
Short-Term Blockade of Pro-Inflammatory Alarmin S100A9 Favorably Modulates Left Ventricle Proteome and Related Signaling Pathways Involved in Post-Myocardial Infarction Recovery

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
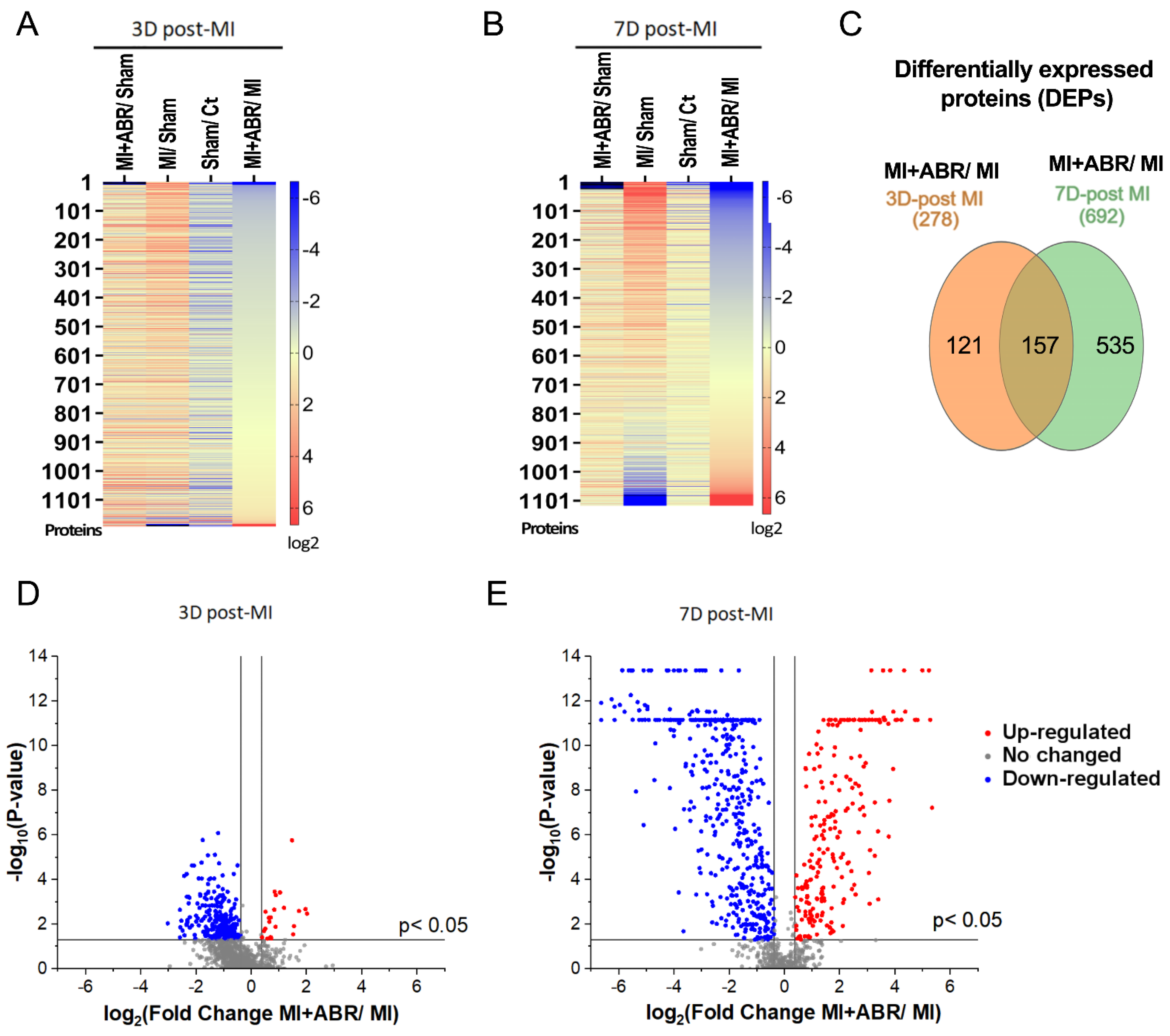
2.1. Identification of Differentially Expressed Proteins (DEPs) in the Infarcted Left Ventricle by Mass Spectrometry
2.2. DEP Gene Ontology Analysis Revealed the Significant Enrichment of Key Cellular Processes Involved in Post-MI Recovery
2.3. S100A9 Blockade Downregulates the Expression of Proteins Involved in Leukocyte Cell–Cell Adhesion during the Inflammatory Phase Post-MI
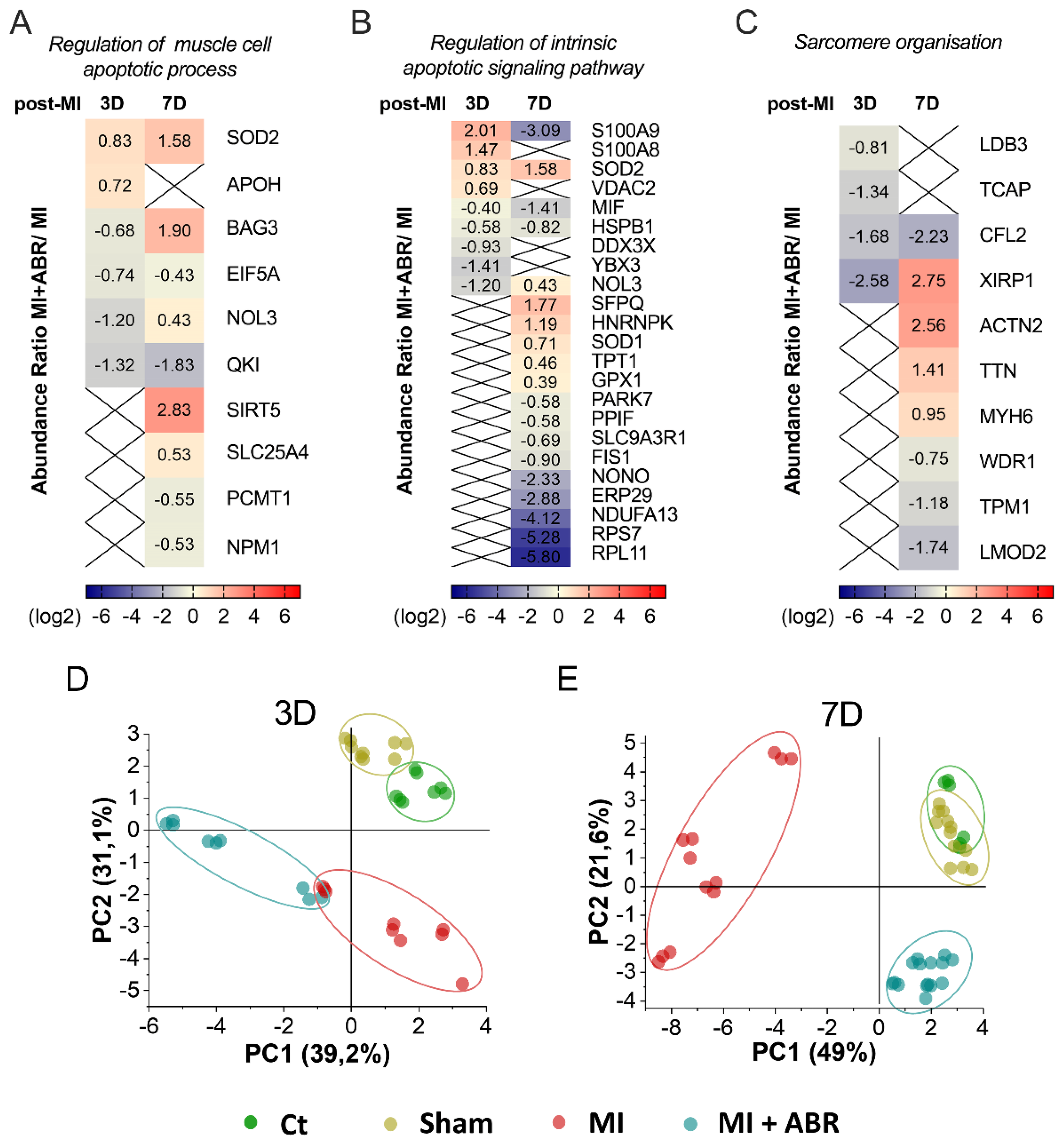
2.4. S100A9 Blockade Modulates the Apoptotic Process and Sarcomere Organization in Cardiomyocytes
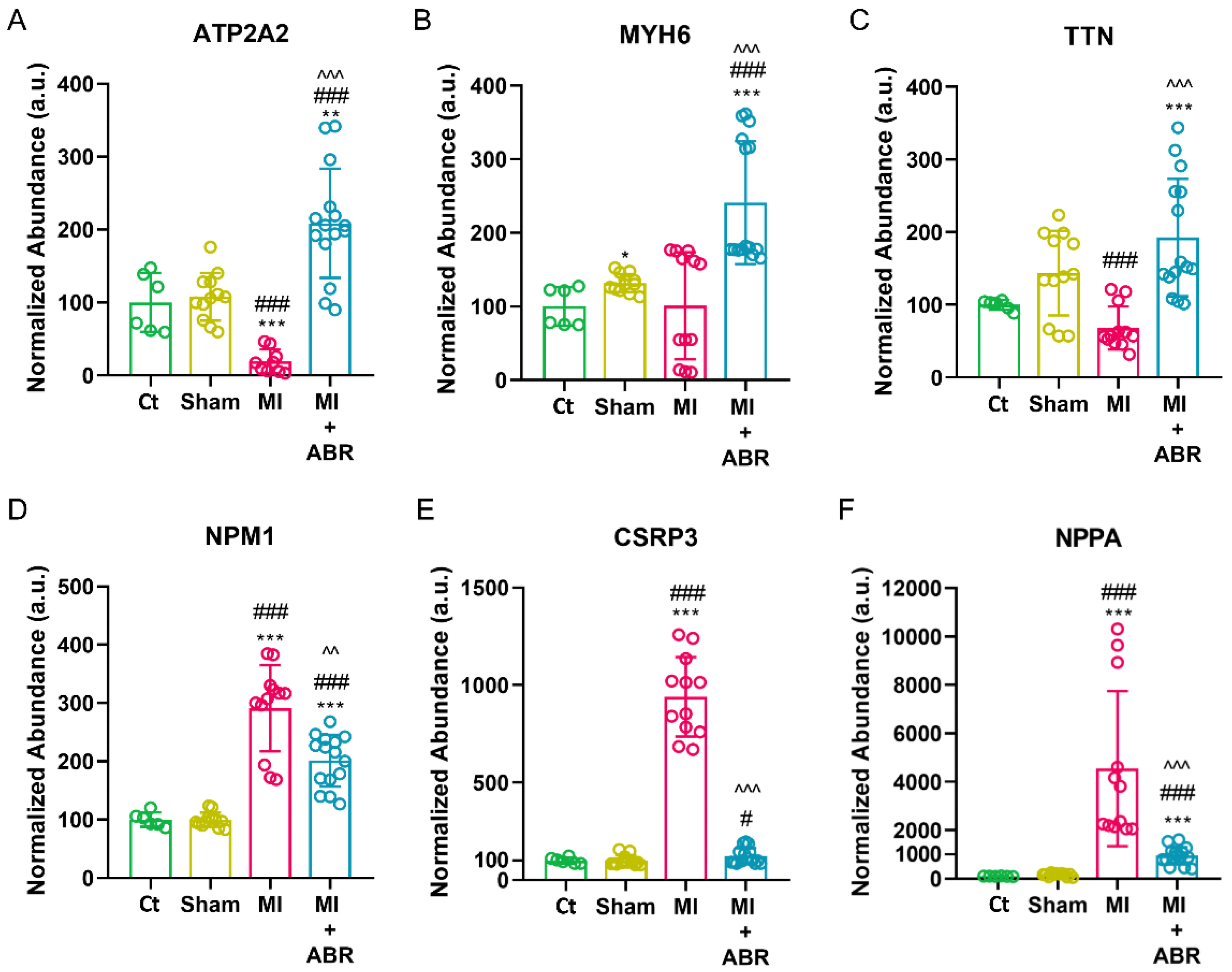
2.5. S100A9 Blockade Regulates Cardiac Hypertrophy Starting at Day 7 Post-MI
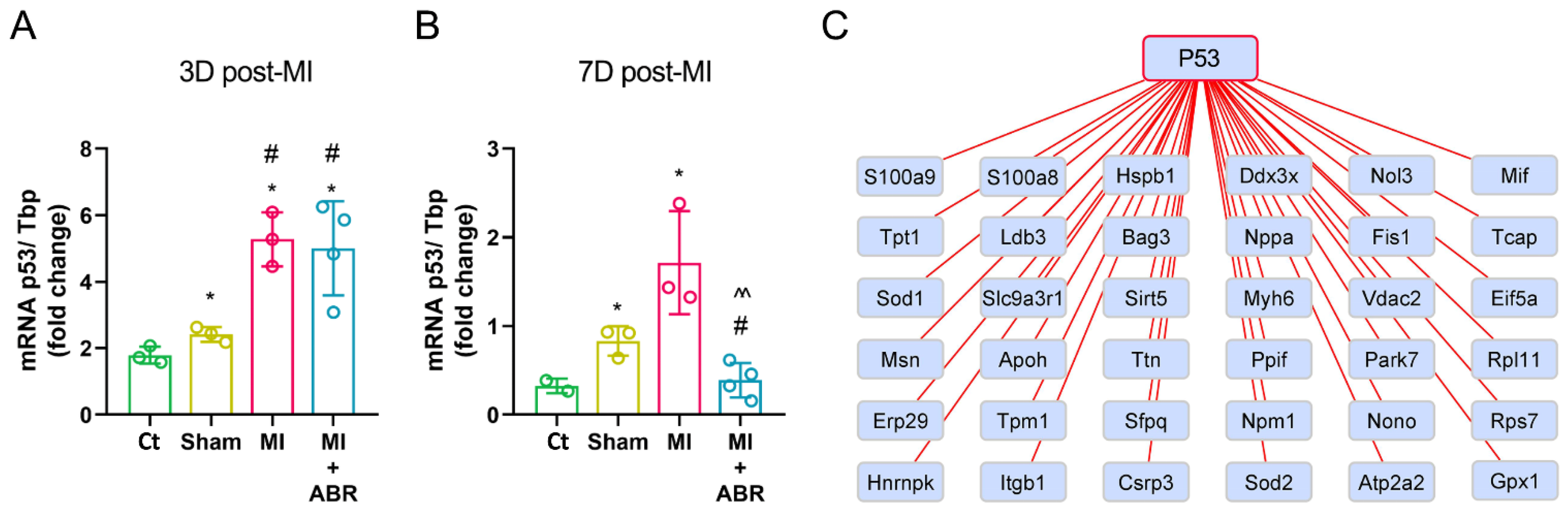
2.6. The Short-Term S100A9 Blockade Significantly Modulates the Cellular Tumor Antigen p53
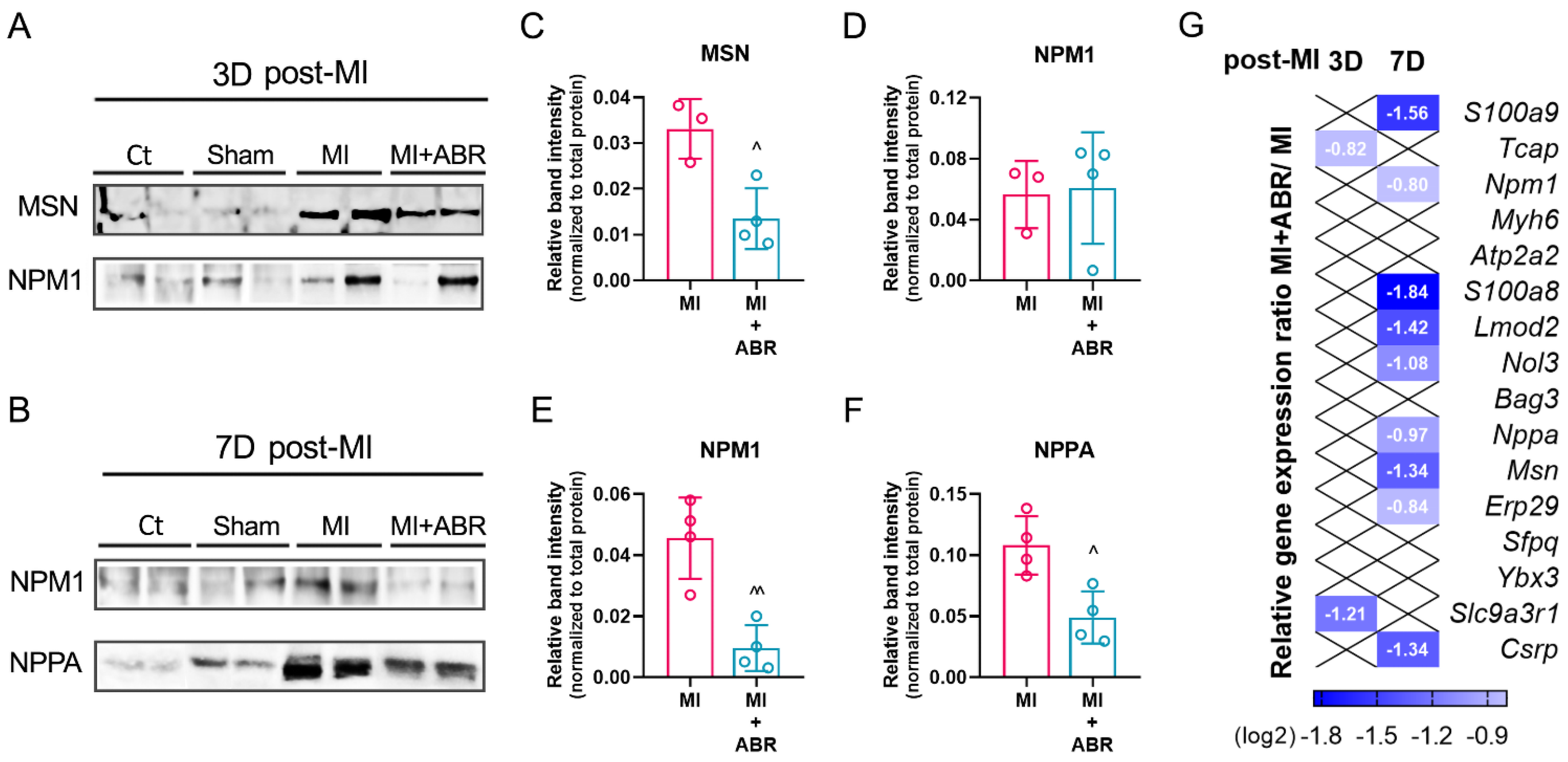
2.7. Western Blot Validation of the LC-MS/MS Data
2.8. Gene Expression Analysis of Differentially Expressed Proteins (DEPs)
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Mouse Model of Myocardial Infarction
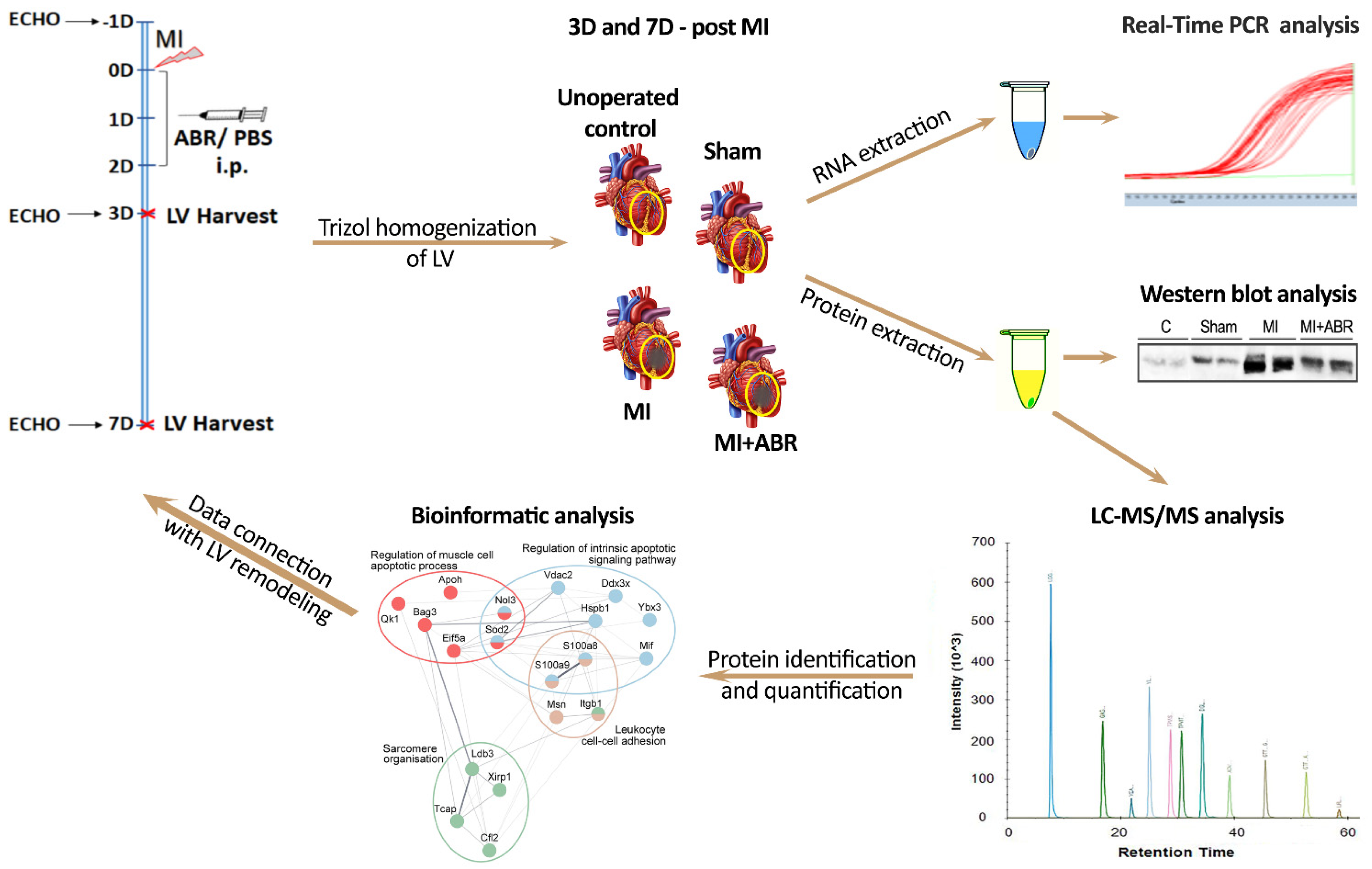
4.3. Experimental Design
4.4. RNA and Protein Extraction
4.5. Liquid Chromatography Coupled to Mass Spectrometry (LC-MS/MS) Analysis
4.6. Data Processing
4.7. Bioinformatic Analysis
4.8. Real-Time PCR
4.9. Western Blot Assays
4.10. Statistics
5. Conclusions
- (i)
- downregulates the expression of proteins involved in leukocytes recruitment and has important beneficial consequences on post-ischemic myocardial repair and recovery;
- (ii)
- favorably modulates the levels of multiple anti-apoptotic and pro-apoptotic proteins involved in p53-related pathways;
- (iii)
- maintains higher expression levels of cardiac structural proteins and inhibits several mediators previously found to drive abnormal compensatory cardiac recovery;
- (iv)
- reduces the markers of cardiac stress, such as NPM1, CSRP3, NPPA, TPT1 and NOL3.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sutton, M.G.; Sharpe, N. Left ventricular remodeling after myocardial infarction: Pathophysiology and therapy. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; Rivera, O.J.; Lindsey, M.L. Myocardial infarction remodeling that progresses to heart failure: A signaling misunderstanding. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H71–H79. [Google Scholar] [CrossRef] [PubMed]
- Bejjani, A.T.; Saab, S.A.; Muhieddine, D.H.; Habeichi, N.J.; Booz, G.W.; Zouein, F.A. Spatiotemporal dynamics of immune cells in early left ventricular remodeling after acutemyocardial infarction in mice. J. Cardiovasc. Pharmacol. 2020, 75, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Shinagawa, H.; Frantz, S. Cellular Immunity and Cardiac Remodeling After Myocardial Infarction: Role of Neutrophils, Monocytes, and Macrophages. Curr. Heart Fail Rep. 2015, 12, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Halmosi, R.; Deres, L.; Gal, R.; Eros, K.; Sumegi, B.; Toth, K. PARP inhibition and postinfarction myocardial remodeling. Int. J. Cardiol. 2016, 217, S52–S59. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, B.; Yang, X.; Zhang, C.; Jiao, Y.; Li, P.; Liu, Y.; Li, Z.; Qiao, B.; Lau, W.B.; et al. S100a8/a9 Signaling Causes Mitochondrial Dysfunction and Cardiomyocyte Death in Response to Ischemic/ReperfusionInjury. Circulation 2019, 140, 751–764. [Google Scholar] [CrossRef]
- Marinković, G.; Koenis, D.S.; De Camp, L.; Jablonowski, R.; Graber, N.; De Waard, V.; De Vries, C.J.; Goncalves, I.; Nilsson, J.; Jovinge, S.; et al. S100A9 links inflammation and repair in myocardial infarction. Circ. Res. 2020, 127, 664–676. [Google Scholar] [CrossRef]
- Marinković, G.; Grauen Larsen, H.; Yndigegn, T.; Szabo, I.A.; Mares, R.G.; De Camp, L.; Weiland, M.; Toma, L.; Goncalves, I.; Nilsson, J.; et al. Inhibition of pro-inflammatory myeloid cell responses by short-term S100A9 blockade improves cardiac function after myocardial infarction. Eur. Heart J. 2019, 40, 2713–2723. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The biological basis for cardiac repair after myocardial infarction. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- García-Ortiz, A.; Serrador, J.M. Erm proteins at the crossroad of leukocyte polarization, migration and intercellular adhesion. Int. J. Mol. Sci. 2020, 21, 1502. [Google Scholar] [CrossRef] [Green Version]
- Teringova, E.; Tousek, P. Apoptosis in ischemic heart disease. J. Transl. Med. 2017, 15, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carluccio, E.; Tommasi, S.; Bentivoglio, M.; Buccolieri, M.; Filippucci, L.; Prosciutti, L.; Corea, L. Prognostic value of left ventricular hypertrophy and geometry in patients with a first, uncomplicated myocardial infarction. Int. J. Cardiol. 2000, 74, 177–183. [Google Scholar] [CrossRef]
- Mak, T.W.; Hauck, L.; Grothe, D.; Billia, F. P53 Regulates the Cardiac Transcriptome. Proc. Natl. Acad. Sci. USA 2017, 114, 2331–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, H.J.; Gao, C.B.; Gong, J.L.; Li, X.J.; Sun, B.; Li, X.N. Comparative proteomic analysis in left ventricular remodeling following myocardial infarction in rats. Biomed. Environ. Sci. 2012, 25, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Rüdebusch, J.; Benkner, A.; Poesch, A.; Dörr, M.; Völker, U.; Grube, K.; Hammer, E.; Felix, S.B. Dynamic adaptation of myocardial proteome during heart failure development. PLoS ONE 2017, 12, e0185915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yajima, Y.; Hiratsuka, T.; Kakimoto, Y.; Ogawa, S.; Shima, K.; Yamazaki, Y.; Yoshikawa, K.; Tamaki, K.; Tsuruyama, T. Region of Interest analysis using mass spectrometry imaging of mitochondrial and sarcomeric proteins in acute cardiac infarction tissue. Sci. Rep. 2018, 8, 7493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, D.; Xia, Y.; Chen, Z.; Chen, A.; Wu, Y.; Jia, J.; Sun, A.; Zou, Y.; Qian, J.; Ge, J. Cardiac proteome profiling in ischemic and dilated cardiomyopathy mouse models. Front. Physiol. 2019, 10, 750. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Li, T.; Ma, L.; Fan, F.; Jin, Y.; Shen, J. The whole transcriptome and proteome changes in the early stage of myocardial infarction. Cell Death Discov. 2019, 5, 73. [Google Scholar] [CrossRef]
- Bouma, G.; Lam-Tse, W.K.; Wierenga-Wolf, A.F.; Drexhage, H.A.; Versnel, M.A. Increased serum levels of MRP-8/14 in type 1 diabetes induce an increased expression of CD11b and an enhanced adhesion of circulating monocytes to fibronectin. Diabetes 2004, 53, 1979–1986. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.J.; Tessier, P.; Poulsom, R.; Hogg, N. The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J. Biol. Chem. 2002, 277, 3658–3665. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Opavsky, M.A.; Stewart, D.J.; Rabinovitch, M.; Dawood, F.; Wen, W.H.; Liu, P.P. Temporal response and localization of integrins β1 and β3 in the heart after myocardial infarction: Regulation by cytokines. Circulation 2003, 107, 1046–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panicker, S.R.; Yago, T.; Shao, B.; McEver, R.P. Neutrophils lacking ERM proteins polarize and crawl directionally but have decreased adhesion strength. Blood Adv. 2020, 4, 3559–3571. [Google Scholar] [CrossRef] [PubMed]
- Rey-Gallardo, A.; Tomlins, H.; Joachim, J.; Rahman, I.; Kitscha, P.; Frudd, K.; Parsons, M.; Ivetic, A. Sequential binding of ezrin and moesin to L-selectin regulates monocyte protrusive behaviour during transendothelial migration. J. Cell Sci. 2018, 131, jcs215541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyawaki, A.; Mitsuhara, Y.; Orimoto, A.; Nakayasu, Y.; Tsunoda, S.I.; Obana, M.; Maeda, M.; Nakayama, H.; Yoshioka, Y.; Tsutsumi, Y.; et al. Moesin is activated in cardiomyocytes in experimental autoimmune myocarditis and mediates cytoskeletal reorganization with protrusion formation. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H476–H486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Chen, H.; Ding, F.; Zhang, Y.; Luo, A.; Wang, M.; Liu, Z. A novel p53 target gene, S100A9, induces p53-dependent cellular apoptosis and mediates the p53 apoptosis pathway. Biochem. J. 2009, 422, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Van Remmen, H.; Williams, M.D.; Guo, Z.; Estlack, L.; Yang, H.; Carlson, E.J.; Epstein, C.J.; Huang, T.T.; Richardson, A. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1422–H1432. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Che, W.; Zheng, C.; Liu, W.; Wen, J.; Fu, H.; Tang, K.; Zhang, J.; Xu, Y. SIRT5: A safeguard against oxidative stress-induced apoptosis in cardiomyocytes. Cell. Physiol. Biochem. 2013, 32, 1050–1059. [Google Scholar] [CrossRef]
- Shen, X.; Zheng, S.; Metreveli, N.S.; Epstein, P.N. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes 2006, 55, 798–805. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, K.A.; Abraham, D.M.; Martin, A.S.; Mao, L.; Liu, J.; Gu, H.; Locasale, J.W.; Hirschey, M.D. Sirtuin 5 is required for mouse survival in response to cardiac pressure overload. J. Biol. Chem. 2017, 292, 19767–19781. [Google Scholar] [CrossRef] [Green Version]
- Myers, V.D.; McClung, J.M.; Wang, J.F.; Tahrir, F.G.; Gupta, M.K.; Gordon, J.; Kontos, C.H.; Khalili, K.; Cheung, J.Y.; Feldman, A.M. The Multifunctional Protein BAG3: A Novel Therapeutic Target in Cardiovascular Disease. JACC Basic Transl. Sci. 2018, 3, 122–131. [Google Scholar] [CrossRef]
- Donath, S.; Li, P.; Willenbockel, C.; Al-Saadi, N.; Gross, V.; Willnow, T.; Bader, M.; Martin, U.; Bauersachs, J.; Wollert, K.C.; et al. Apoptosis repressor with caspase recruitment domain is required for cardioprotection in response to biomechanical and ischemic stress. Circulation 2006, 113, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.Z.; Lu, D.Y.; Tan, W.Q.; Wang, J.X.; Li, P.F. p53 initiates apoptosis by transcriptionally targeting the antiapoptotic protein ARC. Mol. Cell. Biol. 2008, 28, 564–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klumpe, I.; Savvatis, K.; Westermann, D.; Tschöpe, C.; Rauch, U.; Landmesser, U.; Schultheiss, H.P.; Dörner, A. Transgenic overexpression of adenine nucleotide translocase 1 protects ischemic hearts against oxidative stress. J. Mol. Med. 2016, 94, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.N.; Balasubramanian, K.; Ramoth, J.A.; Schroit, A.J. Beta-2-glycoprotein 1-dependent macrophage uptake of apoptotic cells. J. Biol. Chem. 2008, 283, 3761–3766. [Google Scholar] [CrossRef] [Green Version]
- Li, A.L.; Li, H.Y.; Jin, B.F.; Ye, Q.N.; Zhou, T.; Yu, X.D.; Pan, X.; Man, J.H.; He, K.; Yu, M.; et al. A novel eIF5A complex functions as a regulator of p53 and p53-dependent apoptosis. J. Biol. Chem. 2004, 279, 49251–49258. [Google Scholar] [CrossRef] [Green Version]
- Pilotte, J.; Larocque, D.; Richard, S. Nuclear translocation controlled by alternatively spliced isoforms inactivates the QUAKING apoptotic inducer. Genes Dev. 2001, 15, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Yan, G.; Qin, Q.; Yi, B.; Chuprun, K.; Sun, H.; Huang, S.; Sun, J. Protein-L-isoaspartate (D-aspartate) O-methyltransferase protects cardiomyocytes against hypoxia induced apoptosis through inhibiting proapoptotic kinase Mst1. Int. J. Cardiol. 2013, 168, 3291–3299. [Google Scholar] [CrossRef] [Green Version]
- Dhar, S.K.; St Clair, D.K. Nucleophosmin blocks mitochondrial localization of p53 and apoptosis. J. Biol. Chem. 2009, 284, 16409–16418. [Google Scholar] [CrossRef] [Green Version]
- Avitabile, D.; Bailey, B.; Cottage, C.T.; Sundararaman, B.; Joyo, A.; McGregor, M.; Gude, N.; Truffa, S.; Zarrabi, A.; Konstandin, M.; et al. Nucleolar stress is an early response to myocardial damage involving nucleolar proteins nucleostemin and nucleophosmin. Proc. Natl. Acad. Sci. USA 2011, 108, 6145–6150. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, N.; Sussman, M.A. Stressing on the nucleolus in cardiovascular disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 798–801. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Rajapurohitam, V.; Kilić, A.; Hunter, J.C.; Zeidan, A.; Said Faruq, N.; Escobales, N.; Karmazyn, M. Expression of mitochondrial fusion-fission proteins during post-infarction remodeling: The effect of NHE-1 inhibition. Basic Res. Cardiol. 2011, 106, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Dayawansa, N.H.; Gao, X.M.; White, D.A.; Dart, A.M.; Du, X.J. Role of MIF in myocardial ischaemia and infarction: Insight from recent clinical and experimental findings. Clin. Sci. 2014, 127, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.Y.; Hausenloy, D.J.; Arjun, S.; Price, A.N.; Davidson, S.M.; Lythgoe, M.F.; Yellon, D.M. Mitochondrial cyclophilin-D as a potential therapeutic target for post-myocardial infarction heart failure. J. Cell. Mol. Med. 2011, 15, 2443–2451. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Nan, J.; Sun, Y.; Zhu, D.; Xiao, C.; Wang, Y.; Zhu, L.; Wu, Y.; Zhao, J.; Wu, R.; et al. Electron leak from NDUFA13 within mitochondrial complex I attenuates ischemia-reperfusion injury via dimerized STAT3. Proc. Natl. Acad. Sci. USA 2017, 114, 11908–11913. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Liu, X. Identification of key genes involved in myocardial infarction. Eur. J. Med. Res. 2019, 24, 22. [Google Scholar] [CrossRef] [Green Version]
- Cooke, A.; Schwarzl, T.; Huppertz, I.; Kramer, G.; Mantas, P.; Alleaume, A.M.; Huber, W.; Krijgsveld, J.; Hentze, M.W. The RNA-binding protein YBX3 controls amino acid levels by regulating SLC mRNA abundance. Cell Rep. 2019, 27, 3097–3106. [Google Scholar] [CrossRef] [Green Version]
- Hosokawa, M.; Takeuchi, A.; Tanihata, J.; Iida, K.; Takeda, S.; Hagiwara, M. Loss of RNA-binding protein Sfpq causes long-gene transcriptopathy in skeletal muscle and severe muscle mass reduction with metabolic myopathy. iScience 2019, 13, 229–242. [Google Scholar] [CrossRef] [Green Version]
- Swiatkowska, A.; Dutkiewicz, M.; Machtel, P.; Janecki, D.M.; Kabacinska, M.; Żydowicz-Machtel, P.; Ciesiołka, J. Regulation of the p53 expression profile by hnRNP K under stress conditions. RNA Biol. 2020, 17, 1402–1415. [Google Scholar] [CrossRef]
- Wang, P.; Chen, H.; Qin, H.; Sankarapandi, S.; Becher, M.W.; Wong, P.C.; Zweier, J.L. Overexpression of human copper,zinc-superoxide dismutase (SOD1) prevents postischemic injury. Proc. Natl. Acad. Sci. USA 1998, 95, 4556–4560. [Google Scholar] [CrossRef] [Green Version]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Steenbergen, C.; Murphy, E. Does the voltage dependent anion channel modulate cardiac ischemia-reperfusion injury? Biochim. Biophys. Acta 2012, 1818, 1451–1456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Fujita, T.; Hidaka, Y.; Jin, H.; Suita, K.; Shigeta, M.; Kiyonari, H.; Umemura, M.; Yokoyama, U.; Sadoshima, J.; et al. Translationally controlled tumor protein (TCTP) plays a pivotal role in cardiomyocyte survival through a Bnip3-dependent mechanism. Cell Death Dis. 2019, 10, 549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirion, C.; Stucka, R.; Mendel, B.; Gruhler, A.; Jaksch, M.; Nowak, K.J.; Binz, N.; Laing, N.G.; Lochmïller, H. Characterization of human muscle type cofilin (CFL2) in normal and regenerating muscle. Eur. J. Biochem. 2001, 268, 3473–3482. [Google Scholar] [CrossRef] [PubMed]
- Rodal, A.A.; Tetreault, J.W.; Lappalainen, P.; Drubin, D.G.; Amberg, D.C. Aip1p interacts with cofilin to disassemble actin filaments. J. Cell Biol. 1999, 145, 1251–1264. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.; Chau, V.Q.; Mauro, A.G.; Durrant, D.; Toldo, S.; Abbate, A.; Das, A.; Salloum, F.N. Hydrogen sulfide therapy suppresses cofilin-2 and attenuates ischemic heart failure in a mouse model of myocardial infarction. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 472–483. [Google Scholar] [CrossRef]
- Bai, F.; Wang, L.; Kawai, M. A study of tropomyosin’s role in cardiac function and disease using thin-filament reconstituted myocardium. J. Muscle Res. Cell. Motil. 2013, 34, 295–310. [Google Scholar] [CrossRef] [Green Version]
- Mi-Mi, L.; Farman, G.P.; Mayfield, R.M.; Strom, J.; Chu, M.; Pappas, C.T.; Gregorio, C.C. In vivo elongation of thin filaments results in heart failure. PLoS ONE 2020, 15, e0226138. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Jiao, L.; Sun, L.; Li, Y.; Gao, Y.; Xu, C.; Shao, Y.; Li, M.; Li, C.; Lu, Y.; et al. LncRNA ZFAS1 as a SERCA2a inhibitor to cause intracellular Ca2+ overload and contractile dysfunction in a mouse model of myocardial infarction. Circ. Res. 2018, 122, 1354–1368. [Google Scholar] [CrossRef]
- Lipskaia, L.; Chemaly, E.R.; Hadri, L.; Lompre, A.M.; Hajjar, R.J. Sarcoplasmic reticulum Ca2+ ATPase as a therapeutic target for heart failure. Expert Opin. Biol. Ther. 2010, 10, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Boateng, S.Y.; Belin, R.J.; Geenen, D.L.; Margulies, K.B.; Martin, J.L.; Hoshijima, M.; De Tombe, P.P.; Russell, B. Cardiac dysfunction and heart failure are associated with abnormalities in the subcellular distribution and amounts of oligomeric muscle LIM protein. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H259–H269. [Google Scholar] [CrossRef]
- Song, W.; Wang, H.; Wu, Q. Atrial natriuretic peptide in cardiovascular biology and disease (NPPA). Gene 2015, 569, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, E.; Lei, Y.H.; Shang, X.; Huang, Z.M.; Zuo, L.; Boucher, M.; Fan, Q.; Chuprun, J.K.; Ma, X.L.; Koch, W.J. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 2010, 107, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Heberle, H.; Meirelles, V.G.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; Hewapathirana, S.; García-Seisdedos, D.; Kamatchinathan, S.; Kundu, D.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A Hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3 Days Post-MI | 7 Days Post-MI | ||||
|---|---|---|---|---|---|
| Uniprot Accession Key | Protein Description | # Unique Peptides | Sequest Score | # Unique Peptides | Sequest Score |
| P62082 | 40S ribosomal protein S7 | - | - | 9 | 346.06 |
| Q9CXW4 | 60S ribosomal protein L11 | - | - | 7 | 192.95 |
| P48962 | ADP/ATP translocase 1 | - | - | 10 | 1226.26 |
| Q9JI91 | Alpha-actinin-2 | - | - | 11 | 266.49 |
| Q62167 | ATP-dependent RNA helicase DDX3X | 3 | 80.25 | - | - |
| Q9JLV1 | BAG family molecular chaperone regulator 3 | 25 | 2680.89 | 30 | 4404.55 |
| Q01339 | Beta-2-glycoprotein 1 | 24 | 2855.16 | - | - |
| P45591 | Cofilin-2 | 8 | 1206.06 | 14 | 1043.4 |
| P50462 | Cysteine and glycine-rich protein 3 | - | - | 23 | 5298.01 |
| P57759 | Endoplasmic reticulum resident protein 29 | - | - | 5 | 105.24 |
| P63242 | Eukaryotic translation initiation factor 5A-1 | 10 | 851.31 | 13 | 1115.78 |
| O09164 | Extracellular superoxide dismutase [Cu-Zn] | - | - | 11 | 404.16 |
| P11352 | Glutathione peroxidase 1 | - | - | 10 | 966.72 |
| P14602 | Heat shock protein beta-1 | 19 | 6349.35 | 20 | 8314.62 |
| P61979 | Heterogeneous nuclear ribonucleoprotein K | - | - | 28 | 3133.52 |
| P09055 | Integrin beta-1 | 2 | 1287.76 | - | - |
| Q3UHZ5 | Leiomodin-2 | - | - | 4 | 64.35 |
| P34884 | Macrophage Migration inhibitory factor | 4 | 649.78 | 6 | 1206.71 |
| Q9CQ92 | Mitochondrial fission 1 protein | - | - | 4 | 125,56 |
| P26041 | Moesin | 2 | 13.65 | - | - |
| Q02566 | Myosin-6 | - | - | 121 | 14,283.36 |
| P70441-1 | Na(+)/H(+) exchange regulatory cofactor NHE-RF1 | - | - | 4 | 49.87 |
| Q8K2C6 | NAD-dependent protein deacylase sirtuin-5, mitochondrial | - | - | 3 | 73.49 |
| Q9ERS2 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 13 | - | - | 6 | 533.68 |
| P05125 | Natriuretic peptides A | - | - | 7 | 558.71 |
| Q99K48 | Non-POU domain-containing octamer-binding protein | - | - | 5 | 45.36 |
| Q9D1X0 | Nucleolar protein 3 | 7 | 1464.15 | 10 | 1288.64 |
| Q61937 | Nucleophosmin | - | - | 15 | 4324.81 |
| Q99KR7 | Peptidyl-prolyl cis-trans isomerase F, mitochondrial | - | - | 16 | 3735.42 |
| Q9QYS9 | Protein quaking | 4 | 60.89 | 6 | 217.52 |
| P27005 | Protein S100-A8 | 4 | 2554.06 | - | - |
| P31725 | Protein S100-A9 | 6 | 1466.6 | 8 | 1068.22 |
| Q99LX0 | Protein/nucleic acid deglycase DJ-1 | - | - | 13 | 2671.72 |
| P23506 | Protein-L-isoaspartate(D-aspartate) O-methyltransferase | - | - | 9 | 798.88 |
| O55143-1 | Sarcoplasmic/endoplasmic reticulum calcium ATPase 2 | - | - | 9 | 168.54 |
| Q8VIJ6 | Splicing factor, proline- and glutamine-rich | - | - | 10 | 612.84 |
| P08228 | Superoxide dismutase [Cu-Zn] | - | - | 21 | 8472.76 |
| P09671 | Superoxide dismutase [Mn], mitochondrial | 11 | 4230.63 | 15 | 3949.1 |
| O70548 | Telethonin | 9 | 1360.44 | - | - |
| A2ASS6 | Titin | - | - | 7 | 47.42 |
| P63028 | Translationally-controlled tumor protein | - | - | 9 | 2521.48 |
| P58771-1 | Tropomyosin alpha-1 chain | - | - | 7 | 74,845.96 |
| Q60930 | Voltage-dependent anion-selective channel protein 2 | 11 | 1464.28 | - | - |
| O88342 | WD repeat-containing protein 1 | - | - | 4 | 21.46 |
| O70373 | Xin actin-binding repeat-containing protein 1 | 9 | 298.66 | 25 | 1030.8 |
| Q9JKB3-1 | Y-box-binding protein 3 | 5 | 1618.17 | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boteanu, R.M.; Suica, V.-I.; Uyy, E.; Ivan, L.; Cerveanu-Hogas, A.; Mares, R.G.; Simionescu, M.; Schiopu, A.; Antohe, F. Short-Term Blockade of Pro-Inflammatory Alarmin S100A9 Favorably Modulates Left Ventricle Proteome and Related Signaling Pathways Involved in Post-Myocardial Infarction Recovery. Int. J. Mol. Sci. 2022, 23, 5289. https://doi.org/10.3390/ijms23095289
Boteanu RM, Suica V-I, Uyy E, Ivan L, Cerveanu-Hogas A, Mares RG, Simionescu M, Schiopu A, Antohe F. Short-Term Blockade of Pro-Inflammatory Alarmin S100A9 Favorably Modulates Left Ventricle Proteome and Related Signaling Pathways Involved in Post-Myocardial Infarction Recovery. International Journal of Molecular Sciences. 2022; 23(9):5289. https://doi.org/10.3390/ijms23095289
Chicago/Turabian StyleBoteanu, Raluca Maria, Viorel-Iulian Suica, Elena Uyy, Luminita Ivan, Aurel Cerveanu-Hogas, Razvan Gheorghita Mares, Maya Simionescu, Alexandru Schiopu, and Felicia Antohe. 2022. "Short-Term Blockade of Pro-Inflammatory Alarmin S100A9 Favorably Modulates Left Ventricle Proteome and Related Signaling Pathways Involved in Post-Myocardial Infarction Recovery" International Journal of Molecular Sciences 23, no. 9: 5289. https://doi.org/10.3390/ijms23095289