefined MS/MS-Responsive Equimolar Ratio
2.2. Mix24X Datasets
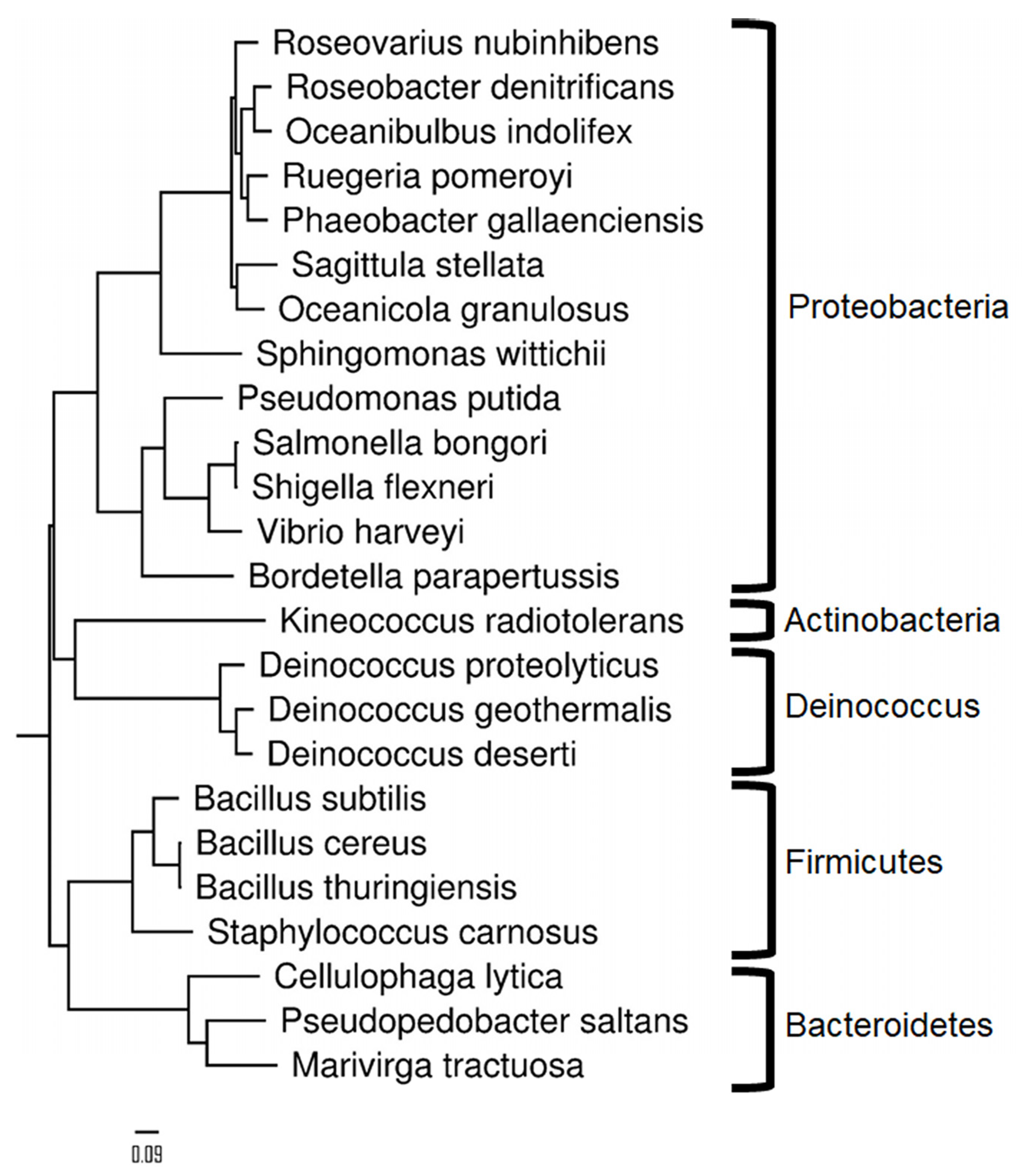
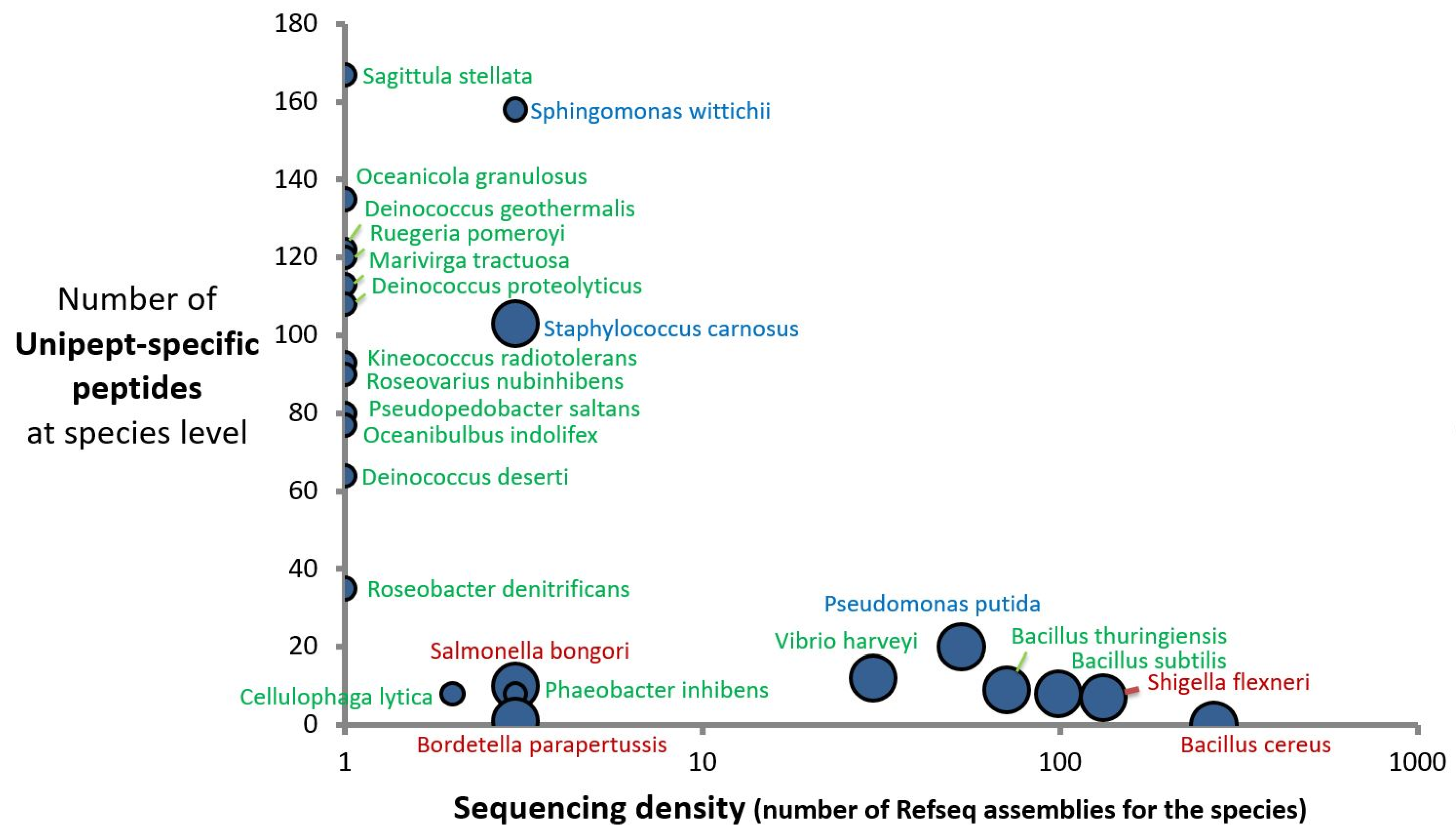
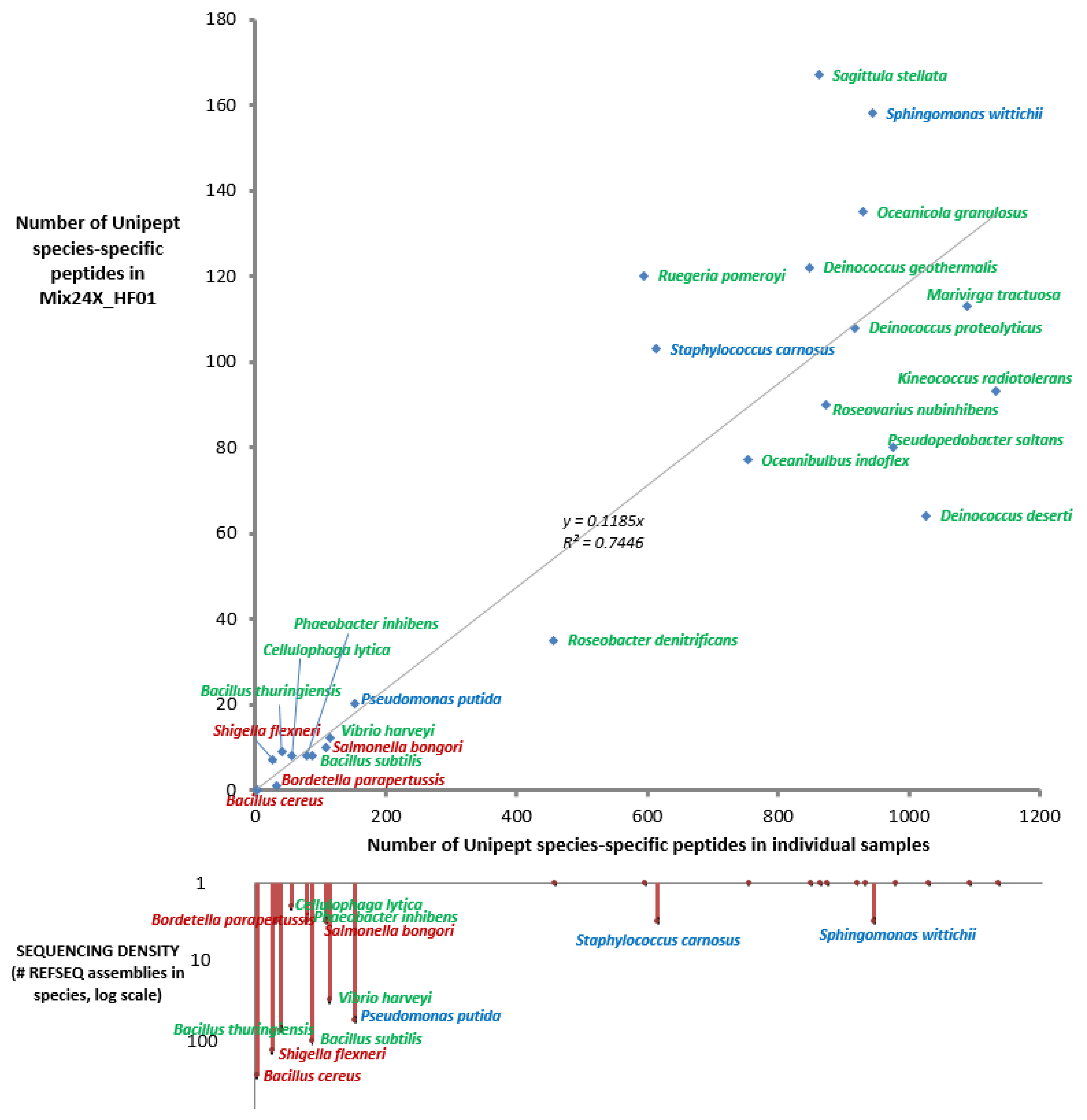
2.3. Taxonomical Characterization Using Species-Specific Peptides
2.4. Identification of Genus and then Species with a Cascade Search
3. Discussion
4. Materials and Methods
4.1. Microbial Cultures and Samples
4.2. Protein Extraction and Trypsin Proteolysis
4.3. NanoLC-MS/MS Analysis
4.4. MS/MS Spectrum Assignment and Protein Identification
4.5. Evaluation of Global Ion Intensity for Each of the 24 Peptide Digests for Mix24X Assembly
4.6. Taxonomical and Functional Data Analysis
4.7. Data Repository
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suarez, S.; Ferroni, A.; Lotz, A.; Jolley, K.A.; Guérin, P.; Leto, J.; Dauphin, B.; Jamet, A.; Maiden, M.C.; Nassif, X.; et al. Ribosomal proteins as biomarkers for bacterial identification by mass spectrometry in the clinical microbiology laboratory. J. Microbiol. Methods 2013, 94, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Grenga, L.; Pible, O.; Armengaud, J. Pathogen proteotyping: A rapidly developing application of mass spectrometry to address clinical concerns. Clin. Mass Spectrom. 2019, 14 Pt A, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, R.; Gonzales-Siles, L.; Boulund, F.; Svensson-Stadler, L.; Skovbjerg, S.; Karlsson, A.; Davidson, M.; Hulth, S.; Kristiansson, E.; Moore, E.R. Proteotyping: Proteomic characterization, classification and identification of microorganisms—A prospectus. Syst. Appl. Microbiol. 2015, 38, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, R.; Davidson, M.; Svensson-Stadler, L.; Karlsson, A.; Olesen, K.; Carlsohn, E.; Moore, E.R. Strain-Level Typing and Identification of Bacteria Using Mass Spectrometry-Based Proteomics. J. Proteome Res. 2012, 11, 2710–2720. [Google Scholar] [CrossRef]
- Hayoun, K.; Pible, O.; Petit, P.; Allain, F.; Jouffret, V.; Culotta, K.; Rivasseau, C.; Armengaud, J.; Alpha-Bazin, B. Proteotyping Environmental Microorganisms by Phylopeptidomics: Case Study Screening Water from a Radioactive Material Storage Pool. Microorganisms 2020, 8, 1525. [Google Scholar] [CrossRef]
- Lozano, C.; Kielbasa, M.; Gaillard, J.-C.; Miotello, G.; Pible, O.; Armengaud, J. Identification and Characterization of Marine Microorganisms by Tandem Mass Spectrometry Proteotyping. Microorganisms 2022, 10, 719. [Google Scholar] [CrossRef]
- Pible, O.; Petit, P.; Steinmetz, G.; Rivasseau, C.; Armengaud, J. Taxonomical composition and functional analysis of biofilms sampled from a nuclear storage pool. Front. Microbiol. 2023, 14, 1148976. [Google Scholar] [CrossRef]
- Petit, P.C.M.; Pible, O.; Van Eesbeeck, V.; Alban, C.; Steinmetz, G.; Mysara, M.; Monsieurs, P.; Armengaud, J.; Rivasseau, C. Direct Meta-Analyses Reveal Unexpected Microbial Life in the Highly Radioactive Water of an Operating Nuclear Reactor Core. Microorganisms 2020, 8, 1857. [Google Scholar] [CrossRef]
- Hayoun, K.; Gouveia, D.; Grenga, L.; Pible, O.; Armengaud, J.; Alpha-Bazin, B. Evaluation of Sample Preparation Methods for Fast Proteotyping of Microorganisms by Tandem Mass Spectrometry. Front. Microbiol. 2019, 10, 1985. [Google Scholar] [CrossRef]
- Hayoun, K.; Gaillard, J.-C.; Pible, O.; Alpha-Bazin, B.; Armengaud, J. High-throughput proteotyping of bacterial isolates by double barrel chromatography-tandem mass spectrometry based on microplate paramagnetic beads and phylopeptidomics. J. Proteom. 2020, 226, 103887. [Google Scholar] [CrossRef]
- Mappa, C.; Alpha-Bazin, B.; Pible, O.; Armengaud, J. Evaluation of the Limit of Detection of Bacteria by Tandem Mass Spectrometry Proteotyping and Phylopeptidomics. Microorganisms 2023, 11, 1170. [Google Scholar] [CrossRef]
- Witt, N.; Andreotti, S.; Busch, A.; Neubert, K.; Reinert, K.; Tomaso, H.; Meierhofer, D. Rapid and Culture Free Identification of Francisella in Hare Carcasses by High-Resolution Tandem Mass Spectrometry Proteotyping. Front. Microbiol. 2020, 11, 636. [Google Scholar] [CrossRef] [PubMed]
- Bourdin, V.; Charlier, P.; Crevat, S.; Slimani, L.; Chaussain, C.; Kielbasa, M.; Pible, O.; Armengaud, J. Deep Paleoproteotyping and Microtomography Revealed No Heart Defect nor Traces of Embalming in the Cardiac Relics of Blessed Pauline Jaricot. Int. J. Mol. Sci. 2023, 24, 3011. [Google Scholar] [CrossRef] [PubMed]
- Rüther, P.L.; Husic, I.M.; Bangsgaard, P.; Gregersen, K.M.; Pantmann, P.; Carvalho, M.; Godinho, R.M.; Friedl, L.; Cascalheira, J.; Taurozzi, A.J.; et al. SPIN enables high throughput species identification of archaeological bone by proteomics. Nat. Commun. 2022, 13, 2458. [Google Scholar] [CrossRef]
- Oumarou Hama, H.; Chenal, T.; Pible, O.; Miotello, G.; Armengaud, J.; Drancourt, M. An ancient coronavirus from individuals in France, circa 16th century. Int. J. Infect. Dis. 2023, 131, 7–12. [Google Scholar] [CrossRef]
- Mesuere, B.; Devreese, B.; Debyser, G.; Aerts, M.; Vandamme, P.; Dawyndt, P. Unipept: Tryptic Peptide-Based Biodiversity Analysis of Metaproteome Samples. J. Proteome Res. 2012, 11, 5773–5780. [Google Scholar] [CrossRef]
- Boulund, F.; Karlsson, R.; Gonzales-Siles, L.; Johnning, A.; Karami, N.; Al-Bayati, O.; Åhrén, C.; Moore, E.R.B.; Kristiansson, E. Typing and Characterization of Bacteria Using Bottom-up Tandem Mass Spectrometry Proteomics. Mol. Cell. Proteom. 2017, 16, 1052–1063. [Google Scholar] [CrossRef]
- Mooradian, A.D.; Van Der Post, S.; Naegle, K.M.; Held, J.M. ProteoClade: A taxonomic toolkit for multi-species and metaproteomic analysis. PLoS Comput. Biol. 2020, 16, e1007741. [Google Scholar] [CrossRef]
- Kuhring, M.; Doellinger, J.; Nitsche, A.; Muth, T.; Renard, B.Y. TaxIt: An Iterative Computational Pipeline for Untargeted Strain-Level Identification Using MS/MS Spectra from Pathogenic Single-Organism Samples. J. Proteome Res. 2020, 19, 2501–2510. [Google Scholar] [CrossRef]
- Alves, G.; Wang, G.; Ogurtsov, A.Y.; Drake, S.K.; Gucek, M.; Sacks, D.B.; Yu, Y.-K. Rapid Classification and Identification of Multiple Microorganisms with Accurate Statistical Significance via High-Resolution Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2018, 29, 1721–1737. [Google Scholar] [CrossRef]
- Alves, G.; Ogurtsov, A.; Karlsson, R.; Jaén-Luchoro, D.; Piñeiro-Iglesias, B.; Salvà-Serra, F.; Andersson, B.; Moore, E.R.B.; Yu, Y.-K. Identification of Antibiotic Resistance Proteins via MiCId’s Augmented Workflow. A Mass Spectrometry-Based Proteomics Approach. J. Am. Soc. Mass Spectrom. 2022, 33, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Alves, G.; Yu, Y.-K. Robust Accurate Identification and Biomass Estimates of Microorganisms via Tandem Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2020, 31, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, J. Metaproteomics to understand how microbiota function: The crystal ball predicts a promising future. Environ. Microbiol. 2023, 25, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Herbst, F.-A.; Lünsmann, V.; Kjeldal, H.; Jehmlich, N.; Tholey, A.; Von Bergen, M.; Nielsen, J.L.; Hettich, R.L.; Seifert, J.; Nielsen, P.H. Enhancing metaproteomics—The value of models and defined environmental microbial systems. Proteomics 2016, 16, 783–798. [Google Scholar] [CrossRef]
- Muth, T.; Behne, A.; Heyer, R.; Kohrs, F.; Benndorf, D.; Hoffmann, M.; Lehtevä, M.; Reichl, U.; Martens, L.; Rapp, E. The MetaProteomeAnalyzer: A Powerful Open-Source Software Suite for Metaproteomics Data Analysis and Interpretation. J. Proteome Res. 2015, 14, 1557–1565. [Google Scholar] [CrossRef]
- Tanca, A.; Palomba, A.; Deligios, M.; Cubeddu, T.; Fraumene, C.; Biosa, G.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. Evaluating the Impact of Different Sequence Databases on Metaproteome Analysis: Insights from a Lab-Assembled Microbial Mixture. PLoS ONE 2013, 8, e82981. [Google Scholar] [CrossRef]
- Tanca, A.; Palomba, A.; Pisanu, S.; Deligios, M.; Fraumene, C.; Manghina, V.; Pagnozzi, D.; Addis, M.F.; Uzzau, S. A straightforward and efficient analytical pipeline for metaproteome characterization. Microbiome 2014, 2, 49. [Google Scholar] [CrossRef]
- Kuharev, J.; Navarro, P.; Distler, U.; Jahn, O.; Tenzer, S. In-depth evaluation of software tools for data-independent acquisition based label-free quantification. Proteomics 2015, 15, 3140–3151. [Google Scholar] [CrossRef]
- Navarro, P.; Kuharev, J.; Gillet, L.C.; Bernhardt, O.M.; MacLean, B.; Röst, H.L.; Tate, S.A.; Tsou, C.-C.; Reiter, L.; Distler, U.; et al. A multicenter study benchmarks software tools for label-free proteome quantification. Nat. Biotechnol. 2016, 34, 1130–1136. [Google Scholar] [CrossRef]
- Kleiner, M.; Thorson, E.; Sharp, C.E.; Dong, X.; Liu, D.; Li, C.; Strous, M. Assessing species biomass contributions in microbial communities via metaproteomics. Nat. Commun. 2017, 8, 1558. [Google Scholar] [CrossRef]
- Helgason, E.; Okstad, O.A.; Caugant, D.A.; Johansen, H.A.; Fouet, A.; Mock, M.; Hegna, I.; Kolsto, A.B. Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis--one species on the basis of genetic evidence. Appl. Environ. Microbiol. 2000, 66, 2627–2630. [Google Scholar] [CrossRef] [PubMed]
- Rasko, D.A.; Altherr, M.R.; Han, C.S.; Ravel, J. Genomics of the Bacillus cereus group of organisms. FEMS Microbiol. Rev. 2005, 29, 303–329. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, M.D.S.; Roca, A.; Molina-Santiago, C.; Duque, E.; Armengaud, J.; Gómez-Garcia, M.R.; Ramos, J.L. Understanding butanol tolerance and assimilation in P seudomonas putida BIRD -1: An integrated omics approach. Microb. Biotechnol. 2016, 9, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Rubiano-Labrador, C.; Bland, C.; Miotello, G.; Guérin, P.; Pible, O.; Baena, S.; Armengaud, J. Proteogenomic insights into salt tolerance by a halotolerant alpha-proteobacterium isolated from an Andean saline spring. J. Proteom. 2014, 97, 36–47. [Google Scholar] [CrossRef]
- Denef, V.J.; Shah, M.B.; VerBerkmoes, N.C.; Hettich, R.L.; Banfield, J.F. Implications of Strain- and Species-Level Sequence Divergence for Community and Isolate Shotgun Proteomic Analysis. J. Proteome Res. 2007, 6, 3152–3161. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Zoun, R.; Becher, B.; Saake, G.; Benndorf, D. Challenges and perspectives of metaproteomic data analysis. J. Biotechnol. 2017, 261, 24–36. [Google Scholar] [CrossRef]
- Muth, T.; Renard, B.Y.; Martens, L. Metaproteomic data analysis at a glance: Advances in computational microbial community proteomics. Expert Rev. Proteom. 2016, 13, 757–769. [Google Scholar] [CrossRef]
- Kleikamp, H.B.; Pronk, M.; Tugui, C.; Guedes da Silva, L.; Abbas, B.; Lin, Y.M.; van Loosdrecht, M.C.; Pabst, M. Database-independent de novo metaproteomics of complex microbial communities. Cell Syst. 2021, 12, 375–383.e375. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Mitchell, H.D.; Burnet, M.C.; Wu, R.; Jenson, S.C.; Merkley, E.D.; Nakayasu, E.S.; Nicora, C.D.; Jansson, J.K.; Burnum-Johnson, K.E.; et al. Uncovering Hidden Members and Functions of the Soil Microbiome Using De Novo Metaproteomics. J. Proteome Res. 2022, 21, 2023–2035. [Google Scholar] [CrossRef]
- Van Den Bossche, T.; Arntzen, M.O.; Becher, D.; Benndorf, D.; Eijsink, V.G.H.; Henry, C.; Jagtap, P.D.; Jehmlich, N.; Juste, C.; Kunath, B.J.; et al. The Metaproteomics Initiative: A coordinated approach for propelling the functional characterization of microbiomes. Microbiome 2021, 9, 243. [Google Scholar] [CrossRef]
- Liu, Y.-X.; Qin, Y.; Chen, T.; Lu, M.; Qian, X.; Guo, X.; Bai, Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 2021, 12, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Soppa, J. Polyploidy and community structure. Nat. Microbiol. 2017, 2, 16261. [Google Scholar] [CrossRef] [PubMed]
- Gohl, D.M.; Vangay, P.; Garbe, J.; MacLean, A.; Hauge, A.; Becker, A.; Gould, T.J.; Clayton, J.B.; Johnson, T.J.; Hunter, R.; et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat. Biotechnol. 2016, 34, 942–949. [Google Scholar] [CrossRef]
- Klappenbach, J.A.; Dunbar, J.M.; Schmidt, T.M. rRNA Operon Copy Number Reflects Ecological Strategies of Bacteria. Appl. Environ. Microbiol. 2000, 66, 1328–1333. [Google Scholar] [CrossRef]
- Hartmann, E.M.; Allain, F.; Gaillard, J.-C.; Pible, O.; Armengaud, J. Taking the Shortcut for High-Throughput Shotgun Proteomic Analysis of Bacteria. Methods Mol. Biol. 2014, 1197, 275–285. [Google Scholar] [CrossRef]
- Clair, G.; Armengaud, J.; Duport, C. Restricting Fermentative Potential by Proteome Remodeling: An adaptive strategy evidenced in Bacillus cereus. Mol. Cell. Proteom. 2012, 11, 013102. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Mesuere, B.; Debyser, G.; Aerts, M.; Devreese, B.; Vandamme, P.; Dawyndt, P. The Unipept metaproteomics analysis pipeline. Proteomics 2015, 15, 1437–1442. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Strain | Gram Staining a | Source b | Growth Condition c |
|---|---|---|---|
| Bacillus cereus ATCC 14579 | + | UMR408 | LB, 24 h, 30 °C |
| Bacillus subtilis ATCC 6633 | + | ATCC | BHI, 24 h, 30 °C |
| Bacillus thuringiensis DSM 5815 | + | DSMZ | LB, 24 h, 30 °C |
| Bordetella parapertussis Bpp5 | − | Pasteur institute | BHI, 48 h, 30 °C |
| Cellulophaga lytica DSM 7489 | − | DSMZ | MB, 24 h, 30 °C |
| Deinococcus deserti VCD115 | ~ | BIAM1 | diluted TSB, 24 h, 30 °C |
| Deinococcus geothermalis DSM 11300 | ~ | BIAM1 | LB, 48 h, 37 °C |
| Deinococcus proteolyticus DSM 20540 | ~ | BIAM1 | LB, 24 h, 30 °C |
| Kineococcus radiotolerans SRS30216 | + | DSMZ | PTYG, 72 h, 30 °C |
| Marivirga tractuosa DSM 4126 | − | DSMZ | MB, 48 h, 30 °C |
| Oceanibulbus indolifex HEL-45 | − | DSMZ | MB, 48 h, 30 °C |
| Oceanicola granulosus HTCC2516 | − | DSMZ | MB, 48 h, 30 °C |
| Phaeobacter inhibens DSM 17395 | − | DSMZ | MB, 48 h, 30 °C |
| Pseudomonas putida mt-2 KT2440 | − | DSMZ | LB, 24 h, 30 °C |
| Pseudopedobacter saltans DSM 12145 | − | DSMZ | TSB and extracts, 24 h, 26 °C |
| Roseobacter denitrificans OCh 114 | − | DSMZ | MB, 48 h, 30 °C |
| Roseovarius nubinhibens ISM | − | DSMZ | MB, 24 h, 30 °C |
| Ruegeria pomeroyi DSS-3 | − | DSMZ | MB, 48 h, 30 °C |
| Sagittula stellata E 37 | − | DSMZ | MB, 48 h, 30 °C |
| Salmonella bongori NCTC 12419 | − | Pasteur institute | TSB, 24 h, 37 °C |
| Shigella flexneri 2a 2457T | − | Pasteur institute | TSB, 24 h, 30 °C |
| Sphingomonas wittichii RW1 | − | DSMZ | LB, 120 h, 30 °C |
| Staphylococcus carnosus TM300 | + | DSMZ | TSB, 24 h, 37 °C |
| Vibrio harveyi ATCC 14126 | − | BIAM2 | PB, 24 h, 26 °C |
| Reference | Gradient Time (min) | MS/MS Platform | MS/MS Spectra | PSMs | Peptide Sequences | Cumulated PSMs a | Cumulated Peptide Sequences b |
|---|---|---|---|---|---|---|---|
| Mix24X_XL01 | 180 | LTQ Orbitrap XL | 20,641 | 2464 | 1242 | 2464 | 1242 |
| Mix24X_XL02 | 180 | LTQ Orbitrap XL | 19,664 | 2358 | 1143 | 4822 | 1503 |
| Mix24X_XL03 | 180 | LTQ Orbitrap XL | 19,085 | 2145 | 1075 | 6967 | 1642 |
| Mix24X_HF01 | 60 | Q-Exactive HF | 40,768 | 8363 | 6201 | 8363 | 6201 |
| Mix24X_HF02 | 60 | Q-Exactive HF | 38,464 | 8275 | 6129 | 16,638 | 8043 |
| Mix24X_HF03 | 60 | Q-Exactive HF | 38,471 | 8303 | 6151 | 24,941 | 9106 |
| Species | HF01 Specific Peptides a | HF01 SC b | HF01 + HF02 + HF03 Specific Peptides a | HF01 + HF02 + HF03 SC b |
|---|---|---|---|---|
| Bacillus cereus | 0 | 0 | 1 | 1 |
| Bacillus subtilis | 8 | 9 | 10 | 24 |
| Bacillus thuringiensis | 9 | 8 | 12 | 27 |
| Bordetella parapertussis | 1 | 1 | 3 | 5 |
| Cellulophaga lytica | 8 | 8 | 12 | 21 |
| Deinococcus deserti | 64 | 83 | 99 | 275 |
| Deinococcus geothermalis | 122 | 147 | 180 | 428 |
| Deinococcus proteolyticus | 108 | 141 | 153 | 414 |
| Kineococcus radiotolerans | 93 | 90 | 143 | 279 |
| Marivirga tractuosa | 113 | 126 | 156 | 377 |
| Oceanibulbus indolifex | 77 | 108 | 116 | 312 |
| Oceanicola granulosus | 135 | 137 | 191 | 379 |
| Phaeobacter inhibens | 8 | 12 | 14 | 40 |
| Pseudomonas putida | 20 | 18 | 25 | 54 |
| Pseudopedobacter saltans | 80 | 69 | 128 | 211 |
| Roseobacter denitrificans | 35 | 36 | 49 | 101 |
| Roseovarius nubinhibens | 90 | 108 | 126 | 287 |
| Ruegeria pomeroyi | 120 | 148 | 173 | 449 |
| Sagittula stellata | 167 | 194 | 242 | 559 |
| Salmonella bongori | 10 | 11 | 14 | 37 |
| Shigella flexneri | 7 | 7 | 9 | 17 |
| Sphingomonas wittichii | 158 | 182 | 208 | 506 |
| Staphylococcus carnosus | 103 | 91 | 159 | 278 |
| Vibrio harveyi | 12 | 9 | 23 | 33 |
| OTHER BACTERIA c | 17 (17) | 15 (17) | 39 (38) | 43 (38) |
| ARCHAEA c | 1 (1) | 1 (1) | 1 (1) | 1 (1) |
| EUKARYOTA c,d | 8 (8) | 7 (8) | 17 (16) | 22 (16) |
| Genus | HF01 Specific Peptides a | HF01 SC b | H01 + HF02 + HF03 Specific Peptides a | H01 + HF02 + HF03 SC b |
|---|---|---|---|---|
| Bacillus | 38 | 40 | 50 | 124 |
| Bordetella | 83 | 84 | 120 | 247 |
| Cellulophaga | 121 | 123 | 168 | 333 |
| Deinococcus | 420 | 505 | 624 | 1546 |
| Kineococcus | 93 | 90 | 143 | 279 |
| Marivirga | 113 | 126 | 156 | 377 |
| Oceanibulbus | 77 | 108 | 116 | 312 |
| Oceanicola | 135 | 137 | 191 | 379 |
| Phaeobacter | 73 | 92 | 103 | 262 |
| Pseudomonas | 52 | 58 | 74 | 175 |
| Pseudopedobacter | 80 | 69 | 128 | 211 |
| Roseobacter | 77 | 73 | 85 | 208 |
| Roseovarius | 94 | 112 | 133 | 292 |
| Ruegeria | 125 | 150 | 179 | 454 |
| Sagittula | 167 | 194 | 242 | 559 |
| Salmonella | 27 | 30 | 35 | 95 |
| Shigella | 10 | 11 | 13 | 31 |
| Sphingomonas | 167 | 191 | 223 | 537 |
| Staphylococcus | 162 | 153 | 236 | 459 |
| Vibrio | 108 | 104 | 173 | 329 |
| OTHER BACTERIA c | 17 (16) | 14 (16) | 39 (37) | 39 (37) |
| ARCHAEA c | 1 (1) | 1 (1) | 1 (1) | 1 (1) |
| EUKARYOTA c,d | 8 (8) | 7 (8) | 18 (17) | 23 (17) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mappa, C.; Alpha-Bazin, B.; Pible, O.; Armengaud, J. Mix24X, a Lab-Assembled Reference to Evaluate Interpretation Procedures for Tandem Mass Spectrometry Proteotyping of Complex Samples. Int. J. Mol. Sci. 2023, 24, 8634. https://doi.org/10.3390/ijms24108634
Mappa C, Alpha-Bazin B, Pible O, Armengaud J. Mix24X, a Lab-Assembled Reference to Evaluate Interpretation Procedures for Tandem Mass Spectrometry Proteotyping of Complex Samples. International Journal of Molecular Sciences. 2023; 24(10):8634. https://doi.org/10.3390/ijms24108634
Chicago/Turabian StyleMappa, Charlotte, Béatrice Alpha-Bazin, Olivier Pible, and Jean Armengaud. 2023. "Mix24X, a Lab-Assembled Reference to Evaluate Interpretation Procedures for Tandem Mass Spectrometry Proteotyping of Complex Samples" International Journal of Molecular Sciences 24, no. 10: 8634. https://doi.org/10.3390/ijms24108634