
Novel Divergent Members of the Kitrinoviricota Discovered through Metagenomics in the Intestinal Contents of Red-Backed Voles (Clethrionomys gapperi)

 , ,
, ,
Abstract
:1. Introduction
2. Results
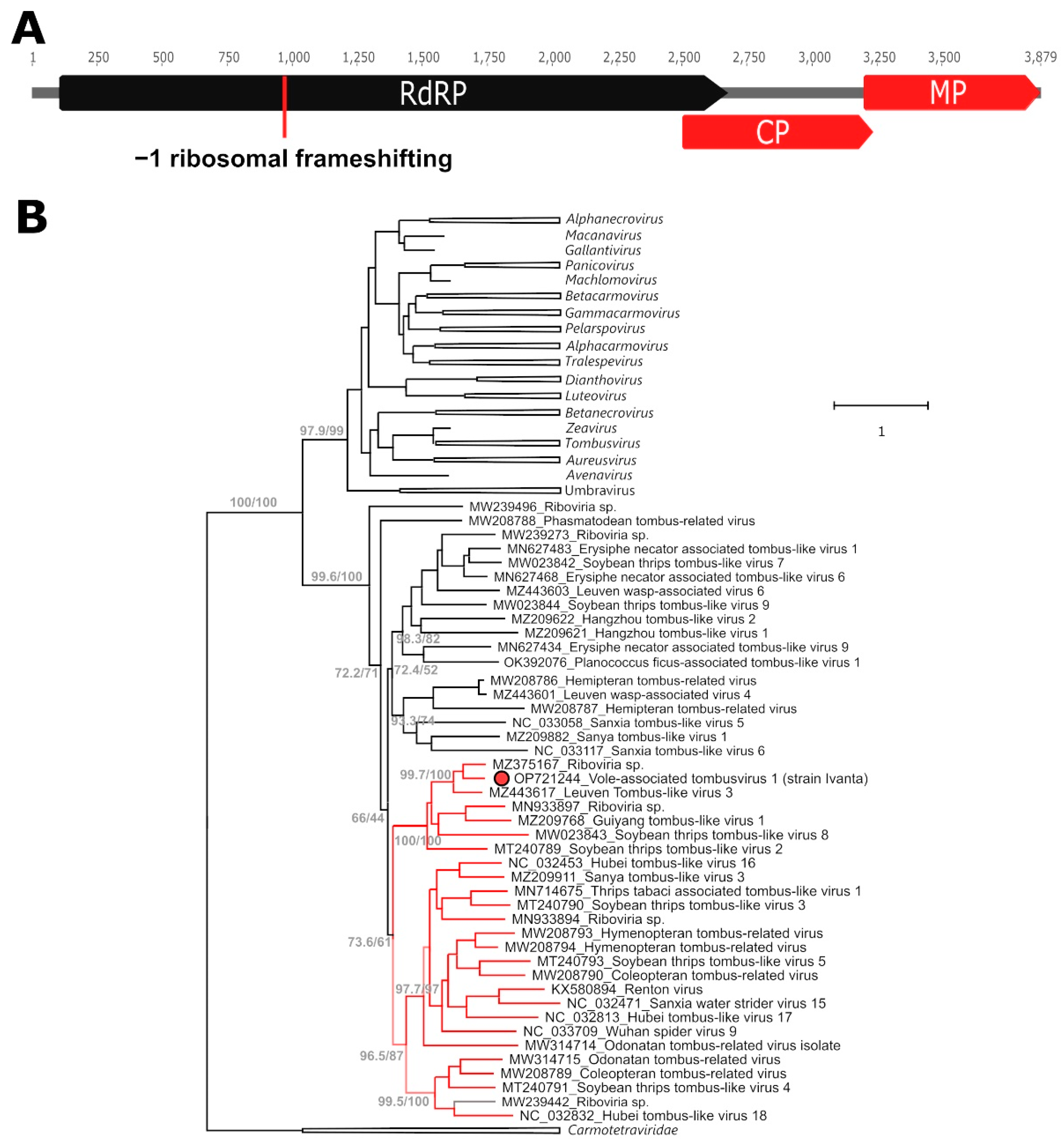
2.1. A Novel Tombusvirus
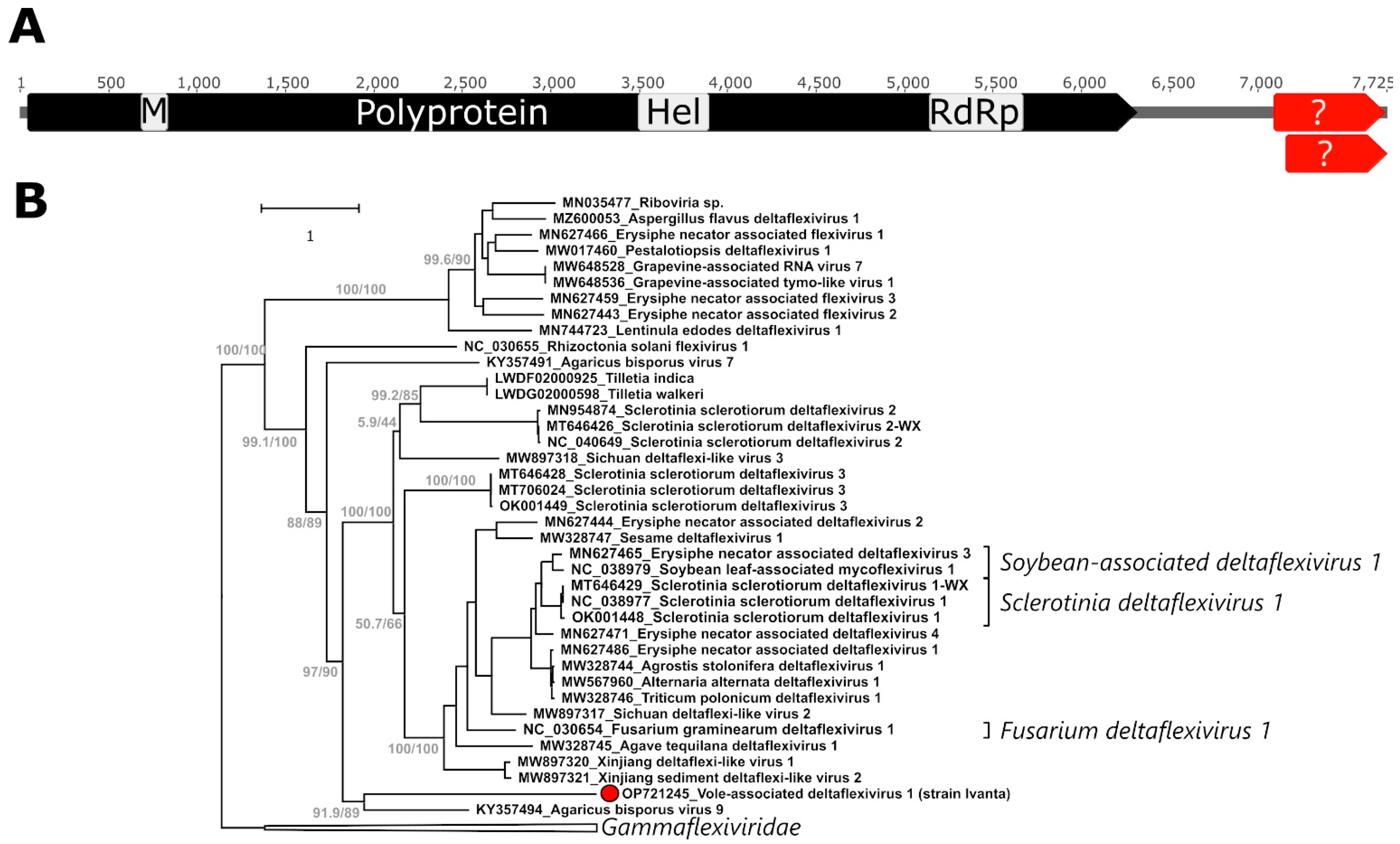
2.2. A Novel Deltaflexivirus
3. Discussion
4. Materials and Methods
4.1. Virus Discovery
4.2. Sequence Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delwart, E.L. Viral Metagenomics. Rev. Med. Virol. 2007, 17, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L. A Decade of RNA Virus Metagenomics Is (Not) Enough. Virus Res. 2018, 244, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L.; et al. The Evolutionary History of Vertebrate RNA Viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus Taxonomy in the Age of Metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, M.; Kuhn, J.H. Create a Megataxonomic Framework, Filling All Principal Taxonomic Ranks, for Realm Riboviria. ICTV [International Committee for Taxonomy of Viruses] Proposal No. 2019.006G. 2019.
- Anthony, S.J.; Epstein, J.H.; Murray, K.A.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Solovyov, A.; Ojeda-Flores, R.; Arrigo, N.C.; Islam, A.; Ali Khan, S.; et al. A Strategy to Estimate Unknown Viral Diversity in Mammals. mBio 2013, 4, e00598-00513. [Google Scholar] [CrossRef] [Green Version]
- King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. (Eds.) Family—Tombusviridae. In Virus Taxonomy; Elsevier: San Diego, CA, USA, 2012; pp. 1111–1138. ISBN 978-0-12-384684-6. [Google Scholar]
- Tahir, M.N.; Bolus, S.; Grinstead, S.C.; McFarlane, S.A.; Mollov, D. A New Virus of the Family Tombusviridae Infecting Sugarcane. Arch. Virol. 2021, 166, 961–965. [Google Scholar] [CrossRef]
- Chen, X.; He, H.; Yang, X.; Zeng, H.; Qiu, D.; Guo, L. The Complete Genome Sequence of a Novel Fusarium Graminearum RNA Virus in a New Proposed Family within the Order Tymovirales. Arch. Virol. 2016, 161, 2899–2903. [Google Scholar] [CrossRef]
- Li, K.; Zheng, D.; Cheng, J.; Chen, T.; Fu, Y.; Jiang, D.; Xie, J. Characterization of a Novel Sclerotinia Sclerotiorum RNA Virus as the Prototype of a New Proposed Family within the Order Tymovirales. Virus Res. 2016, 219, 92–99. [Google Scholar] [CrossRef]
- Marzano, S.-Y.L.; Domier, L.L. Novel Mycoviruses Discovered from Metatranscriptomics Survey of Soybean Phyllosphere Phytobiomes. Virus Res. 2016, 213, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutilh, B.E.; Varsani, A.; Tong, Y.; Simmonds, P.; Sabanadzovic, S.; Rubino, L.; Roux, S.; Muñoz, A.R.; Lood, C.; Lefkowitz, E.J.; et al. Perspective on Taxonomic Classification of Uncultivated Viruses. Curr. Opin. Virol. 2021, 51, 207–215. [Google Scholar] [CrossRef]
- Merritt, J.F. Clethrionomys Gapperi. Mamm. Species 1981, 146, 1–9. [Google Scholar] [CrossRef]
- Canuti, M.; van der Hoek, L. Virus Discovery: Are We Scientists or Genome Collectors? Trends Microbiol. 2014, 22, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, J.T.P.; Canuti, M.; Munro, H.J.; Dufour, S.C.; Lang, A.S. ViDiT-CACTUS: An Inexpensive and Versatile Library Preparation and Sequence Analysis Method for Virus Discovery and Other Microbiology Applications. Can. J. Microbiol. 2018, 64, 761–773. [Google Scholar] [CrossRef]
- Canuti, M.; Verhoeven, J.T.P.; Munro, H.J.; Roul, S.; Ojkic, D.; Robertson, G.J.; Whitney, H.G.; Dufour, S.C.; Lang, A.S. Investigating the Diversity and Host Range of Novel Parvoviruses from North American Ducks Using Epidemiology, Phylogenetics, Genome Structure, and Codon Usage Analysis. Viruses 2021, 13, 193. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Wilson, L.; Bowes, V.; Redford, T.; Dufour, S.C.; Lang, A.S.; Verhoeven, J.T.P. A Novel Calicivirus Discovered in Trumpeter Swans (Cygnus Buccinator) Expands the Richness of Known Avian Caliciviruses. Curr. Res. Microb. Sci. 2022, 3, 100169. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.-J.; Weber, N.; Schuster, S.C. Integrative Analysis of Environmental Sequences Using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef]
- Kashnikov, A.Y.; Epifanova, N.V.; Novikova, N.A. Picobirnaviruses: Prevalence, Genetic Diversity, Detection Methods. Vavilovskii Zhurnal Genet. Sel. 2020, 24, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Contigs | Closest Relative | |||
|---|---|---|---|---|
| Number | Length Range | Virus Name | Family, Genus | Percent Identity |
| 7 | 257–526 nt | Hubei virga-like virus 11 | Virgaviridae, unclassified | 67–82 |
| 6 | 285–428 nt | various | Permutotetraviridae, unclassified | 61–69 |
| 4 | 238–1024 nt | various | Partitiviridae, unclassified | 60–82 |
| 3 | 211–218 nt | Blueberry shock virus | Bromoviridae, Ilarvirus | 80–86 |
| 1 | 243 nt | Israeli acute paralysis virus | Dicistroviridae, Aparavirus | 65 |
| 1 | 3879 nt | Riboviria sp. strain 1PE-RDRP-18 | Tombusviridae, unclassified | 67 |
| 1 | 7725 nt | Agaricus bisporus virus 9 | Deltaflexiviridae, unclassified | 69 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canuti, M.; Rodrigues, B.; Lang, A.S.; Dufour, S.C.; Verhoeven, J.T.P. Novel Divergent Members of the Kitrinoviricota Discovered through Metagenomics in the Intestinal Contents of Red-Backed Voles (Clethrionomys gapperi). Int. J. Mol. Sci. 2023, 24, 131. https://doi.org/10.3390/ijms24010131
Canuti M, Rodrigues B, Lang AS, Dufour SC, Verhoeven JTP. Novel Divergent Members of the Kitrinoviricota Discovered through Metagenomics in the Intestinal Contents of Red-Backed Voles (Clethrionomys gapperi). International Journal of Molecular Sciences. 2023; 24(1):131. https://doi.org/10.3390/ijms24010131
Chicago/Turabian StyleCanuti, Marta, Bruce Rodrigues, Andrew S. Lang, Suzanne C. Dufour, and Joost T. P. Verhoeven. 2023. "Novel Divergent Members of the Kitrinoviricota Discovered through Metagenomics in the Intestinal Contents of Red-Backed Voles (Clethrionomys gapperi)" International Journal of Molecular Sciences 24, no. 1: 131. https://doi.org/10.3390/ijms24010131