
Transcriptomic Data Meta-Analysis Sheds Light on High Light Response in Arabidopsis thaliana L.

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
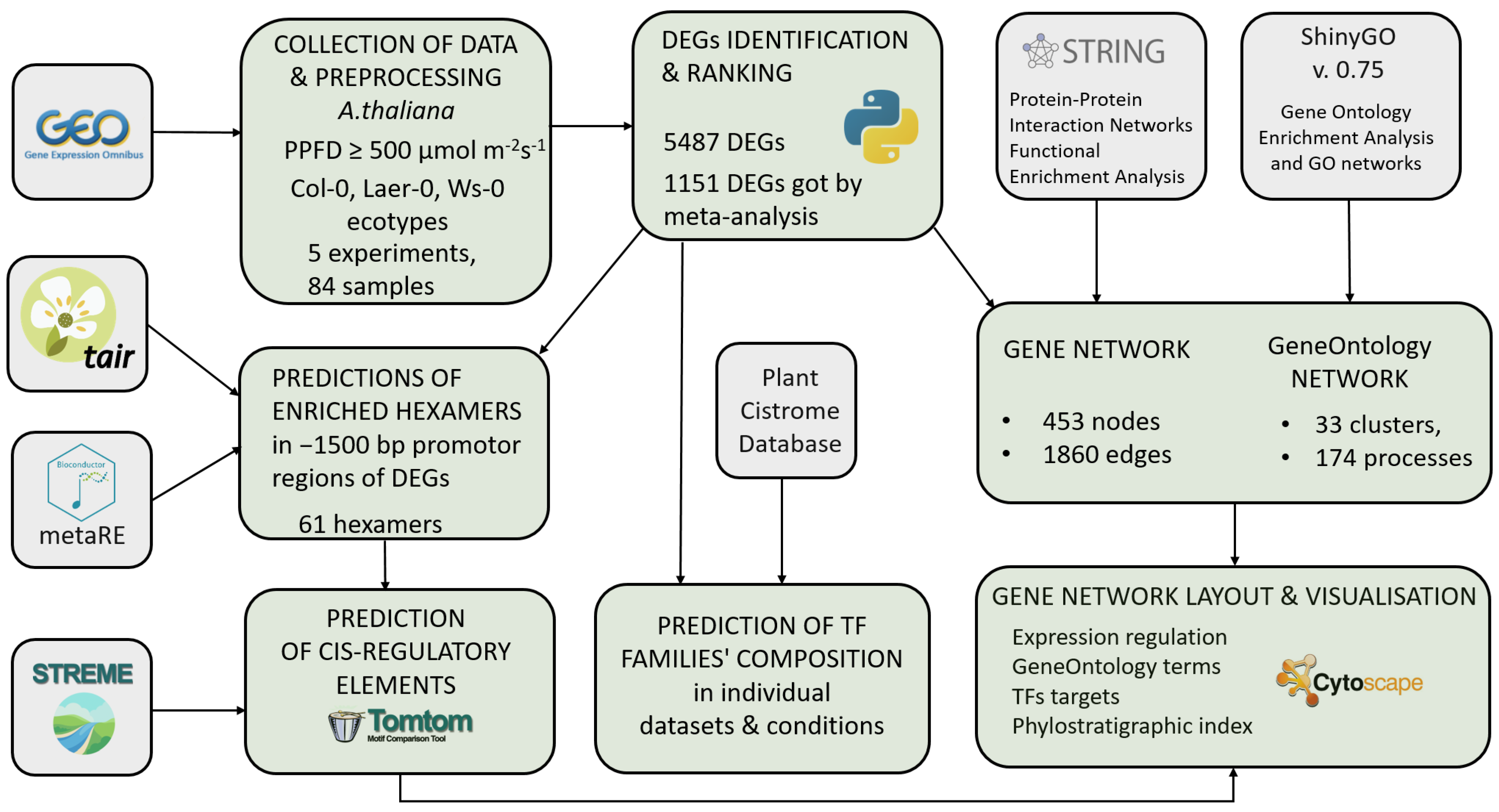
2.1. A Pipeline for Large-Scale Systematic Analysis of the High Light Response Regulation at Different Levels
2.2. The High Light Response Differentially Expressed Genes
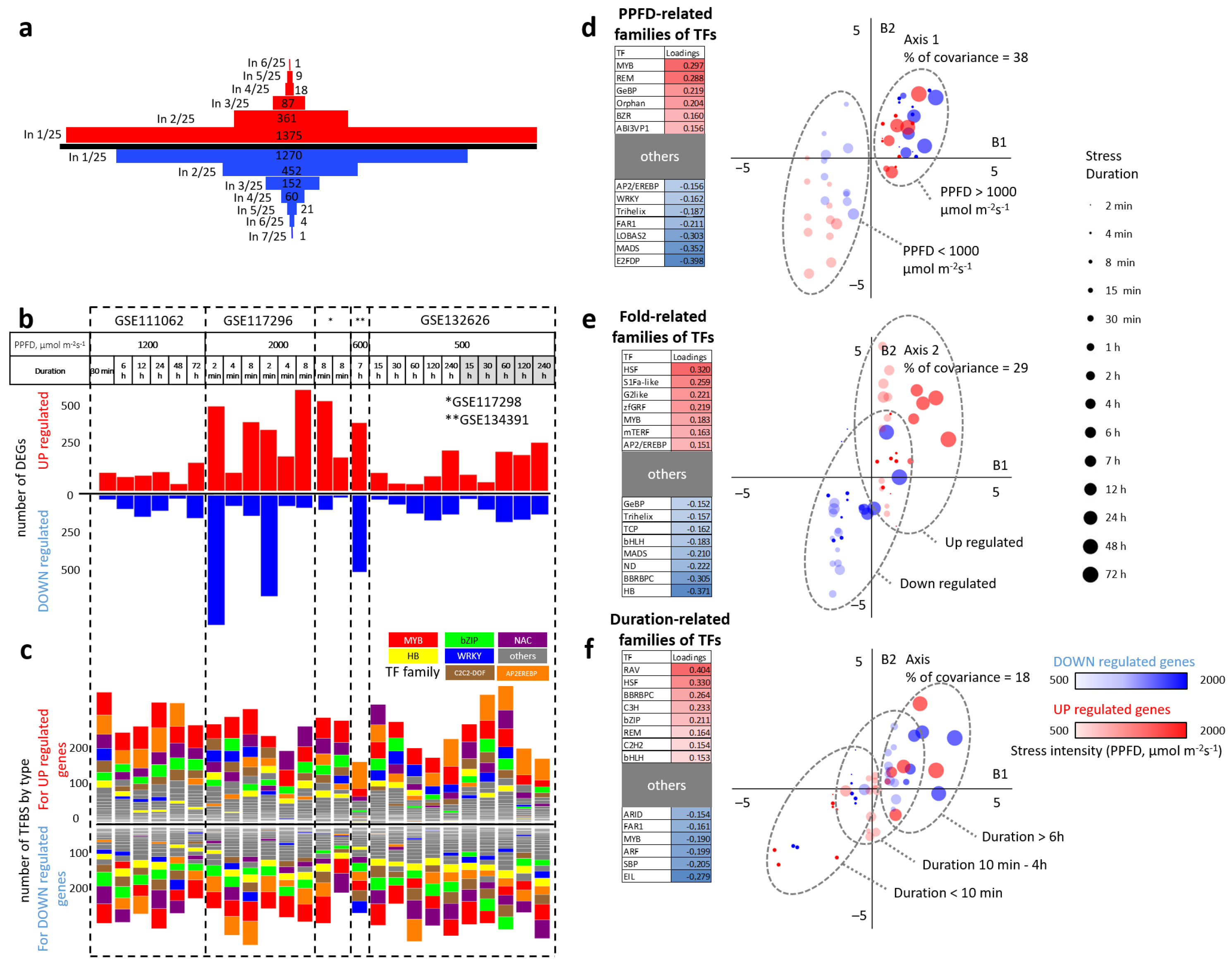
2.2.1. Sets of DEGs Predicted from Individual Transcriptomic Datasets
2.2.2. Sets of Enriched TFs Families for DEGs in Individual Datasets Considering the Direction of the Expression Change
2.2.3. Prediction of TFs Families Mediating the Direction of Expression Change and Response to Varying Intensity and Duration of High Light Exposure
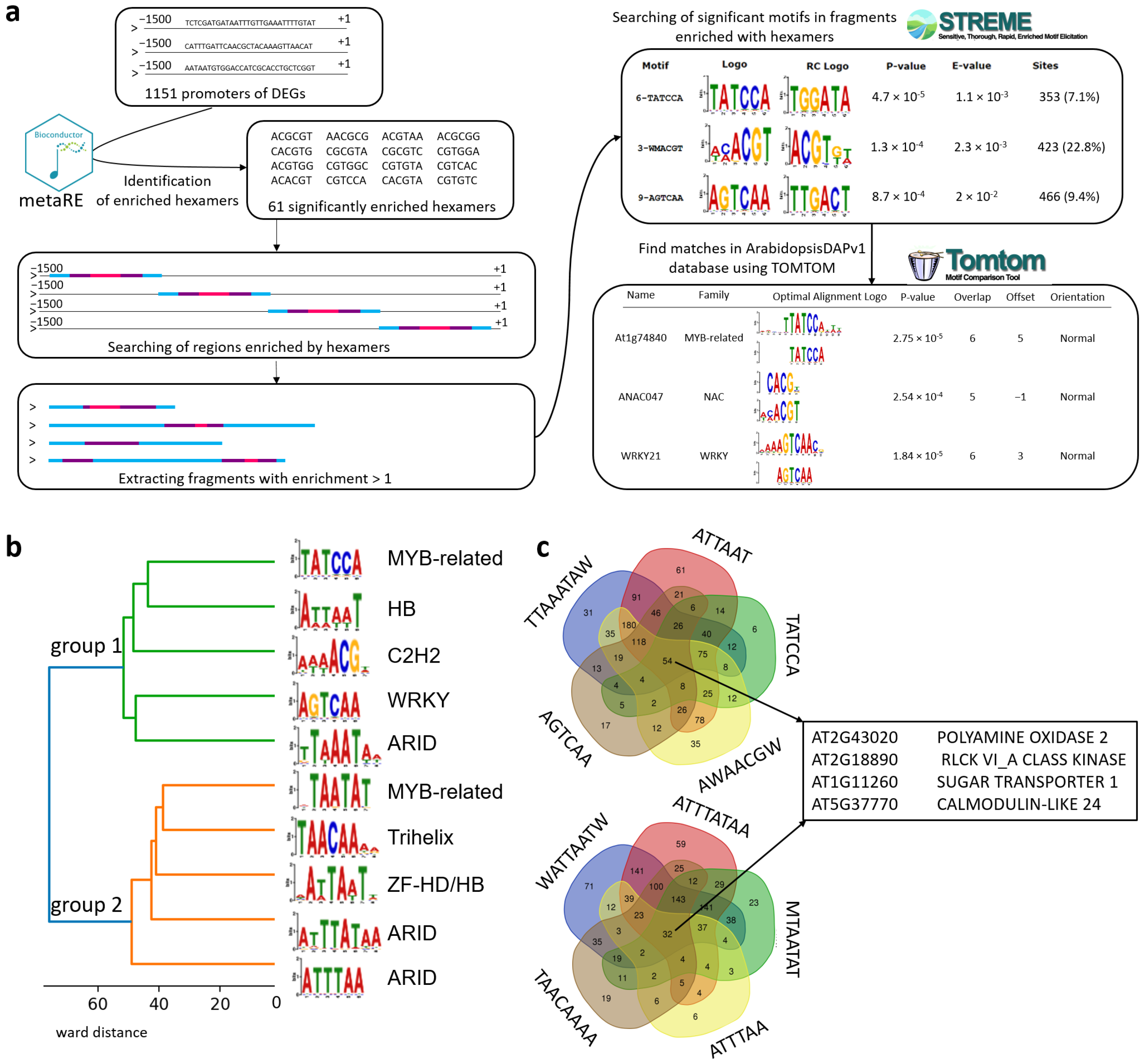
2.2.4. Set of the Most Significant DEGs Predicted by Meta-Analysis of Several Transcriptomic Experiments
2.2.5. Set of TFs Motifs Enriched in Promoter Regions of the Most Significant DEGs
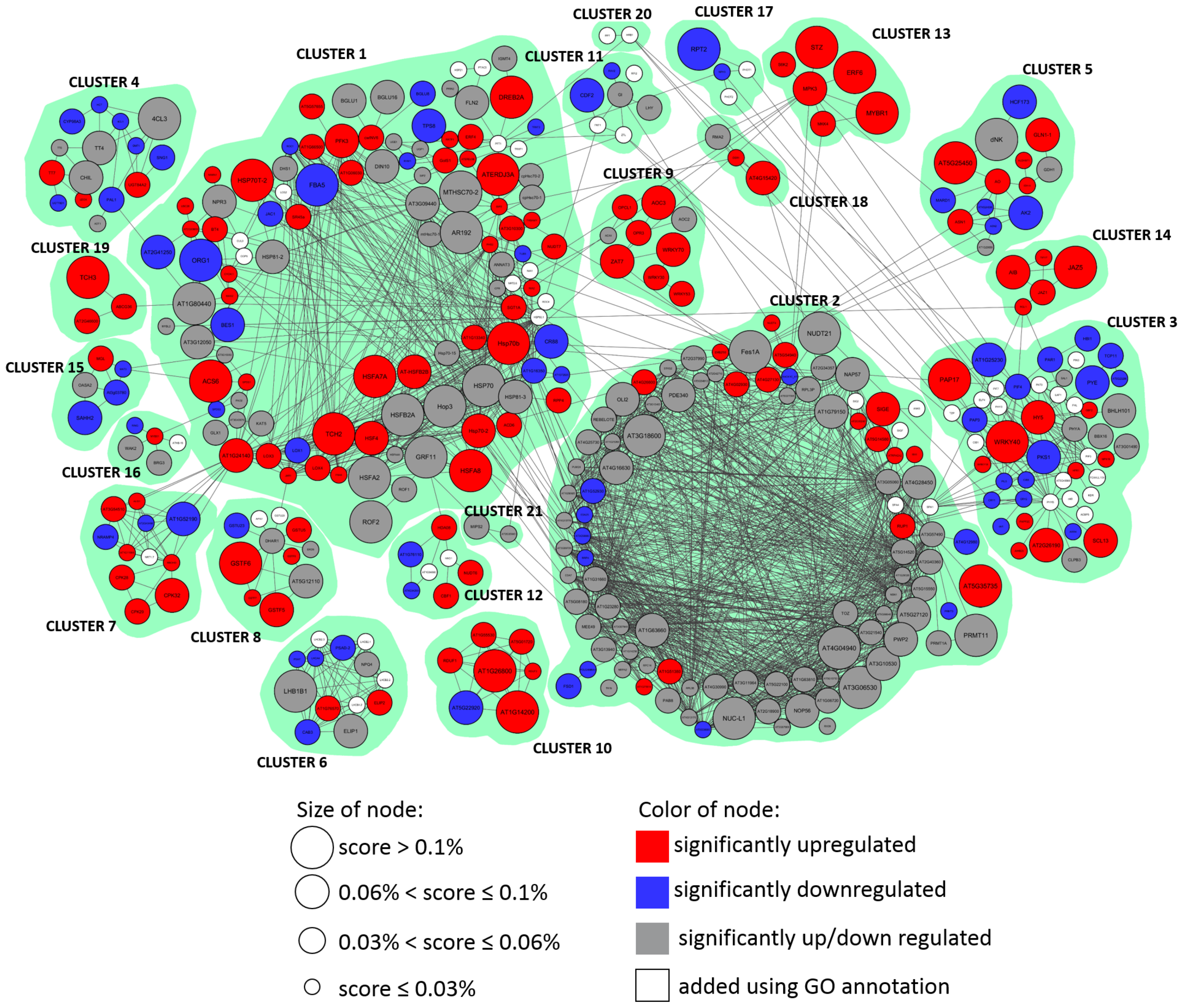
2.3. The High Light Response Core Gene Network
2.3.1. General Structure of Functional Modules
- Cluster 1:
- Heat-shock proteins (>20), genes with catalytic activities (7), regulators of the cellular metabolic process (5), lipoxygenases (3), cysteine and methionine metabolism (3), ethylene signaling pathway (2);
- Cluster 2:
- Genes of ribosomal biogenesis (25), circadian rhythm regulators (3), sigma factors (3), FE superoxide dismutase 1 (FSD1), ANK6, which promotes the anthocyanin accumulator;
- Cluster 3:
- Genes responding to light stimulus (7), PIF TFs (4), phosphoproteins (4), phytochromes (3), cryptochromes (2), cryptochrome-interacting basic-helix-loop-helix (2), WRKY (2);
- Cluster 4:
- Genes of flavonoid biosynthesis (8), genes of phenylalanine biosynthesis (4), genes of the phenylpropanoid synthesis pathway (2), cytochrome P450 (CYP98A3);
- Cluster 5:
- Genes of arginine biosynthesis (3), asparagine synthetases (2), aspartate kinase;
- Cluster 6:
- Subunits of the LHCB complex (7), proteins of chlorophyll A-B binding family (4), subunits of photosystem (2);
- Cluster 7:
- Calcium-dependent protein kinases (3), respiratory burst oxidase (RBOHD);
- Cluster 8:
- Genes of glutathione transferases (9), dehydroascorbate reductase 1 (DHAR1), ascorbate peroxidase 2 (APX2);
- Cluster 9:
- Genes of the jasmonic acid biosynthetic process (4), WRKY regulators (3), acyl-coenzyme A oxidase 4 (ACX4), ZAT7;
- Cluster 10:
- Genes with ubiquitin protein ligase activity (4);
- Cluster 11:
- Genes of circadian rhythm (5), E3 ubiquitin ligase component complex (2), ethylene response DNA binding factor 2 (RAV2);
- Cluster 12:
- Transcription factors (5);
- Cluster 13:
- MAPK signaling pathway (2), myb domain protein r1 (MYBR1), 40S ribosomal protein S6 kinase (S6K2), ethylene responsive element binding factor 6 (ERF6);
- Cluster 14:
- Regulators of the jasmonic acid mediated signaling pathway (3);
- Cluster 15:
- Genes related to cysteine and methionine metabolism (5);
- Cluster 16:
- Wall-associated receptor kinase 2 (WAK2), transcription factor MYB51;
- Cluster 17:
- Phototropins (PHOT1/PHOT2, nonphototropic hypocotyl 3 (NPH3), root phototropism protein 2 (RPT2));
- Cluster 18:
- C3HC4-RING finger E3 ubiquitin ligase (AtAIRP4), Derlin-2/3 (DER1), ubiquitin fusion degradation protein 1 (UFD1);
- Cluster 19:
- Calcium-binding EF-hand family protein (AT2G46600), ATP-binding cassete G36 (ABCG36), calmodulin-like 4 (TCH3);
- Cluster 20:
- Hypersensitive to red and blue 1 (HRB1), serine/threonine phosphatase 7 (PP7),;
- Cluster 21:
- AICARFT/IMPCHase bienzyme family protein (AT2G35040), inositol 3-phosphate synthase (MIPS2).
2.3.2. Distribution of TFs Motifs over the Network
2.3.3. Distribution of PAI over the Network
3. Discussion
4. Materials and Methods
4.1. Transcriptomic Data Search and Pre-Processing
| Organism: | Arabidopsis thaliana [porgn:__txid3702] |
| Study type: | Expression profiling by high throughput sequencing |
| Text fiters: | “high light” OR “hyperinsolation” |
4.2. Prediction of Differentially Expressed Genes in Individual Datasets
- 1.
- Preferential selection of genes by the level of their changes in single experiments:where
- 2.
- Preferential selection of genes by their presence in different experiments
- 3.
- Preferential selection of genes by combined Fisher’s p-value above all detected experiments and their summary change in their detection in experiments
4.3. Prediction of TFs Families Enriched in Individual Datasets and Matching Them with Experimental Conditions
4.4. Identification of Enriched Motifs in Promoters of Selected DEGs
4.5. Gene Ontology Enrichment Analysis
4.6. Reconstruction of the Gene Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 2B-PLS | Two-block partial least squares |
| ABA | Abscisic acid |
| AP2 | APETALA2 |
| ARID | AT-rich interaction domain |
| bZIP | Basic leucine zipper domain |
| BZR | Brassinazole-resistant family |
| C2H2 | Cys2His2 |
| CPM | Counts per million |
| DAP-seq | DNA affinity purification sequencing |
| DEG | Differentially expressed gene |
| ELIP | Early light-induced protein |
| EREBP | Ethylene-responsive element binding protein |
| FDR | False discovery rate |
| GEO NCBI | Gene Expression Omnibus National Center for Biotechnology Information |
| GO | Gene ontology |
| HB | Homeobox family |
| HSF | Heat stress transcription factor family |
| JA | Jasmonic acid |
| PAI | Phylostratigraphic age index |
| PIF | Phytochrome interacting factor |
| PPFD | Photosynthetic photon flux density |
| ROS | Reactive oxygen species |
| TF | Transcription factor |
| UV-B | Ultraviolet B |
| UVR8 | UV-B resistance 8 |
| ZF-HD | Zinc finger homeobox family protein |
References
- Jing, Y.; Lin, R. Transcriptional regulatory network of the light signaling pathways. New Phytol. 2020, 227, 683–697. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wakao, S.; Fischer, B.B.; Niyogi, K.K. Sensing and responding to excess light. Annu. Rev. Plant Biol. 2009, 60, 239–260. [Google Scholar] [CrossRef]
- Zuo, Z.; Liu, H.; Liu, B.; Liu, X.; Lin, C. Blue light-dependent interaction of CRY2 with SPA1 regulates COP1 activity and floral initiation in Arabidopsis. Curr. Biol. 2011, 21, 841–847. [Google Scholar] [CrossRef] [Green Version]
- Suetsugu, N.; Wada, M. Evolution of three LOV blue light receptor families in green plants and photosynthetic stramenopiles: Phototropin, ZTL/FKF1/LKP2 and aureochrome. Plant Cell Physiol. 2013, 54, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Ouyang, X.; Deng, X.W. Beyond repression of photomorphogenesis: Role switching of COP/DET/FUS in light signaling. Curr. Opin. Plant Biol. 2014, 21, 96–103. [Google Scholar] [CrossRef]
- Ni, W.; Xu, S.L.; Tepperman, J.M.; Stanley, D.J.; Maltby, D.A.; Gross, J.D.; Burlingame, A.L.; Wang, Z.Y.; Quail, P.H. A mutually assured destruction mechanism attenuates light signaling in Arabidopsis. Science 2014, 344, 1160–1164. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Song, Y.H.; Imaizumi, T. LOV domain-containing F-box proteins: Light-dependent protein degradation modules in Arabidopsis. Mol. Plant 2012, 5, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Fujiwara, S.; Suh, S.S.; Kim, J.; Kim, Y.; Han, L.; David, K.; Putterill, J.; Nam, H.G.; Somers, D.E. ZEITLUPE is a circadian photoreceptor stabilized by GIGANTEA in blue light. Nature 2007, 449, 356–360. [Google Scholar] [CrossRef]
- Sawa, M.; Nusinow, D.A.; Kay, S.A.; Imaizumi, T. FKF1 and GIGANTEA complex formation is required for day-length measurement in Arabidopsis. Science 2007, 318, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.H.; Estrada, D.A.; Johnson, R.S.; Kim, S.K.; Lee, S.Y.; MacCoss, M.J.; Imaizumi, T. Distinct roles of FKF1, GIGANTEA, and ZEITLUPE proteins in the regulation of CONSTANS stability in Arabidopsis photoperiodic flowering. Proc. Natl. Acad. Sci. USA 2014, 111, 17672–17677. [Google Scholar] [CrossRef] [Green Version]
- Tilbrook, K.; Arongaus, A.B.; Binkert, M.; Heijde, M.; Yin, R.; Ulm, R. The UVR8 UV-B photoreceptor: Perception, signaling and response. Arab. Book/Am. Soc. Plant Biol. 2013, 11, e0164. [Google Scholar] [CrossRef] [Green Version]
- Tissot, N.; Ulm, R. Cryptochrome-mediated blue-light signalling modulates UVR8 photoreceptor activity and contributes to UV-B tolerance in Arabidopsis. Nat. Commun. 2020, 11, 1323. [Google Scholar] [CrossRef]
- Osterlund, M.T.; Hardtke, C.S.; Wei, N.; Deng, X.W. Targeted destabilization of HY5 during light-regulated development of Arabidopsis. Nature 2000, 405, 462–466. [Google Scholar] [CrossRef]
- Laubinger, S.; Marchal, V.; Gentilhomme, J.; Wenkel, S.; Adrian, J.; Jang, S.; Kulajta, C.; Braun, H.; Coupland, G.; Hoecker, U. Arabidopsis SPA proteins regulate photoperiodic flowering and interact with the floral inducer CONSTANS to regulate its stability. Development 2006, 133, 3213–3222. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Marchal, V.; Panigrahi, K.C.; Wenkel, S.; Soppe, W.; Deng, X.W.; Valverde, F.; Coupland, G. Arabidopsis COP1 shapes the temporal pattern of CO accumulation conferring a photoperiodic flowering response. EMBO J. 2008, 27, 1277–1288. [Google Scholar] [CrossRef] [Green Version]
- Mittler, R.; Vanderauwera, S.; Gollery, M.; Van Breusegem, F. Reactive oxygen gene network of plants. Trends Plant Sci. 2004, 9, 490–498. [Google Scholar] [CrossRef]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [Green Version]
- Maruta, T.; Noshi, M.; Tanouchi, A.; Tamoi, M.; Yabuta, Y.; Yoshimura, K.; Ishikawa, T.; Shigeoka, S. H2O2-triggered retrograde signaling from chloroplasts to nucleus plays specific role in response to stress. J. Biol. Chem. 2012, 287, 11717–11729. [Google Scholar] [CrossRef] [Green Version]
- Brunetti, C.; Guidi, L.; Sebastiani, F.; Tattini, M. Isoprenoids and phenylpropanoids are key components of the antioxidant defense system of plants facing severe excess light stress. Environ. Exp. Bot. 2015, 119, 54–62. [Google Scholar] [CrossRef]
- Asada, K. Production and scavenging of reactive oxygen species in chloroplasts and their functions. Plant Physiol. 2006, 141, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Foyer, C.H.; Noctor, G. Redox regulation in photosynthetic organisms: Signaling, acclimation, and practical implications. Antioxidants Redox Signal. 2009, 11, 861–905. [Google Scholar] [CrossRef]
- Borisova, M.M.M.; Kozuleva, M.A.; Rudenko, N.N.; Naydov, I.A.; Klenina, I.B.; Ivanov, B.N. Photosynthetic electron flow to oxygen and diffusion of hydrogen peroxide through the chloroplast envelope via aquaporins. Biochim. Biophys. Acta (BBA)-Bioenerg. 2012, 1817, 1314–1321. [Google Scholar] [CrossRef] [Green Version]
- Laloi, C.; Przybyla, D.; Apel, K. A genetic approach towards elucidating the biological activity of different reactive oxygen species in Arabidopsis thaliana. J. Exp. Bot. 2006, 57, 1719–1724. [Google Scholar] [CrossRef] [Green Version]
- Baxter, A.; Mittler, R.; Suzuki, N. ROS as key players in plant stress signalling. J. Exp. Bot. 2014, 65, 1229–1240. [Google Scholar] [CrossRef]
- Choudhury, F.K.; Rivero, R.M.; Blumwald, E.; Mittler, R. Reactive oxygen species, abiotic stress and stress combination. Plant J. 2017, 90, 856–867. [Google Scholar] [CrossRef]
- Awad, J.; Stotz, H.U.; Fekete, A.; Krischke, M.; Engert, C.; Havaux, M.; Berger, S.; Mueller, M.J. 2-cysteine peroxiredoxins and thylakoid ascorbate peroxidase create a water-water cycle that is essential to protect the photosynthetic apparatus under high light stress conditions. Plant Physiol. 2015, 167, 1592–1603. [Google Scholar] [CrossRef] [Green Version]
- Foyer, C.H.; Noctor, G. Managing the cellular redox hub in photosynthetic organisms. Plant Cell Environ. 2012, 35, 199–201. [Google Scholar] [CrossRef]
- Foyer, C.H.; Noctor, G. Defining robust redox signalling within the context of the plant cell. Plant Cell Environ. 2015, 38, 239. [Google Scholar] [CrossRef]
- Kato, Y.; Sun, X.; Zhang, L.; Sakamoto, W. Cooperative D1 degradation in the photosystem II repair mediated by chloroplastic proteases in Arabidopsis. Plant Physiol. 2012, 159, 1428–1439. [Google Scholar] [CrossRef] [Green Version]
- Devireddy, A.R.; Zandalinas, S.I.; Gómez-Cadenas, A.; Blumwald, E.; Mittler, R. Coordinating the overall stomatal response of plants: Rapid leaf-to-leaf communication during light stress. Sci. Signal. 2018, 11, eaam9514. [Google Scholar] [CrossRef] [Green Version]
- Mustafin, Z.S.; Zamyatin, V.I.; Konstantinov, D.K.; Doroshkov, A.V.; Lashin, S.A.; Afonnikov, D.A. Phylostratigraphic analysis shows the earliest origination of the abiotic stress associated genes in Arabidopsis thaliana. Genes 2019, 10, 963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umezawa, T.; Nakashima, K.; Miyakawa, T.; Kuromori, T.; Tanokura, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Molecular basis of the core regulatory network in ABA responses: Sensing, signaling and transport. Plant Cell Physiol. 2010, 51, 1821–1839. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hu, R.; Sreedasyam, A.; Garcia, T.M.; Lipzen, A.; Wang, M.; Yerramsetty, P.; Liu, D.; Ng, V.; Schmutz, J.; et al. Light-responsive expression atlas reveals the effects of light quality and intensity in Kalanchoë fedtschenkoi, a plant with crassulacean acid metabolism. GigaScience 2020, 9, giaa018. [Google Scholar] [CrossRef] [PubMed]
- Cherenkov, P.; Novikova, D.; Omelyanchuk, N.; Levitsky, V.; Grosse, I.; Weijers, D.; Mironova, V. Diversity of cis-regulatory elements associated with auxin response in Arabidopsis thaliana. J. Exp. Bot. 2018, 69, 329–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, D.; Jouannet, V.; Agustí, J.; Kaul, V.; Levitsky, V.; Sanchez, P.; Mironova, V.V.; Greb, T. Tissue-specific transcriptome profiling of the Arabidopsis inflorescence stem reveals local cellular signatures. Plant Cell 2021, 33, 200–223. [Google Scholar] [CrossRef] [PubMed]
- Ermakov, A.; Bobrovskikh, A.; Zubairova, U.; Konstantinov, D.; Doroshkov, A. Stress-induced changes in the expression of antioxidant system genes for rice (Oryza sativa L.) and bread wheat (Triticum aestivum L.). PeerJ 2019, 7, e7791. [Google Scholar] [CrossRef]
- O’Malley, R.C.; Huang, S.s.C.; Song, L.; Lewsey, M.G.; Bartlett, A.; Nery, J.R.; Galli, M.; Gallavotti, A.; Ecker, J.R. Cistrome and epicistrome features shape the regulatory DNA landscape. Cell 2016, 165, 1280–1292. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhao, X.; Chory, J. The Arabidopsis transcriptome responds specifically and dynamically to high light stress. Cell Rep. 2019, 29, 4186–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandalinas, S.I.; Sengupta, S.; Burks, D.; Azad, R.K.; Mittler, R. Identification and characterization of a core set of ROS wave-associated transcripts involved in the systemic acquired acclimation response of Arabidopsis to excess light. Plant J. 2019, 98, 126–141. [Google Scholar] [CrossRef]
- Balfagón, D.; Sengupta, S.; Gómez-Cadenas, A.; Fritschi, F.B.; Azad, R.K.; Mittler, R.; Zandalinas, S.I. Jasmonic acid is required for plant acclimation to a combination of high light and heat stress. Plant Physiol. 2019, 181, 1668–1682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weise, S.E.; Liu, T.; Childs, K.L.; Preiser, A.L.; Katulski, H.M.; Perrin-Porzondek, C.; Sharkey, T.D. Transcriptional regulation of the glucose-6-phosphate/phosphate translocator 2 is related to carbon exchange across the chloroplast envelope. Front. Plant Sci. 2019, 10, 827. [Google Scholar] [CrossRef]
- Sharma, R.; Singh, G.; Bhattacharya, S.; Singh, A. Comparative transcriptome meta-analysis of Arabidopsis thaliana under drought and cold stress. PLoS ONE 2018, 13, e0203266. [Google Scholar] [CrossRef]
- Sellaro, R.; Pacín, M.; Casal, J.J. Meta-analysis of the transcriptome reveals a core set of shade-avoidance genes in Arabidopsis. Photochem. Photobiol. 2017, 93, 692–702. [Google Scholar] [CrossRef]
- Zemlyanskaya, E.V.; Wiebe, D.S.; Omelyanchuk, N.A.; Levitsky, V.G.; Mironova, V.V. Meta-analysis of transcriptome data identified TGTCNN motif variants associated with the response to plant hormone auxin in Arabidopsis thaliana L. J. Bioinform. Comput. Biol. 2016, 14, 1641009. [Google Scholar] [CrossRef]
- Lotkowska, M.E.; Tohge, T.; Fernie, A.R.; Xue, G.P.; Balazadeh, S.; Mueller-Roeber, B. The Arabidopsis transcription factor MYB112 promotes anthocyanin formation during salinity and under high light stress. Plant Physiol. 2015, 169, 1862–1880. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, K.; Xu, Z.; El-Kereamy, A.; Casaretto, J.A.; Rothstein, S.J. The Arabidopsis transcription factor ANAC032 represses anthocyanin biosynthesis in response to high sucrose and oxidative and abiotic stresses. Front. Plant Sci. 2016, 7, 1548. [Google Scholar] [CrossRef]
- Choi, H.; Jeong, S.; Kim, D.S.; Na, H.J.; Ryu, J.S.; Lee, S.S.; Nam, H.G.; Lim, P.O.; Woo, H.R. The homeodomain-leucine zipper ATHB23, a phytochrome B-interacting protein, is important for phytochrome B-mediated red light signaling. Physiol. Plant. 2014, 150, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Perotti, M.F.; Ribone, P.A.; Cabello, J.V.; Ariel, F.D.; Chan, R.L. AtHB23 participates in the gene regulatory network controlling root branching, and reveals differences between secondary and tertiary roots. Plant J. 2019, 100, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Robson, F.; Okamoto, H.; Patrick, E.; Harris, S.R.; Wasternack, C.; Brearley, C.; Turner, J.G. Jasmonate and phytochrome A signaling in Arabidopsis wound and shade responses are integrated through JAZ1 stability. Plant Cell 2010, 22, 1143–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Figueroa, P.; Browse, J. Characterization of JAZ-interacting bHLH transcription factors that regulate jasmonate responses in Arabidopsis. J. Exp. Bot. 2011, 62, 2143–2154. [Google Scholar] [CrossRef] [Green Version]
- Stracke, R.; Favory, J.J.; Gruber, H.; Bartelniewoehner, L.; Bartels, S.; Binkert, M.; Funk, M.; Weisshaar, B.; Ulm, R. The Arabidopsis bZIP transcription factor HY5 regulates expression of the PFG1/MYB12 gene in response to light and ultraviolet-B radiation. Plant Cell Environ. 2010, 33, 88–103. [Google Scholar] [CrossRef]
- Morishita, T.; Kojima, Y.; Maruta, T.; Nishizawa-Yokoi, A.; Yabuta, Y.; Shigeoka, S. Arabidopsis NAC transcription factor, ANAC078, regulates flavonoid biosynthesis under high-light. Plant Cell Physiol. 2009, 50, 2210–2222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Y.; Yang, H.; Ma, L.; Sun, N.; Yu, H.; Liu, T.; Gao, Y.; Gu, H.; Chen, Z.; Wada, M.; et al. A genome-wide analysis of blue-light regulation of Arabidopsis transcription factor gene expression during seedling development. Plant Physiol. 2003, 133, 1480–1493. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Jia, Q.; Guo, J.; Zhu, Z.; Shao, M.; Wang, J.; Li, W.; Dai, J.; Guo, M.; Li, R.; et al. The High Chlorophyll Fluorescence 244 (HCF244) Is Potentially Involved in Glutathione Peroxidase 7-regulated High Light Stress in Arabidopsis thaliana. Environ. Exp. Bot. 2022, 195, 104767. [Google Scholar] [CrossRef]
- Fisher, R.A. Statistical Methods for Research Workers; Springer: Berlin/Heidelberg, Germany, 1992; pp. 66–70. [Google Scholar]
- Wilkinson, B. A statistical consideration in psychological research. Psychol. Bull. 1951, 48, 156. [Google Scholar] [CrossRef]
- Griffith, O.L.; Melck, A.; Jones, S.J.; Wiseman, S.M. Meta-analysis and meta-review of thyroid cancer gene expression profiling studies identifies important diagnostic biomarkers. J. Clin. Oncol. 2006, 24, 5043–5051. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. Quantitative trait loci analysis using the false discovery rate. Genetics 2005, 171, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohlf, F.J.; Corti, M. Use of two-block partial least-squares to study covariation in shape. Syst. Biol. 2000, 49, 740–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Novikova, D.; Cherenkov, P.; Sizentsova, Y.; Mironova, V. metaRE R Package for Meta-Analysis of Transcriptome Data to Identify the cis-Regulatory Code behind the Transcriptional Reprogramming. Genes 2020, 11, 634. [Google Scholar] [CrossRef]
- Franco-Zorrilla, J.M.; López-Vidriero, I.; Carrasco, J.L.; Godoy, M.; Vera, P.; Solano, R. DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc. Natl. Acad. Sci. USA 2014, 111, 2367–2372. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Nepusz, T.; Yu, H.; Paccanaro, A. Detecting overlapping protein complexes in protein-protein interaction networks. Nat. Methods 2012, 9, 471–472. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO ID | Ecotype | Sampled Tissue | Plant Age | PPFD, μmol m s | Treatment Duration | Control/ Treatment Samples | DEGs 1 | Related Article |
|---|---|---|---|---|---|---|---|---|
| GSE111062 | Col-0 | seedling | 7 days | 1200 | 0.5–72 h | 12/12 | 595 | [42] |
| GSE117296 | Col-0 | leaf | 4–5 weeks | 2000 | 2, 4, 8 min | 6/18 | 2654 | [43] |
| GSE117298 | Col-0 | leaf | 4–5 weeks | 2000 | 8 min | 6/6 | 633 | [43] |
| GSE134391 | Col-0 | leaf | 30 days | 600 | 7 h | 3/3 | 492 | [44] |
| GSE132626 | Col-0, Ws-0 | leaf | NA 2 | 500 | 15–240 h | 3/15 | 1113 | [45] |
| ID | Vertices | Edges | ↑ 1 | ↓ 2 | ↑↓ 3 | Added |
|---|---|---|---|---|---|---|
| Cluster 1 | 108 | 420 | 43 | 16 | 38 | 11 |
| Cluster 2 | 97 | 983 | 15 | 13 | 67 | 5 |
| Cluster 3 | 48 | 87 | 11 | 15 | 6 | 16 |
| Cluster 4 | 15 | 22 | 3 | 7 | 4 | 0 |
| Cluster 5 | 14 | 20 | 6 | 5 | 3 | 0 |
| Cluster 6 | 13 | 59 | 2 | 4 | 3 | 4 |
| Cluster 7 | 11 | 30 | 7 | 3 | 0 | 1 |
| Cluster 8 | 11 | 14 | 5 | 1 | 3 | 2 |
| Cluster 9 | 9 | 8 | 7 | 0 | 2 | 0 |
| Cluster 10 | 7 | 21 | 6 | 1 | 0 | 0 |
| Cluster 11 | 7 | 7 | 0 | 2 | 2 | 3 |
| Cluster 12 | 7 | 7 | 3 | 2 | 0 | 2 |
| Cluster 13 | 6 | 6 | 6 | 0 | 0 | 0 |
| Cluster 14 | 5 | 6 | 5 | 0 | 0 | 0 |
| Cluster 15 | 5 | 5 | 1 | 3 | 1 | 0 |
| Cluster 16 | 5 | 4 | 1 | 1 | 2 | 0 |
| Cluster 17 | 4 | 3 | 0 | 2 | 0 | 2 |
| Cluster 18 | 3 | 2 | 2 | 0 | 1 | 0 |
| Cluster 19 | 3 | 2 | 3 | 0 | 0 | 0 |
| Cluster 20 | 2 | 1 | 0 | 0 | 0 | 2 |
| Cluster 21 | 2 | 1 | 0 | 0 | 2 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobrovskikh, A.V.; Zubairova, U.S.; Bondar, E.I.; Lavrekha, V.V.; Doroshkov, A.V. Transcriptomic Data Meta-Analysis Sheds Light on High Light Response in Arabidopsis thaliana L. Int. J. Mol. Sci. 2022, 23, 4455. https://doi.org/10.3390/ijms23084455
Bobrovskikh AV, Zubairova US, Bondar EI, Lavrekha VV, Doroshkov AV. Transcriptomic Data Meta-Analysis Sheds Light on High Light Response in Arabidopsis thaliana L. International Journal of Molecular Sciences. 2022; 23(8):4455. https://doi.org/10.3390/ijms23084455
Chicago/Turabian StyleBobrovskikh, Aleksandr V., Ulyana S. Zubairova, Eugeniya I. Bondar, Viktoriya V. Lavrekha, and Alexey V. Doroshkov. 2022. "Transcriptomic Data Meta-Analysis Sheds Light on High Light Response in Arabidopsis thaliana L." International Journal of Molecular Sciences 23, no. 8: 4455. https://doi.org/10.3390/ijms23084455