
The Transcriptome and Metabolome Reveal the Potential Mechanism of Lodging Resistance in Intergeneric Hybrids between Brassica napus and Capsella bursa-pastoris

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Anatomical Structure and Lignocellulose Analysis of ZY821 and YG689
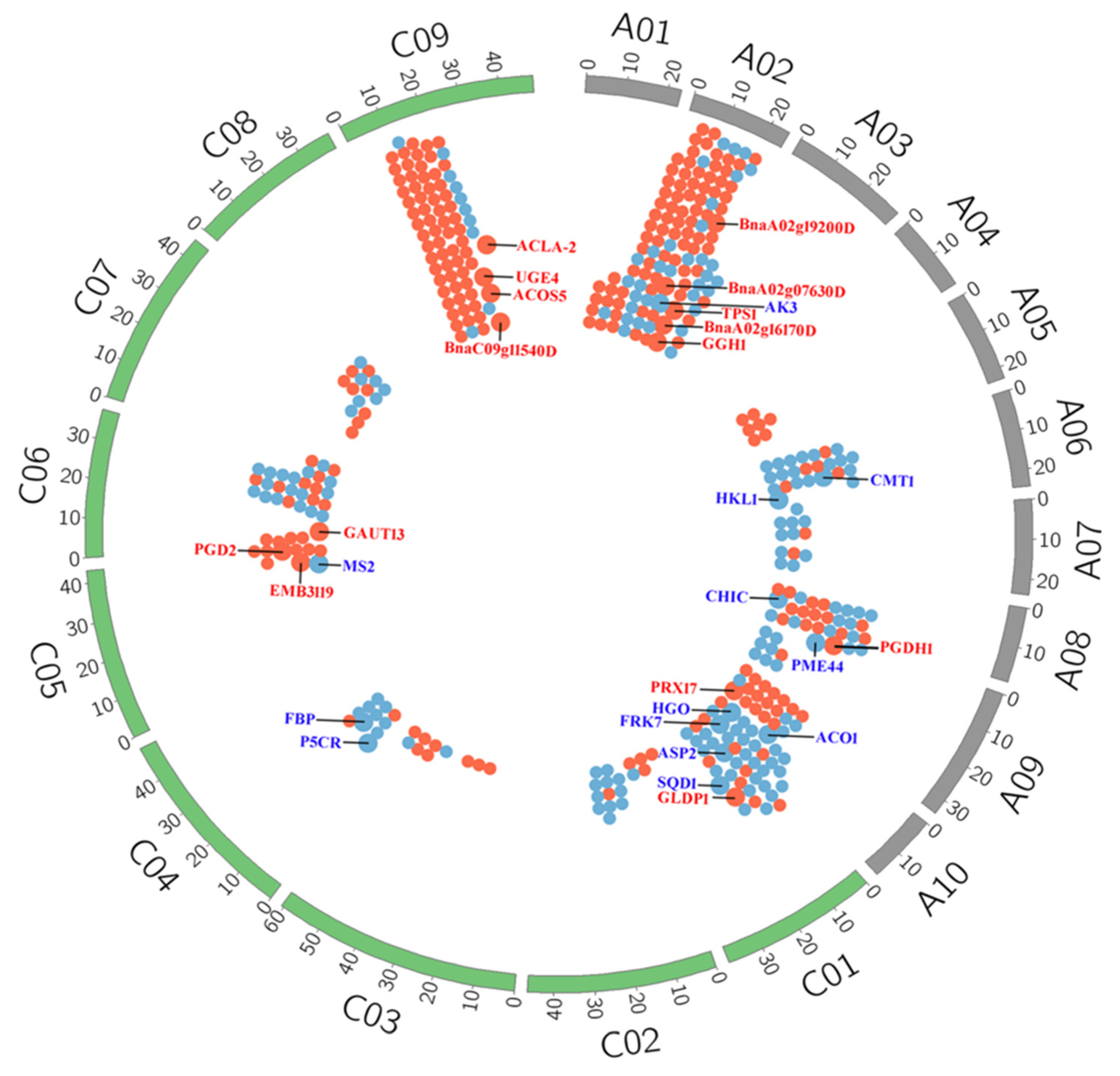
2.2. Transcriptome Analysis of YG689 and ZY821
2.3. Metabolome Analysis of Stem of YG689 and ZY821
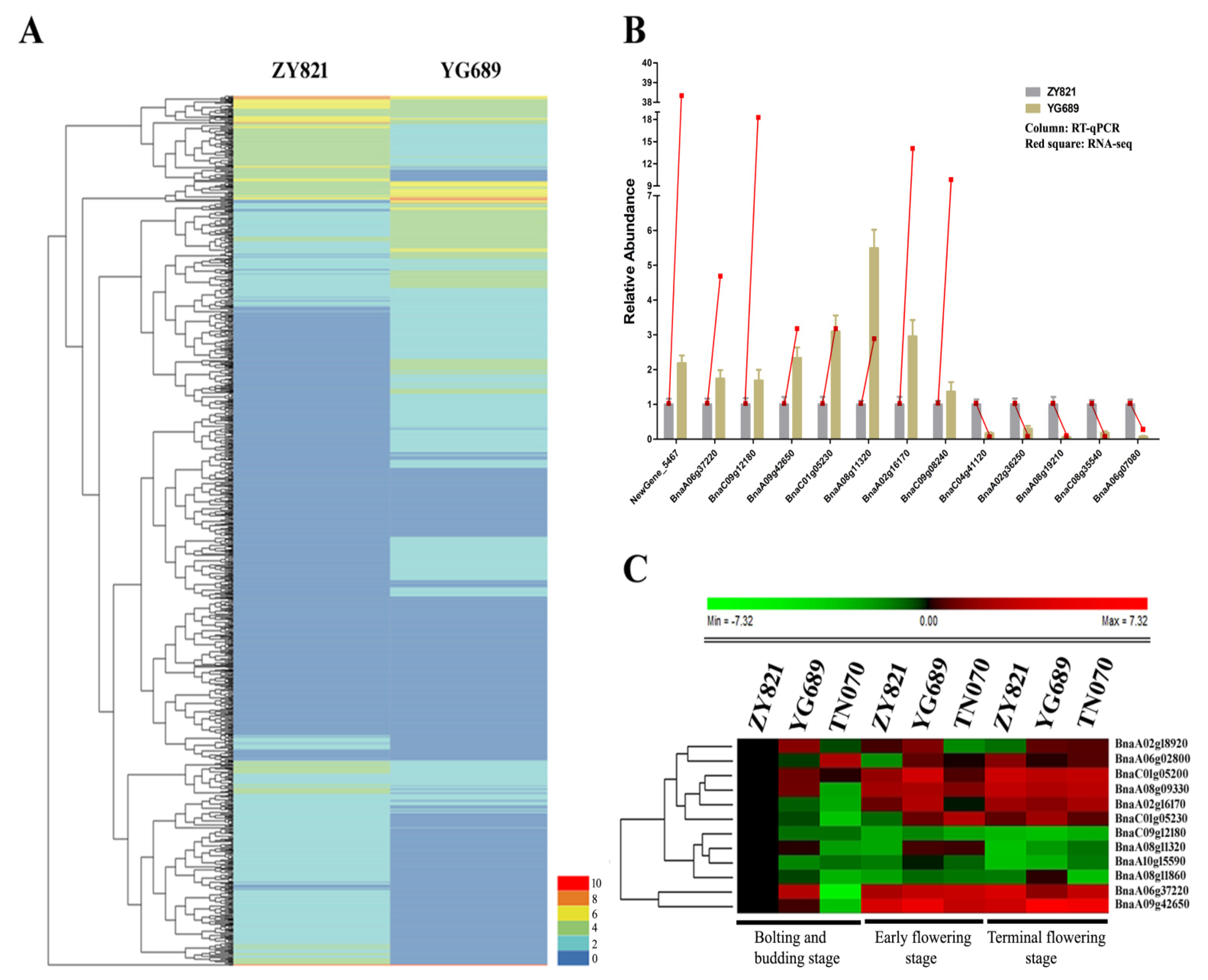
2.4. The Validation of DEGs Regulating the Metabolic Pathway of Lignocellulose Synthesis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Anatomical Structure and Lignocellulose Content Analysis of YG689 and ZY821
4.3. RNA Extraction and Transcriptome Sequencing
4.4. Differentially Genes Expression and Function Enrichment Analysis
4.5. Metabolome Analysis of YG689 and ZY821
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berry, P.M.; Spink, J.H.; Gay, A.P.; Craigon, J. A comparison of root and stem lodging risks among winter wheat cultivars. J. Agric. Sci. 2003, 141, 191–202. [Google Scholar] [CrossRef]
- Zuber, U.; Winzeler, H.; Messmer, M.M.; Keller, M.; Stampet, P. Morphological traits associated with lodging resistance of spring wheat (Triticumae stivum L.). J. Agron. Crop Sci 1999, 182, 17–24. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, P.; Zhang, X.; Zheng, Q.; Chen, M.; Ge, F.; Li, Z.; Sun, W.; Guan, Z.; Liang, T.; et al. Multi-Locus Genome-Wide Association Study Reveals the Genetic Architecture of Stalk Lodging Resistance-Related Traits in Maize. Front. Plant Sci. 2018, 9, 611. [Google Scholar] [CrossRef]
- Kashiwagi, T.; Ishimaru, K. Identification and functional analysis of a locus for improvement of lodging resistance in rice. Plant. Physiol. 2004, 134, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Pinthus, M.J. Lodging in wheat, barley and oats: The phenomenon, its causes and preventative measures. Adv. Agron. 1973, 25, 210–263. [Google Scholar]
- Ma, J.F.; Yamaji, N. Silicon uptake and accumulation in higher plants. Trends Plant Sci. 2006, 11, 392–397. [Google Scholar] [CrossRef]
- Islam, M.S.; Peng, S.; Visperas, R.M.; Ereful, N.; Bhuiya, M.; Julfiquar, A.W. Lodging-related morphological traits of hybrid rice in a tropical irrigated ecosystem. Field Crop Res. 2007, 101, 240–248. [Google Scholar] [CrossRef]
- Ma, Q.H.; Xu, Y.; Lin, Z.B.; He, P. Cloning of cDNA encoding COMT from wheat which is differentially expressed in lodging-sensitive and-resistant cultivars. J. Exp. Bot. 2002, 53, 2281–2282. [Google Scholar] [CrossRef] [Green Version]
- Ookawa, T.; Inoue, K.; Matsuoka, M.; Ebitani, T.; Takarada, T.; Yamamoto, T.; Ueda, T.; Yokoyama, T.; Sugiyama, C.; Nakaba, S.; et al. Increased lodging resistance in long- culm, low-lignin gh2 rice for improved feed and bioenergy production. Sci. Rep. 2014, 4, 6567. [Google Scholar] [CrossRef] [Green Version]
- Ookawa, T.; Ishihara, K. Varietal differences of physical characteristics of the culm related to lodging in paddy rice. Jpn. J. Crop Sci. 1992, 61, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Kashiwagi, T.; Madoka, Y.; Hirotsu, N.; Ishimaru, K. Locus prl5 improves lodging resistance of rice by delaying senescence and increasing carbohydrate reaccumulation. Plant Physiol. Biochem. 1992, 44, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.; Chen, X.; Yin, Y.; Lu, K.; Yang, W.; Tang, Y.; Wang, Z. Lodging resistance of winter wheat (Triticumae stivum L.) lignin accumulation and its related enzymes activities due to the application of paclobutrazol or gibberellin acid. Field Crop Res. 2014, 157, 1–7. [Google Scholar] [CrossRef]
- Tan, H.; Xie, Q.; Xiang, X.; Li, J.; Zheng, S.; Xu, X.; Guo, H.; Ye, W. Dynamic metabolic profiles and tissue specific source effects on the metabolome of developing seeds of Brassica napus. PLoS ONE 2015, 10, e0124794. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.; Xiang, X.; Tang, J.; Wang, X. Nutritional functions of the funiculus in Brassica napus seed maturation revealed by transcriptome and dynamic metabolite profile analyses. Plant Mol. Biol. 2016, 92, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisjuk, L.; Neuberger, T.; Schwender, J.; Heinzel, N.; Sunderhaus, S.; Fuchs, J.; Hay, J.O.; Tschiersch, H.; Braun, H.P.; Denolf, P.; et al. Seed architecture shapes embryo metabolism in oilseed rape. Plant Cell. 2013, 25, 1625–1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortesniemi, M.; Vuorinen, A.L.; Sinkkonen, J.; Yang, B.; Rajala, A.; Kallio, H. NMR metabolomics of ripened and developing oilseed rape (Brassica napus) and turnip rape (Brassica rapa). Food Chem. 2015, 172, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Jian, H.; Lu, K.; Yin, N.; Wang, J.; Duan, X.; Li, W.; Liu, L.; Xu, X.; Wang, R.; et al. Genetic and transcriptomic analyses of lignin- and lodging-related traits in Brassica napus. Theor. Appl. Genet. 2017, 130, 1961–1973. [Google Scholar] [CrossRef]
- Li, H.; Cheng, X.; Zhang, L.; Hu, J.; Zhang, F.; Chen, B.; Xu, K.; Gao, G.; Li, H.; Li, L.; et al. An Integration of Genome-Wide Association Study and Gene Co-expression Network Analysis Identifies Candidate Genes of Stem Lodging-Related Traits in Brassica napus. Front. Plant Sci. 2018, 9, 796. [Google Scholar] [CrossRef]
- Wei, C.; Zhu, L.; Wen, J.; Yi, B.; Ma, C.; Tu, J.; Shen, J.; Fu, T. Morphological, transcriptomics and biochemical characterization of new dwarf mutant of Brassica napus. Plant Sci. 2018, 270, 97–113. [Google Scholar] [CrossRef]
- Miller, C.N.; Harper, A.L.; Trick, M.; Wellner, N.; Werner, P.; Waldron, K.W.; Bancroft, I. Dissecting the complex regulation of lodging resistance in Brassica napus. Mol. Breed. 2018, 38, 30. [Google Scholar] [CrossRef] [Green Version]
- Kawakatsu, T.; Teramoto, S.; Takayasu, S.; Maruyama, N.; Nishijima, R.; Kitomi, Y.; Uga, Y. The transcriptomic landscapes of rice cultivars with diverse root system architectures grown in upland field conditions. Plant J. 2021, 106, 1177–1190. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolska, I.; Businge, E.; Abreu, I.N.; Moritz, T.; Egertsdotter, U. Metabolome and transcriptome profiling reveal new insights into somatic embryo germination in Norway spruce (Picea abies). Tree Physiol. 2017, 37, 1752–1766. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yao, Q.; Xia, E.; Gao, L. Metabolomics and Transcriptomics Analyses Reveal Nitrogen Influences on the Accumulation of Flavonoids and Amino Acids in Young Shoots of Tea Plant (Camellia sinensis L.) Associated with Tea Flavor. J. Agric. Food Chem. 2018, 66, 9828–9838. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, W.; Feng, Y.; Tu, M.; Wittich, P.E.; Bate, N.J.; Messing, J. Transcriptome and metabolome reveal distinct carbon allocation patterns during internode sugar accumulation in different sorghum genotypes. Plant Biotechnol. J. 2019, 17, 472–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reem, N.T.; Chen, H.Y.; Hur, M.; Zhao, X.; Wurtele, E.S.; Li, X.; Li, L.; Zabotina, O. Comprehensive transcriptome analyses correlated with untargeted metabolome reveal differentially expressed pathways in response to cell wall alterations. Plant Mol. Biol. 2018, 96, 509–529. [Google Scholar] [CrossRef]
- Verhoeven, K.J.F.; Verbon, E.H.; van Gurp, T.P.; Oplaat, C.; Ferreira de Carvalho, J.; Morse, A.M.; Stahl, M.; Macel, M.; McIntyre, L.M. Intergenerational environmental effects: Functional signals in offspring transcriptomes and metabolomes after parental jasmonic acid treatment in apomictic dandelion. New Phytol. 2018, 217, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Gyawali, A.; Morrison, G.D.; Saski, C.A.; Robertson, D.J.; Cook, D.D.; Tharayil, N.; Schaefer, R.J.; Beissinger, T.M.; Sekhon, R.S. Genetic Architecture of Maize Rind Strength Revealed by the Analysis of Divergently Selected Populations. Plant Cell Physiol. 2021, 62, 1199–1214. [Google Scholar] [CrossRef]
- Yu, M.; Wang, M.; Gyalpo, T.; Basang, Y. Stem lodging resistance in hulless barley: Transcriptome and metabolome analysis of lignin biosynthesis pathways in contrasting genotypes. Genomics 2021, 113, 935–943. [Google Scholar] [CrossRef]
- Guo, Q.; Li, X.; Niu, L.; Jameson, P.E.; Zhou, W. Transcription-associated metabolomic adjustments in maize occur during combined drought and cold stress. Plant Physiol. 2021, 186, 677–695. [Google Scholar] [CrossRef]
- Zhao, J.; Buchwaldt, L.; Rimmer, S.R.; Sharpe, A.; McGregor, L.; Bekkaoui, D.; Hegedus, D. Patterns of differential gene expression in Brassica napus cultivars infected with Sclerotinia sclerotiorum. Mol Plant Pathol. 2009, 10, 635–649. [Google Scholar] [CrossRef]
- Park, R.J. The occurrence of mustard oil glucosides in Lepidium hyssopifolium Desv., L. bonariense (L.) and Capsella bursa pastoris (L.) Medic. Aust. J. Chem. 1967, 20, 799–2801. [Google Scholar] [CrossRef]
- Sigareva, M.A.; Earle, E.D. Regeneration of plants from protoplasts of Capsella bursa-pastoris and somatic hybridization with rapid cycling Brassica oleracea. Plant Cell Rep. 1999, 18, 412–417. [Google Scholar] [CrossRef]
- Chen, H.F.; Wang, H.; Li, Z.Y. Production and genetic analysis of partial hybrids in intertribal crosses between Brassica species (B. rapa, B. napus) and Capsella bursa-pastoris. Plant Cell Rep. 2007, 26, 1791–1800. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, X.; Fan, Z.; Pang, Y.; Sun, X.; Wang, X.; Tanga, K. Molecular cloning and characterization of a novel cold-regulated gene from Capsella bursa-pastoris. DNA Seq. 2004, 15, 262–268. [Google Scholar] [CrossRef]
- Shen, Y.; Xiang, Y.; Xu, E.; Ge, X.; Li, Z. Major Co-localized QTL for Plant Height, Branch Initiation Height, Stem Diameter, and Flowering Time in an Alien Introgression Derived Brassica napus DH Population. Front. Plant Sci. 2018, 9, 390. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.J. Genetic and epigenetic mechanisms for gene expression and phenotypic variation in plant polyploids. Ann. Rev. Plant Biol. 2007, 58, 377–406. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, A.; Liang, F.; Yao, X.; Wang, Y.; Liu, X.; Zhang, Y.; Dalelhan, J.; Zhang, B.; Qin, M.; et al. Screening of clubroot-resistant varieties and transfer of clubroot resistance genes to Brassica napus using distant hybridization. Breed Sci. 2018, 68, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Gong, Q.; Dai, C.Y.; Zhang, X.H.; Wang, X.L.; Huang, Z.; Xu, A.X.; Dong, J.G.; Yu, C.Y. Towards breeding of rapeseed (Brassica napus) with alien cytoplasm and powdery mildew resistance from Ethiopian mustard (Brassica carinata). Breed Sci. 2020, 70, 387–395. [Google Scholar] [CrossRef]
- Kendall, S.L.; Holmes, H.; White, C.A.; Clarke, S.M.; Berry, P.M. Quantifying lodging-induced yield losses in oilseed rape. Field Crop Res. 2017, 211, 106–113. [Google Scholar] [CrossRef]
- Long, W.; Dan, D.; Yuan, Z.; Chen, Y.; Jin, J.; Yang, W.; Zhang, Z.; Li, N.; Li, S. Deciphering the Genetic Basis of Lodging Resistance in Wild Rice Oryza longistaminata. Front. Plant Sci. 2020, 11, 628. [Google Scholar] [CrossRef]
- Li, F.; Xie, G.; Huang, J.; Zhang, R.; Li, Y.; Zhang, M.; Wang, Y.; Li, A.; Li, X.; Xia, T.; et al. OsCESA9 conserved-site mutation leads to largely enhanced plant lodging resistance and biomass enzymatic saccharification by reducing cellulose DP and crystallinity in rice. Plant Biotechnol. J. 2017, 15, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Cosio, C.; Ranocha, P.; Francoz, E.; Burlat, V.; Zheng, Y.; Perry, S.E.; Ripoll, J.J.; Yanofsky, M.; Dunand, C. The class III peroxidase PRX17 is a direct target of the MADS-box transcription factor AGAMOUS-LIKE15 (AGL15) and participates in lignified tissue formation. New Phytol. 2017, 213, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Gou, X.; Xun, H.; Bian, Y.; Ma, X.; Li, J.; Li, N.; Gong, L.; Feldman, M.; Liu, B.; et al. Homoeologous exchanges occur through intragenic recombination generating novel transcripts and proteins in wheat and other polyploids. Proc. Natl. Acad. Sci. USA 2020, 117, 14561–14571. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Wang, J.; Wang, R.; Duan, M.; Qiao, C.; Chen, X.; Ma, G.; Zhou, X.; Zhu, M.; Jing, F.; et al. Comparative transcriptomic and metabolomic analyses of carotenoid biosynthesis reveal the basis of white petal color in Brassica napus. Planta 2021, 253, 8. [Google Scholar] [CrossRef]
- Tan, H.; Zhang, J.; Qi, X.; Shi, X.; Zhou, J.; Wang, X.; Xiang, X. Correlation analysis of the transcriptome and metabolome reveals the regulatory network for lipid synthesis in developing Brassica napus embryos. Plant Mol. Biol. 2019, 99, 31–44. [Google Scholar] [CrossRef]
- Wang, W.; Pang, J.; Zhang, F.; Sun, L.; Yang, L.; Zhao, Y.; Yang, Y.; Wang, Y.; Siddique, K.H.M. Integrated transcriptomics and metabolomics analysis to characterize alkali stress responses in canola (Brassica napus L.). Plant Physiol. Biochem. 2021, 166, 605–620. [Google Scholar] [CrossRef]
- Courbet, G.; D’Oria, A.; Maillard, A.; Jing, L.; Pluchon, S.; Arkoun, M.; Pateyron, S.; Paysant Le Roux, C.; Diquélou, S.; Ourry, A.; et al. Comparative Omics Analysis of Brassica napus Roots Subjected to Six Individual Macronutrient Deprivations Reveals Deficiency-Specific Genes and Metabolomic Profiles. Int. J. Mol. Sci. 2021, 22, 1679. [Google Scholar] [CrossRef]
- D’Oria, A.; Jing, L.; Arkoun, M.; Pluchon, S.; Pateyron, S.; Trouverie, J.; Etienne, P.; Diquélou, S.; Ourry, A. Transcriptomic, Metabolomic and Ionomic Analyses Reveal Early Modulation of Leaf Mineral Content in Brassica napus under Mild or Severe Drought. Int. J. Mol. Sci. 2022, 23, 781. [Google Scholar] [CrossRef]
- Raza, A.; Su, W.; Hussain, M.A.; Mehmood, S.S.; Zhang, X.; Cheng, Y.; Zou, X.; Lv, Y. Integrated Analysis of Metabolome and Transcriptome Reveals Insights for Cold Tolerance in Rapeseed (Brassica napus L.). Front. Plant Sci. 2021, 12, 721681. [Google Scholar] [CrossRef]
- Fichtner, F.; Barbier, F.F.; Annunziata, M.G.; Feil, R.; Olas, J.J.; Mueller-Roeber, B.; Stitt, M.; Beveridge, C.A.; Lunn, J.E. Regulation of shoot branching in arabidopsis by trehalose 6-phosphate. New Phytol. 2021, 229, 2135–2151. [Google Scholar] [CrossRef]
- Lyra, D.H.; Griffiths, C.A.; Watson, A.; Joynson, R.; Molero, G.; Igna, A.A.; Hassani-Pak, K.; Reynolds, M.P.; Hall, A.; Paul, M.J. Gene-based mapping of trehalose biosynthetic pathway genes reveals association with source- and sink-related yield traits in a spring wheat panel. Food Energy Secur. 2021, 10, e292. [Google Scholar] [CrossRef]
- Hwang, G.; Kim, S.; Cho, J.Y.; Paik, I.; Kim, J.I.; Oh, E. Trehalose-6-phosphate signaling regulates thermoresponsive hypocotyl growth in Arabidopsis thaliana. EMBO Rep. 2019, 20, e47828. [Google Scholar] [CrossRef] [PubMed]
- Engle, K.A.; Amos, R.A.; Yang, J.Y.; Glushka, J.; Atmodjo, M.A.; Tan, L.; Huang, C.; Moremen, K.W.; Mohnen, D. Multiple Arabidopsis galacturonosyltransferases synthesize polymeric homogalacturonan by oligosaccharide acceptor-dependent or de novo synthesis. Plant J. 2021, 109, 1441–1456. [Google Scholar] [CrossRef] [PubMed]
- Van, A.R.; Vanholme, R.; Storme, V.; Mortimer, J.C.; Dupree, P.; Boerjan, W. Lignin biosynthesis perturbations affect secondary cell wall composition and saccharification yield in Arabidopsis thaliana. Biotechnol. Biofuels. 2013, 6, 46. [Google Scholar]
- Chang, X.F.; Chandra, R.; Berleth, T.; Beatson, R.P. Rapid, microscale, acetylbromide-based method for high-throughput determination of lignin content in Arabidopsis thaliana. J. Agric. Food Chem. 2008, 56, 6825–6834. [Google Scholar] [CrossRef]
- Chen, F.; Tobimatsu, Y.; Jackson, L.; Nakashima, J.; Ralph, J.; Dixon, R.A. Novel seed coat lignins in the Cactaceae: Structure, distribution and implications for the evolution of lignin diversity. Plant J. 2013, 73, 201–211. [Google Scholar] [CrossRef]
- Besseau, S.; Hoffmann, L.; Geoffroy, P.; Lapierre, C.; Pollet, B.; Legrand, M. Flavonoid accumulation in Arabidopsis repressed in lignin synthesis affects auxin transport and plant growth. Plant Cell. 2007, 19, 148–162. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Elling, A.A.; Li, X.; Li, N.; Peng, Z.; He, G.; Sun, H.; Qi, Y.; Liu, X.S.; Deng, X.W. Genome-wide and organ-specific landscapes of epigenetic modifications and their relationships to mRNA and small RNA transcriptomes in maize. Plant Cell. 2009, 21, 1053–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Fu, W.Q.; Zhao, Z.G.; Ge, X.H.; Ding, L.; Li, Z.Y. Anatomy and transcript profiling of gynoecium development in female sterile Brassica napus mediated by one alien chromosome from Orychophragmus violaceus. BMC Genom. 2014, 15, 61. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Dong, C.; Yu, J.; Liu, W.; Jiang, C.; Liu, J.; Hu, Q.; Fang, X.; Wei, W. Transcriptome profile analysis of young floral buds of fertile and sterile plants from the self-pollinated offspring of the hybrid between novel restorer line NR1 and Nsa CMS line in Brassica napus. BMC Genom. 2013, 14, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slotte, T.; Hazzouri, K.M.; Ågren, J.A.; Koenig, D.; Maumus, F.; Guo, Y.L.; Steige, K.; Platts, A.E.; Escobar, J.S.; Newman, L.K.; et al. The Capsella rubella genome and the genomic consequences of rapid mating system evolution. Nat Genet. 2013, 45, 831–835. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.Z.; Wang, D.M.; Wang, B.; Wang, J.K.; Liu, H.Y.; Guan, L.; Liu, J.X. Metabolomics of four biofluids from dairy cows: Potential biomarkers for milk production and quality. J. Proteome Res. 2015, 14, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H.; et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Miao, L.; He, J.; Li, H.; Li, M. The Transcriptome and Metabolome Reveal the Potential Mechanism of Lodging Resistance in Intergeneric Hybrids between Brassica napus and Capsella bursa-pastoris. Int. J. Mol. Sci. 2022, 23, 4481. https://doi.org/10.3390/ijms23094481
Zhang L, Miao L, He J, Li H, Li M. The Transcriptome and Metabolome Reveal the Potential Mechanism of Lodging Resistance in Intergeneric Hybrids between Brassica napus and Capsella bursa-pastoris. International Journal of Molecular Sciences. 2022; 23(9):4481. https://doi.org/10.3390/ijms23094481
Chicago/Turabian StyleZhang, Libin, Liyun Miao, Jianjie He, Huaixin Li, and Maoteng Li. 2022. "The Transcriptome and Metabolome Reveal the Potential Mechanism of Lodging Resistance in Intergeneric Hybrids between Brassica napus and Capsella bursa-pastoris" International Journal of Molecular Sciences 23, no. 9: 4481. https://doi.org/10.3390/ijms23094481