
Rap1a Activity Elevated the Impact of Endogenous AGEs in Diabetic Collagen to Stimulate Increased Myofibroblast Transition and Oxidative Stress


Abstract
:1. Introduction
2. Results
2.1. Diabetic Cardiac Fibroblasts Exhibited Higher Levels of RAGE-Associated Signaling Proteins Compared to Non-Diabetic and Rap1a Fibroblasts
2.2. On Non-Diabetic Collagen, RAGE-Associated Cascade Signal Proteins in Diabetic RKO Fibroblasts Did Not Differ from Non-Diabetic RKO Fibroblasts
2.3. Exposing Fibroblasts to AGEs in Diabetic Collagen and EPAC Increased Expression of RAGE Associated Signaling Proteins
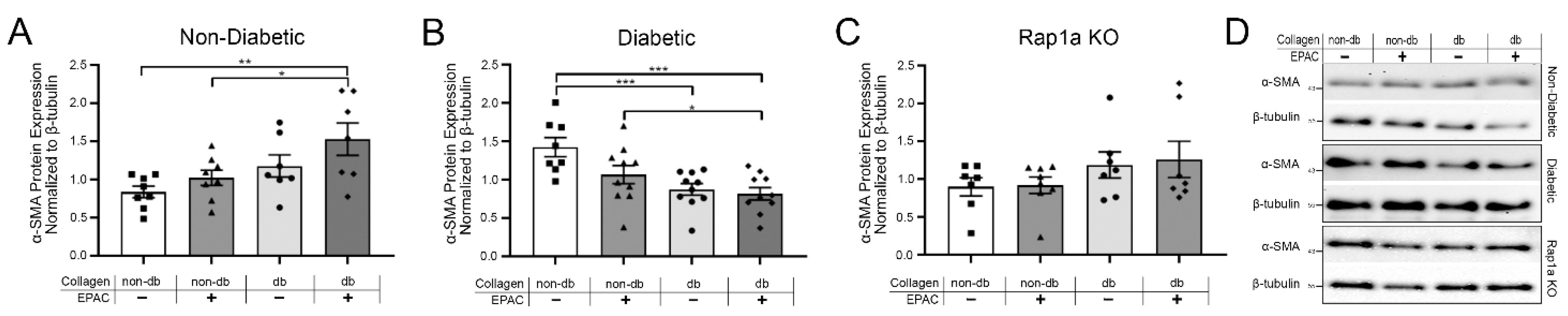
2.4. Endogenous AGEs and Rap1a Activation Altered α-SMA Protein Expression in Both Non-Diabetic and Diabetic Fibroblasts
2.5. Endogenous AGE Exposure and Increased Rap1a Activity Caused Elevated p-NF-κB Protein Expression
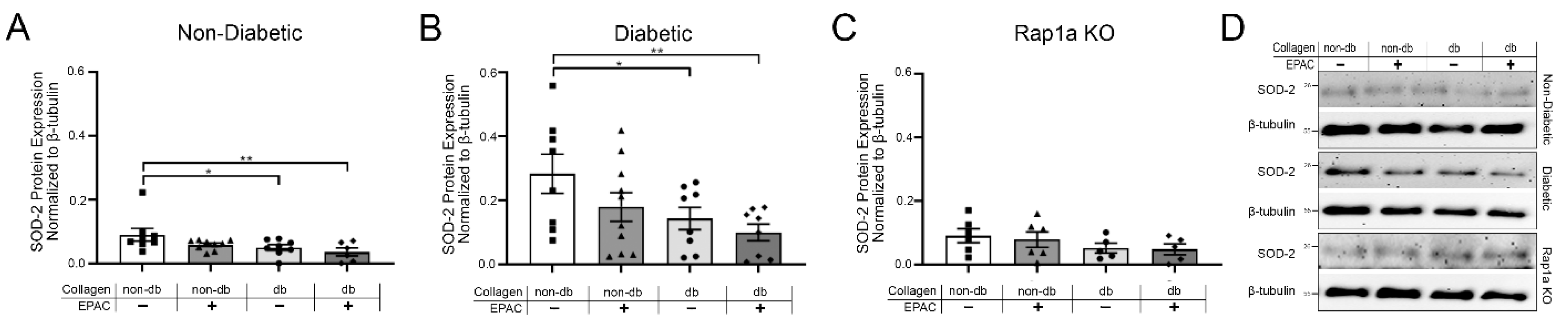
2.6. Treatment with EPAC and Diabetic Collagen Caused Decreased SOD Expression in Cardiac Fibroblasts
2.7. Endogenous AGEs and EPAC Treatment Caused a Decrease in Hydrogen Peroxide in Cardiac Fibroblast with Functional RAGE and Rap1a
3. Discussion
4. Methods
4.1. Animal Models
4.2. Animal Care
4.3. Cardiac Fibroblast Isolation
4.4. Cell Culture and Experimental Treatment
4.5. Collagen Extraction
4.6. Protein Isolation and Western Blot Analysis
4.7. Hydrogen Peroxide Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fowlkes, V.; Clark, J.; Fix, C.; Law, B.A.; Morales, M.O.; Qiao, X.; Ako-Asare, K.; Goldsmith, J.G.; Carver, W.; Murray, D.B.; et al. Type II diabetes promotes a myofibroblast phenotype in cardiac fibroblasts. Life Sci. 2013, 92, 669–676. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, K.R.; Lord, C.K.; West, T.A.; Stewart, J.A. Cardiac Fibroblast-Dependent Extracellular Matrix Accumulation Is Associated with Diastolic Stiffness in Type 2 Diabetes. PLoS ONE 2013, 8, e72080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamhart, P.; Luther, D.; Adapala, R.; Bryant, J.; Petersen, K.; Meszaros, J.; Thodeti, C. Hyperglycemia enhances function and differentiation of adult rat cardiac fibroblasts. Can J Physiol Pharmacol 2014, 92, 598–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burr, S.D.; Stewart, J.A. Extracellular matrix components isolated from diabetic mice alter cardiac fibroblast function through the AGE/RAGE signaling cascade. Life Sci. 2020, 250, 117569. [Google Scholar] [CrossRef]
- Fan, D.; Takawale, A.; Lee, J.; Kassiri, Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis Tissue Repair 2012, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, K.; Hou, L.; Huang, N. Role of extracellular matrix signalling cues in modulating cell fate commitment for cardiovascular tissue engineering. Adv. Healthc. Mater. 2015, 3, 628–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, O.Z.; Galiano, R.D.; Armour, M.; Levine, J.P.; Gurtner, G.C. Cellular dysfunction in the diabetic fibroblast: Impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am. J. Pathol. 2003, 162, 303–312. [Google Scholar] [CrossRef]
- Lijnen, P.; Petrov, V.; Van Pelt, J.; Fagard, R. Inhibition of superoxide dismutase induces collagen production in cardiac fibroblasts. Am. J. Hypertens. 2008, 21, 1129–1136. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Fukuda, K.; Sato, T.; Kawaguchi, H.; Suematsu, M.; Matsuda, S.; Koyasu, S.; Matsui, H.; Yamauchi-Takihara, K.; Harada, M.; et al. ERK and p38 MAPK, but not NF-KB, Are Critically Involved in Reactive Oxygen Species-Mediated Induction of IL-6 by Angiotensin II in Cardiac Fibroblasts. Circ. Res. 2001, 89, 661–669. [Google Scholar] [CrossRef]
- Bai, J.; Zhang, N.; Hua, Y.; Wang, B.; Ling, L. Metformin Inhibits Angiotensin II-Induced Differentiation of Cardiac Fibroblasts into Myofibroblasts. PLoS ONE 2013, 8, e72120. [Google Scholar] [CrossRef] [Green Version]
- Roberts, C.K.; Won, D.; Pruthi, S.; Lin, S.S.; Barnard, R.J. Effect of a diet and exercise intervention on oxidative stress, inflammation and monocyte adhesion in diabetic men. Diabetes Res. Clin. Pract. 2006, 73, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Wang, F.S.; Kuo, Y.R.; Huang, Y.T.; Huang, H.C.; Sun, Y.C.; Kuo, Y.H. Ras modulation of superoxide activates ERK-dependent fibronectin expression in diabetes-induced renal injuries. Kidney Int. 2006, 69, 1593–1600. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Yamashita, T.; Sekiguchi, A.; Tsuneda, T.; Sagara, K.; Takamura, M.; Kaneko, S.; Aizawa, T.; Fu, L.T. AGEs-RAGE system mediates atrial structural remodeling in the diabetic rat. J. Cardiovasc. Electrophysiol. 2008, 19, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Umadevi, S.; Gopi, V.; Elangovan, V. Regulatory mechanism of gallic acid against advanced glycation end products induced cardiac remodeling in experimental rats. Chem. Biol. Interact. 2014, 208, 28–36. [Google Scholar] [CrossRef]
- Zhao, J.; Randive, R.; Stewart, J.A. Molecular mechanisms of AGE/RAGE-mediated fibrosis in the diabetic heart. World J. Diabetes 2014, 5, 860–867. [Google Scholar] [CrossRef]
- Bierhaus, A.; Humpert, P.M.; Morcos, M.; Wendt, T.; Chavakis, T.; Arnold, B.; Stern, D.M.; Nawroth, P.P. Understanding RAGE, the receptor for advanced glycation end products. J. Mol. Med. 2005, 83, 876–886. [Google Scholar] [CrossRef]
- Piperi, C.; Goumenos, A.; Adamopoulos, C.; Papavassiliou, A.G. AGE/RAGE signalling regulation by miRNAs: Associations with diabetic complications and therapeutic potential. Int. J. Biochem. Cell Biol. 2015, 60, 197–201. [Google Scholar] [CrossRef]
- Tikellis, C.; Thomas, M.C.; Harcourt, B.E.; Coughlan, M.T.; Pete, J.; Bialkowski, K.; Tan, A.; Bierhaus, A.; Cooper, M.E.; Forbes, J.M. Cardiac inflammation associated with a Western diet is mediated via activation of RAGE by AGEs. Am. J. Physiol. Endocrinol. Metab. 2008, 295, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Hegab, Z.; Gibbons, S.; Neyses, L.; Mamas, M.A. Role of advanced glycation end products in cardiovascular disease. World J. Cardiol. 2012, 4, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Burr, S.D.; Harmon, M.B.; Stewart, J.A., Jr. The Impact of Diabetic Conditions and AGE/RAGE Signaling on Cardiac Fibroblast Migration. Front. Cell Dev. Biol. 2020, 8, 112. [Google Scholar] [CrossRef]
- Oliveira Volpe, C.M.; Henrique Villar-Delfino, P.; Ferreira Dos Anjos, P.M.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Nitti, M.; Furfaro, A.L.; Traverso, N.; Odetti, P.; Storace, D.; Cottalasso, D.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. PKC delta and NADPH oxidase in AGE-induced neuronal death. Neurosci. Lett. 2007, 416, 261–265. [Google Scholar] [CrossRef]
- Cai, W.; Torreggiani, M.; Zhu, L.; Chen, X.; Cijiang He, J.; Striker, G.E.; Vlassara, H. AGER1 regulates endothelial cell NADPH oxidase-dependent oxidant stress via PKC-: Implications for vascular disease. Am. J. Physiol. Cell. Physiol. 2010, 298, 624–634. [Google Scholar] [CrossRef] [Green Version]
- Jian Guo, Z.; Xin Niu, H.; Fan Hou, F.; Zhang, L.; Fu, N.; Nagai, R.; Lu, X.; Hong Chen, B.; Xin Shan, Y.; Wei Tian, J.; et al. Advanced Oxidation Protein Products Activate Vascular Endothelial Cells via a RAGE-Mediated Signaling Pathway. Antioxid. Redox Signal. 2008, 10, 1699–1712. [Google Scholar] [CrossRef]
- Adamopoulos, C.; Piperi, C.; Gargalionis, A.N.; Dalagiorgou, G.; Spilioti, E.; Korkolopoulou, P.; Diamanti-Kandarakis, E.; Papavassiliou, A.G. Advanced glycation end products upregulate lysyl oxidase and endothelin-1 in human aortic endothelial cells via parallel activation of ERK1/2-NF-??B and JNK-AP-1 signaling pathways. Cell. Mol. Life Sci. 2016, 73, 1685–1698. [Google Scholar] [CrossRef]
- Gąsiorowski, K.; Brokos, B.; Echeverria, V.; Barreto, G.E.; Leszek, J. RAGE-TLR Crosstalk Sustains Chronic Inflammation in Neurodegeneration. Mol. Neurobiol. 2018, 55, 1463–1476. [Google Scholar] [CrossRef]
- Zhong, Y.; Cheng, C.F.; Luo, Y.Z.; Tian, C.W.; Yang, H.; Liu, B.R.; Chen, M.S.; Chen, Y.F.; Liu, S.M. C-reactive protein stimulates RAGE expression in human coronary artery endothelial cells in vitro via ROS generation and ERK/NF-κB activation. Acta Pharmacol. Sin. 2015, 36, 440–447. [Google Scholar] [CrossRef]
- Kelly, K.A.; Hill, M.R.; Kellee, Y.; Wanker, F.; Gimble, J.M. Dimethyl Sulfoxide Modulates NF-KB and Cytokine Activation in Lipopolysaccharide-Treated Murine Macrophages Downloaded from. Infect. Immun. 1994, 62, 3122–3128. [Google Scholar] [CrossRef] [Green Version]
- Tobón-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as Mediator of NF-kB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol. Disord. Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Mondola, P.; Santillo, M.; Serù, R.; Damiano, S.; Alvino, C.; Ruggiero, G.; Formisano, P.; Terrazzano, G.; Secondo, A.; Annunziato, L. Cu,Zn superoxide dismutase increases intracellular calcium levels via a phospholipase C-protein kinase C pathway in SK-N-BE neuroblastoma cells q. Biochem. Biophys. Res. Commun. 2004, 324, 887–892. [Google Scholar] [CrossRef]
- Leuner, B.; Ruhs, S.; Urgen Br€, H.-J.; Bierhaus, A.; Sel, S.; Silber, R.-E.; Somoza, V.; Simm, A.; Nass, N. RAGE-dependent activation of gene expression of superoxide dismutase and vanins by AGE-rich extracts in mice cardiac tissue and murine cardiac fibroblasts. Food Funct. 2012, 3, 1091–1098. [Google Scholar] [CrossRef]
- Li, X.; Weng, H.; Reece, E.A.; Yang, P. SOD1 overexpression in vivo blocks hyperglycemia-induced specific PKC isoforms-substrate activation and consequent lipidperoxidation in diabetic embryopathy. Am J Obs. Gynecol 2011, 205, 84–85. [Google Scholar] [CrossRef] [Green Version]
- Burr, S.D.; Stewart, J.A. Rap1a Overlaps the AGE/RAGE Signaling Cascade to Alter Expression of α-SMA, p-NF-κB, and p-PKC-ζ in Cardiac Fibroblasts Isolated from Type 2 Diabetic Mice. Cells 2021, 10, 557. [Google Scholar] [CrossRef]
- Dong, W.; Yang, Z.; Yang, F.; Wang, J.; Zhuang, Y.; Xu, C.; Zhang, B. Suppression of Rap1 Impairs Cardiac Myofibrils and Conduction System in Zebrafish. PLoS ONE 2012, 7, e50960. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Li, F.; Ingram, D.A.; Quilliam, L.A. Rap1a Is a Key Regulator of Fibroblast Growth Factor 2-Induced Angiogenesis and Together with Rap1b Controls Human Endothelial Cell Functions. Mol. Cell. Biol. 2008, 28, 5803–5810. [Google Scholar] [CrossRef] [Green Version]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Jeyaraj, S.C.; Unger, N.T.; Chotani, M.A. Rap1 GTPases: An emerging role in the cardiovasculature. Life Sci. 2011, 88, 645–652. [Google Scholar] [CrossRef] [Green Version]
- Roberts, O.L.; Kamishima, T.; Barrett-Jolley, R.; Quayle, J.M.; Dart, C. Exchange protein activated by cAMP (Epac) induces vascular relaxation by activating Ca2+-sensitive K+ channels in rat mesenteric artery. J. Physiol. 2013, 591, 5107–5123. [Google Scholar] [CrossRef]
- Wang, H.; Fotheringham, L.; Wittchen, E.S.; Hartnett, M.E. Rap1 GTPase inhibits tumor necrosis factor-α-induced choroidal endothelial migration via NADPH oxidase- and NF-κB-dependent activation of Rac1. Am. J. Pathol. 2015, 185, 3316–3325. [Google Scholar] [CrossRef] [Green Version]
- Panchatcharam, M.; Miriyala, S.; Yang, F.; Leitges, M.; Chrzanowska-Wodnicka, M.; Quilliam, L.A.; Anaya, P.; Morris, A.J.; Smyth, S.S. Enhanced proliferation and migration of vascular smooth muscle cells in response to vascular injury under hyperglycemic conditions is controlled by β3 integrin signaling. Int. J. Biochem. Cell Biol. 2010, 42, 965–974. [Google Scholar] [CrossRef] [Green Version]
- Thallas-bonke, V.; Coughlan, M.T.; Tan, A.L.; Harcourt, B.E.; Morgan, P.E.; Davies, M.J.; Bach, L.A.; Cooper, M.E.; Forbes, J.M.; Thallas-Bonke, V. Targeting the AGE-RAGE axis improves renal function in the context of a healthy diet low in advanced glycation end-product content. Nephrology 2012, 18, 47–56. [Google Scholar] [CrossRef]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef]
- Mott, J.D.; Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 2004, 16, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Whelan, M.C.; Senger, D.R. Collagen I Initiates Endothelial Cell Morphogenesis by Inducing Actin Polymerization through Suppression of Cyclic AMP and Protein Kinase A*. J. Biol. Chem. 2002, 278, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Constien, R.; Forde, A.; Liliensiek, B.; Gröne, H.J.; Nawroth, P.; Hämmerling, G.; Arnold, B. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis 2001, 30, 36–44. [Google Scholar] [CrossRef]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Gröne, H.-J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef]
- Brodeur, M.R.; Bouvet, C.; Bouchard, S.; Moreau, S.; Leblond, J. Reduction of Advanced-Glycation End Products Levels and Inhibition of RAGE Signaling Decreases Rat Vascular Calcification Induced by Diabetes. PLoS ONE 2014, 9, e85922. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yan, J.; De, P.; Chang, H.-C.; Yamauchi, A.; Christopherson II, K.W.; Paranavitana, N.C.; Peng, X.; Kim, C.; Munugalavadla, V.; et al. Rap1a null mice have altered myeloid cell functions suggesting distinct roles for the closely related Rap1a and 1b proteins 1. J. Immunol. 2007, 179, 8322–8331. [Google Scholar] [CrossRef] [Green Version]
- Sorrell, J.M.; Caplan, A.I. Fibroblast heterogeneity: More than skin deep. J. Cell Sci. 2004, 117, 667–675. [Google Scholar] [CrossRef] [Green Version]
- Driskell, R.R.; Watt, F.M. Understanding fibroblast heterogeneity in the skin. Trends Cell Biol. 2015, 25, 92–99. [Google Scholar] [CrossRef]
- Sriram, G.; Bigliardi, P.L.; Bigliardi-Qi, M. Fibroblast heterogeneity and its implications for engineering organotypic skin models in vitro. Eur. J. Cell Biol. 2015, 94, 483–512. [Google Scholar] [CrossRef] [Green Version]
- Rajan, N.; Habermehl, J.; Coté, M.-F.; Doillon, C.J.; Mantovani, D. Preparation of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nat. Protoc. 2007, 1, 2753. [Google Scholar] [CrossRef]
- Somensi, N.; Brum, P.O.; De, V.; Ramos, M.; Gasparotto, J.; Zanotto-Filho, A.; Rostirolla, D.C.; Da, M.; Morrone, S.; Claudio, J.; et al. Cellular Physiology and Biochemistry Cellular Physiology and Biochemistry Extracellular HSP70 Activates ERK1/2, NF-kB and Pro-Inflammatory Gene Transcription Through Binding with RAGE in A549 Human Lung Cancer Cells. Cell Physiol. Biochem. 2017, 42, 2507–2522. [Google Scholar] [CrossRef]
- Serban, A.I.; Stanca, L.; Geicu, O.I.; Munteanu, M.C.; Dinischiotu, A. RAGE and TGF-β1 Cross-Talk Regulate Extracellular Matrix Turnover and Cytokine Synthesis in AGEs Exposed Fibroblast Cells. PLoS ONE 2016, 11, e0152376. [Google Scholar] [CrossRef]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef]
- Simard, E.; Söllradl, T.; Maltais, J.-S.; Boucher, J.; D’orléans-Juste, P.; Grandbois, M. Receptor for Advanced Glycation End-Products Signaling Interferes with the Vascular Smooth Muscle Cell Contractile Phenotype and Function. PLoS ONE 2015, 10, e0128881. [Google Scholar] [CrossRef] [Green Version]
- Vicens-Zygmunt, V.; Estany, S.; Colom, A.; Montes-Worboys, A.; Machahua, C.; Sanabria, A.J.; Llatjos, R.; Escobar, I.; Manresa, F.; Dorca, J.; et al. Fibroblast viability and phenotypic changes within glycated stiffened three-dimensional collagen matrices. Respir. Res. 2015, 16, 82. [Google Scholar] [CrossRef] [Green Version]
- Cukierman, E.; Pankov, R.; Yamada, K.M. Cell interactions with three-dimensional matrices. Curr. Opin. Cell Biol. 2002, 14, 633–639. [Google Scholar] [CrossRef]
- Wang, S.E.; En S Leo Nard, S.P.; Vince Castranova, F.; Vallyathan, V.; Shi, X. The role of superoxide radical in TN F-a induced N F-kB activation*. Ann. Clin. Lab. Sci. 1999, 29, 192–199. [Google Scholar]
- Fleissner, F.; Jazbutyte, V.; Fiedler, J.; Gupta, S.K.; Yin, X.; Xu, Q.; Galuppo, P.; Kneitz, S.; Mayr, M.; Ertl, G.; et al. Asymmetric dimethylarginine impairs angiogenic progenitor cell function in patients with coronary artery disease through a MicroRNA-21-Dependent mechanism. Circ. Res. 2010, 107, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Muratsu-Ikeda, S.; Nangaku, M.; Ikeda, Y.; Tanaka, T.; Wada, T.; Inagi, R. Downregulation of miR-205 Modulates Cell Susceptibility to Oxidative and Endoplasmic Reticulum Stresses in Renal Tubular Cells. PLoS ONE 2012, 7, e41462. [Google Scholar] [CrossRef] [Green Version]
- Pathomthongtaweechai, N.; Chutipongtanate, S. AGE/RAGE signaling-mediated endoplasmic reticulum stress and future prospects in non-coding RNA therapeutics for diabetic nephropathy. Biomed. Pharmacother. 2020, 131, 110655. [Google Scholar] [CrossRef] [PubMed]
- Gospodarowicz, D.; Ill, C. Extracellular Matrix and Control of Proliferation of Vascular Endothelial Cells. J. Clin. Endocrinol. Metab. 1980, 65, 1351–1364. [Google Scholar] [CrossRef] [Green Version]
- Klein, C.E.; Dressel, D.; Steinmayer, T.; Mauch, C.; Eckes, B.; Krieg, T.; Bankert, R.B.; Weber, L. Integrin a2ß1 Is Upregulated in Fibroblasts and Highly Aggressive Melanoma Cells in Three-Dimensional Collagen Lattices and Mediates the Reorganization of Collagen I Fibrils. J. Cell Biol. 1991, 115, 1427–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, D.; Govindaraju, P.; Jacob, M.; Todd, L.; Monslow, J.; Puré, E. Extracellular Matrix Directs Phenotypic Heterogeneity of Activated Fibroblasts. Matrix Biol. 2018, 67, 90–106. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heart Weight (g) | Blood Glucose (mg/dL) | |
|---|---|---|
| Body Weight (g) | ||
| Non-Diabetic (n = 30) | 0.0038 ± 5.362 × 10−5 | 198.9 ± 5.187 |
| Diabetic (n = 16) | 0.0022 ± 7.378 × 10−5 **** | 511.4 ± 27.98 **** |
| Non-Diabetic RKO (n = 24) | 0.0037 ± 7.349 × 10−5 | 193.1 ± 7.148 |
| Diabetic RKO (n = 14) | 0.0022 ± 3.472 × 10−5 **** | 428.2 ± 27.99 **** |
| Rap1a KO (n = 27) | 0.0040 ± 7.297 × 10−5 | 187.8 ± 7.007 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burr, S.D.; Dorroh, C.C.; Stewart, J.A., Jr. Rap1a Activity Elevated the Impact of Endogenous AGEs in Diabetic Collagen to Stimulate Increased Myofibroblast Transition and Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 4480. https://doi.org/10.3390/ijms23094480
Burr SD, Dorroh CC, Stewart JA Jr. Rap1a Activity Elevated the Impact of Endogenous AGEs in Diabetic Collagen to Stimulate Increased Myofibroblast Transition and Oxidative Stress. International Journal of Molecular Sciences. 2022; 23(9):4480. https://doi.org/10.3390/ijms23094480
Chicago/Turabian StyleBurr, Stephanie D., Christopher C. Dorroh, and James A. Stewart, Jr. 2022. "Rap1a Activity Elevated the Impact of Endogenous AGEs in Diabetic Collagen to Stimulate Increased Myofibroblast Transition and Oxidative Stress" International Journal of Molecular Sciences 23, no. 9: 4480. https://doi.org/10.3390/ijms23094480