
Revisiting the Role of Astrocytic MAOB in Parkinson’s Disease

 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. MAOB Expression in the Brain
2. Challenges for Defining the Role of MAOB in Parkinson’s Disease
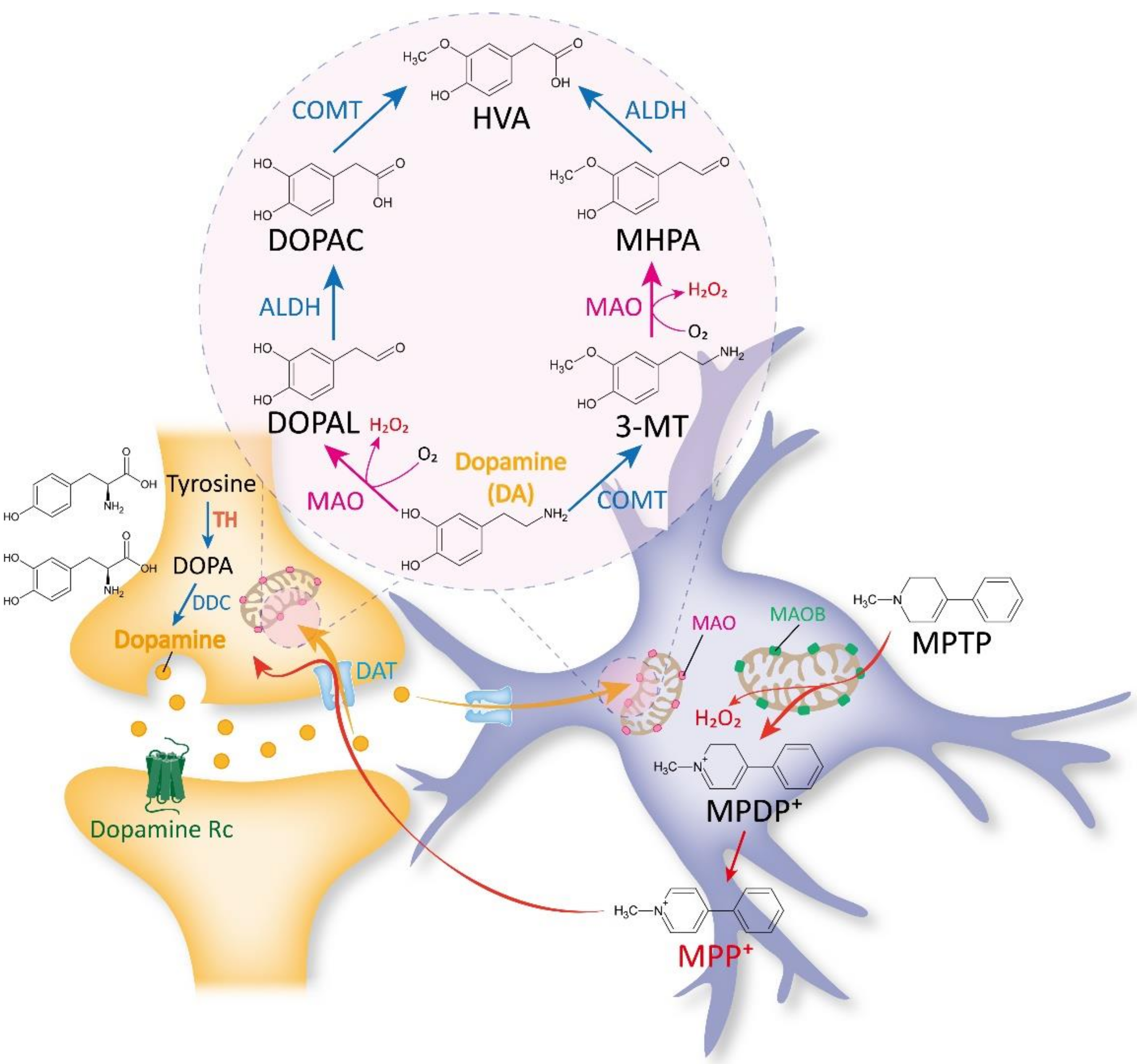
2.1. Traditional Views on MAOB as a DA-Metabolizing Enzyme
2.2. Past and Recent Discoveries against the Belief of MAOB as a DA-Degrading Enzyme
2.3. Traditional Views on MAOB as an MPTP-Metabolizing Enzyme
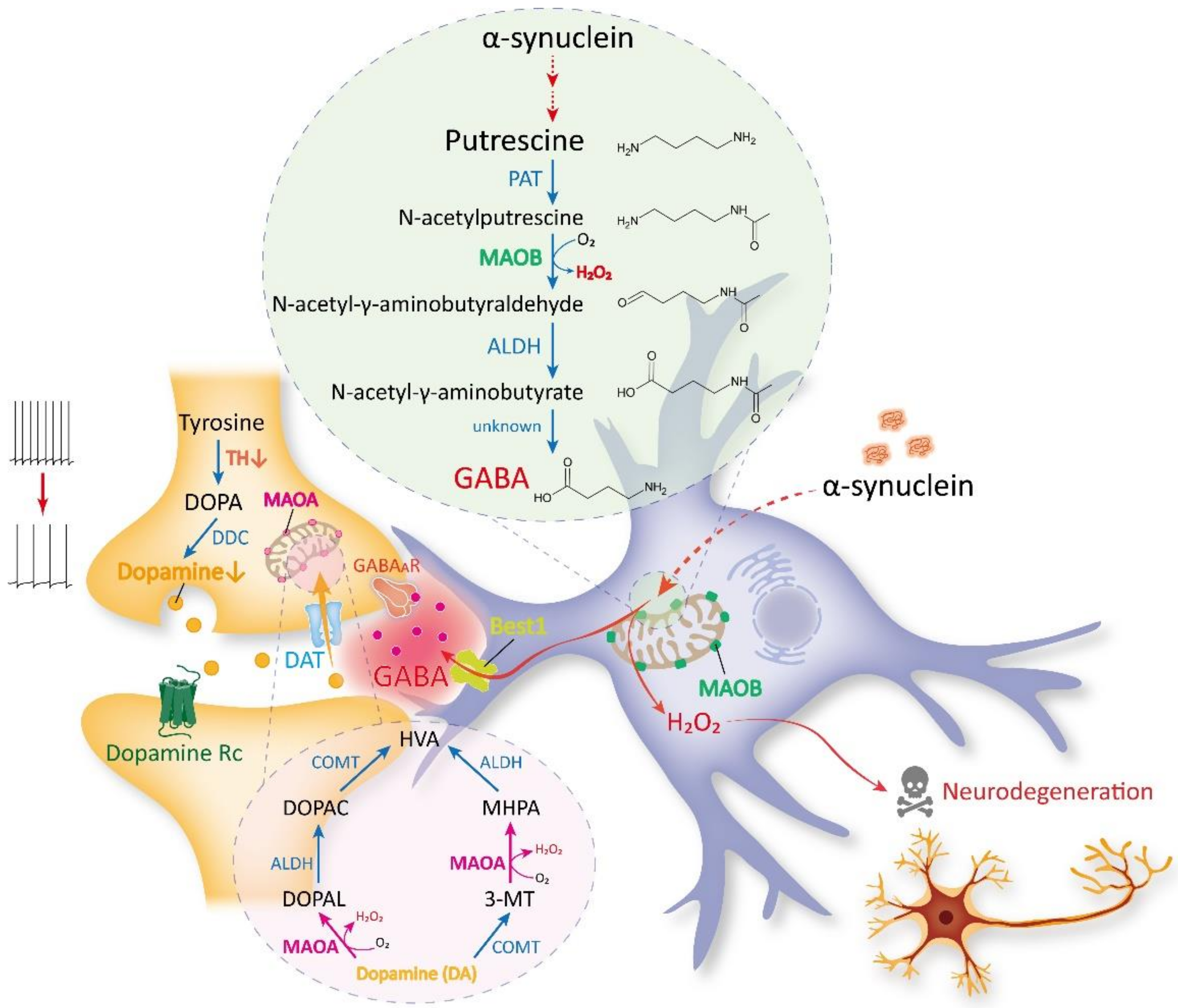
3. Recent Discoveries on MAOB as the Astrocytic GABA- and H2O2-Synthesizing Enzyme
4. MAOB as a Therapeutic Target for PD
4.1. Selegiline
4.2. Rasagiline
4.3. Safinamide
4.4. KDS2010
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Hare, M.L. Tyramine oxidase: A new enzyme system in liver. Biochem. J. 1928, 22, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Rebhun, J.; Feinberg, S.M.; Zeller, E.A. Potentiating effect of iproniazid on action of some sympathicomimetic amines. Proc. Soc. Exp. Biol. Med. 1954, 87, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Gorkin, V.Z. On certain properties of monoaminoxidase in liver and brain mitochondria in rats. Biokhimiia 1959, 24, 826–832. [Google Scholar] [PubMed]
- Nagatsu, T. Progress in monoamine oxidase (MAO) research in relation to genetic engineering. Neurotoxicology 2004, 25, 11–20. [Google Scholar] [CrossRef]
- Youdim, M.B.; Edmondson, D.; Tipton, K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006, 7, 295–309. [Google Scholar] [CrossRef]
- Tong, J.; Meyer, J.H.; Furukawa, Y.; Boileau, I.; Chang, L.J.; Wilson, A.A.; Houle, S.; Kish, S.J. Distribution of monoamine oxidase proteins in human brain: Implications for brain imaging studies. J. Cereb. Blood. Flow. Metab. 2013, 33, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Vitalis, T.; Fouquet, C.; Alvarez, C.; Seif, I.; Price, D.; Gaspar, P.; Cases, O. Developmental expression of monoamine oxidases A and B in the central and peripheral nervous systems of the mouse. J. Comp. Neurol. 2002, 442, 331–347. [Google Scholar] [CrossRef]
- Luque, J.M.; Kwan, S.W.; Abell, C.W.; Da Prada, M.; Richards, J.G. Cellular expression of mRNAs encoding monoamine oxidases A and B in the rat central nervous system. J. Comp. Neurol. 1995, 363, 665–680. [Google Scholar] [CrossRef]
- Riederer, P.; Konradi, C.; Schay, V.; Kienzl, E.; Birkmayer, G.; Danielczyk, W.; Sofic, E.; Youdim, M.B. Localization of MAO-A and MAO-B in human brain: A step in understanding the therapeutic action of L-deprenyl. Adv. Neurol. 1987, 45, 111–118. [Google Scholar]
- Levitt, P.; Pintar, J.E.; Breakefield, X.O. Immunocytochemical demonstration of monoamine oxidase B in brain astrocytes and serotonergic neurons. Proc. Natl. Acad. Sci. USA 1982, 79, 6385–6389. [Google Scholar] [CrossRef] [Green Version]
- Saura, J.; Richards, J.G.; Mahy, N. Differential age-related changes of MAO-A and MAO-B in mouse brain and peripheral organs. Neurobiol. Aging 1994, 15, 399–408. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Munch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahy, N.; Andres, N.; Andrade, C.; Saura, J. Age-related changes of MAO-A and -B distribution in human and mouse brain. Neurobiology 2000, 8, 47–54. [Google Scholar] [PubMed]
- Fowler, J.S.; Volkow, N.D.; Wang, G.J.; Logan, J.; Pappas, N.; Shea, C.; MacGregor, R. Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol. Aging 1997, 18, 431–435. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Park, J.H.; Ju, Y.H.; Choi, J.W.; Song, H.J.; Jang, B.K.; Woo, J.; Chun, H.; Kim, H.J.; Shin, S.J.; Yarishkin, O.; et al. Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer’s disease. Sci. Adv. 2019, 5, eaav0316. [Google Scholar] [CrossRef] [Green Version]
- Chun, H.; Lim, J.; Park, K.D.; Lee, C.J. Inhibition of monoamine oxidase B prevents reactive astrogliosis and scar formation in stab wound injury model. Glia 2022, 70, 354–367. [Google Scholar] [CrossRef]
- Mallajosyula, J.K.; Kaur, D.; Chinta, S.J.; Rajagopalan, S.; Rane, A.; Nicholls, D.G.; Di Monte, D.A.; Macarthur, H.; Andersen, J.K. MAO-B elevation in mouse brain astrocytes results in Parkinson’s pathology. PLoS ONE 2008, 3, e1616. [Google Scholar] [CrossRef] [Green Version]
- Moriguchi, S.; Wilson, A.A.; Miler, L.; Rusjan, P.M.; Vasdev, N.; Kish, S.J.; Rajkowska, G.; Wang, J.; Bagby, M.; Mizrahi, R.; et al. Monoamine Oxidase B Total Distribution Volume in the Prefrontal Cortex of Major Depressive Disorder: An [11C]SL25.1188 Positron Emission Tomography Study. JAMA Psychiatry 2019, 76, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Vieitez, E.; Carter, S.F.; Chiotis, K.; Saint-Aubert, L.; Leuzy, A.; Scholl, M.; Almkvist, O.; Wall, A.; Langstrom, B.; Nordberg, A. Comparison of Early-Phase 11C-Deuterium-l-Deprenyl and 11C-Pittsburgh Compound B PET for Assessing Brain Perfusion in Alzheimer Disease. J. Nucl. Med. 2016, 57, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.Y.; Nam, M.H.; Yoon, H.H.; Kim, J.; Hwang, Y.J.; Won, W.; Woo, D.H.; Lee, J.A.; Park, H.J.; Jo, S.; et al. Aberrant Tonic Inhibition of Dopaminergic Neuronal Activity Causes Motor Symptoms in Animal Models of Parkinson’s Disease. Curr. Biol. 2020, 30, 276–291.e9. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Heo, J.Y.; Lee, C.J.; Nam, M.H. The Pathological Role of Astrocytic MAOB in Parkinsonism Revealed by Genetic Ablation and Over-expression of MAOB. Exp. Neurobiol. 2021, 30, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.; Benabid, A.L.; Sadoul, R.; Verna, J.M. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: Contribution to the apoptotic theory in Parkinson’s disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef]
- Alborghetti, M.; Nicoletti, F. Different Generations of Type-B Monoamine Oxidase Inhibitors in Parkinson’s Disease: From Bench to Bedside. Curr. Neuropharmacol. 2019, 17, 861–873. [Google Scholar] [CrossRef]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [Green Version]
- Westlund, K.N.; Denney, R.M.; Rose, R.M.; Abell, C.W. Localization of distinct monoamine oxidase A and monoamine oxidase B cell populations in human brainstem. Neuroscience 1988, 25, 439–456. [Google Scholar] [CrossRef]
- Thorpe, L.W.; Westlund, K.N.; Kochersperger, L.M.; Abell, C.W.; Denney, R.M. Immunocytochemical localization of monoamine oxidases A and B in human peripheral tissues and brain. J. Histochem. Cytochem. 1987, 35, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Fagervall, I.; Ross, S.B. A and B forms of monoamine oxidase within the monoaminergic neurons of the rat brain. J. Neurochem. 1986, 47, 569–576. [Google Scholar] [CrossRef]
- Bach, A.W.; Lan, N.C.; Johnson, D.L.; Abell, C.W.; Bembenek, M.E.; Kwan, S.W.; Seeburg, P.H.; Shih, J.C. cDNA cloning of human liver monoamine oxidase A and B: Molecular basis of differences in enzymatic properties. Proc. Natl. Acad. Sci. USA 1988, 85, 4934–4938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.P.; Weyler, W.; Chen, S.; Sims, K.B.; Rinehart, W.B.; Utterback, M.C.; Powell, J.F.; Breakefield, X.O. Structural features of human monoamine oxidase A elucidated from cDNA and peptide sequences. J. Neurochem. 1988, 51, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Brannan, T.; Prikhojan, A.; Martinez-Tica, J.; Yahr, M.D. In vivo comparison of the effects of inhibition of MAO-A versus MAO-B on striatal L-DOPA and dopamine metabolism. J. Neural. Transm. Park Dis. Dement. Sect. 1995, 10, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Rathitharan, G.; Meyer, J.H.; Furukawa, Y.; Ang, L.C.; Boileau, I.; Guttman, M.; Hornykiewicz, O.; Kish, S.J. Brain monoamine oxidase B and A in human parkinsonian dopamine deficiency disorders. Brain 2017, 140, 2460–2474. [Google Scholar] [CrossRef]
- Glover, V.; Sandler, M.; Owen, F.; Riley, G.J. Dopamine is a monoamine oxidase B substrate in man. Nature 1977, 265, 80–81. [Google Scholar] [CrossRef]
- Tetrud, J.W.; Langston, J.W. The effect of deprenyl (selegiline) on the natural history of Parkinson’s disease. Science 1989, 245, 519–522. [Google Scholar] [CrossRef]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B. Rasagiline: A novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog. Neurobiol. 2010, 92, 330–344. [Google Scholar] [CrossRef]
- Nam, M.H.; Cho, J.; Kwon, D.H.; Park, J.Y.; Woo, J.; Lee, J.M.; Lee, S.; Ko, H.Y.; Won, W.; Kim, R.G.; et al. Excessive Astrocytic GABA Causes Cortical Hypometabolism and Impedes Functional Recovery after Subcortical Stroke. Cell Rep. 2020, 32, 107975. [Google Scholar] [CrossRef]
- Cho, H.U.; Kim, S.; Sim, J.; Yang, S.; An, H.; Nam, M.H.; Jang, D.P.; Lee, C.J. Redefining differential roles of MAO-A in dopamine degradation and MAO-B in tonic GABA synthesis. Exp. Mol. Med. 2021, 53, 1148–1158. [Google Scholar] [CrossRef]
- Yoon, B.E.; Woo, J.; Chun, Y.E.; Chun, H.; Jo, S.; Bae, J.Y.; An, H.; Min, J.O.; Oh, S.J.; Han, K.S.; et al. Glial GABA, synthesized by monoamine oxidase B, mediates tonic inhibition. J. Physiol. 2014, 592, 4951–4968. [Google Scholar] [CrossRef]
- Nam, M.H.; Park, J.H.; Song, H.J.; Choi, J.W.; Kim, S.; Jang, B.K.; Yoon, H.H.; Heo, J.Y.; Lee, H.; An, H.; et al. KDS2010, a Newly Developed Reversible MAO-B Inhibitor, as an Effective Therapeutic Candidate for Parkinson’s Disease. Neurotherapeutics 2021, 18, 1729–1747. [Google Scholar] [CrossRef] [PubMed]
- Weiner, N. Substrate specificity of brain amine oxidase of several mammals. Proj. Rep. USAF Sch. Aviat. Med. 1960, 60, 1–8. [Google Scholar] [CrossRef]
- Braestrup, C.; Andersen, H.; Randrup, A. The monoamine oxidase B inhibitor deprenyl potentiates phenylethylamine behaviour in rats without inhibition of catecholamine metabolite formation. Eur. J. Pharmacol. 1975, 34, 181–187. [Google Scholar] [CrossRef]
- Waldmeier, P.C.; Delini-Stula, A.; Maitre, L. Preferential deamination of dopamine by an A type monoamine oxidase in rat brain. Naunyn-Schmiedebergs Arch. Pharmacol. 1976, 292, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Green, A.R.; Mitchell, B.D.; Tordoff, A.F.; Youdim, M.B. Evidence for dopamine deamination by both type A and type B monoamine oxidase in rat brain in vivo and for the degree of inhibition of enzyme necessary for increased functional activity of dopamine and 5-hydroxytryptamine. Br. J. Pharmacol. 1977, 60, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrick, N.A.; Murphy, D.L. Species differences in the deamination of dopamine and other substrates for monoamine oxidase in brain. Psychopharmacology 1980, 72, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Schoepp, D.D.; Azzaro, A.J. Specificity of endogenous substrates for types A and B monoamine oxidase in rat striatum. J. Neurochem. 1981, 36, 2025–2031. [Google Scholar] [CrossRef]
- Roth, J.A.; Feor, K. Deamination of dopamine and its 3-O-methylated derivative by human brain monoamine oxidase. Biochem. Pharmacol. 1978, 27, 1606–1608. [Google Scholar] [CrossRef]
- Glover, V.; Elsworth, J.D.; Sandler, M. Dopamine oxidation and its inhibition by (-)-deprenyl in man. J. Neural. Transm. Suppl. 1980, 16, 163–172. [Google Scholar]
- Tipton, K.F.; Houslay, M.D.; Garrett, N.J. Allotopic properties of human brain monoamine oxidase. Nat. New Biol. 1973, 246, 213–214. [Google Scholar] [CrossRef]
- Robakis, D.; Fahn, S. Defining the Role of the Monoamine Oxidase-B Inhibitors for Parkinson’s Disease. CNS Drugs 2015, 29, 433–441. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Murakami, S.; Diaz-Corrales, F.J.; Ogawa, N. Striatal astrocytes act as a reservoir for L-DOPA. PLoS ONE 2014, 9, e106362. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Kopin, I.J.; Goldstein, D.S. Catecholamine metabolism: A contemporary view with implications for physiology and medicine. Pharmacol. Rev. 2004, 56, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal. 2013, 11, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nirenberg, M.J.; Vaughan, R.A.; Uhl, G.R.; Kuhar, M.J.; Pickel, V.M. The dopamine transporter is localized to dendritic and axonal plasma membranes of nigrostriatal dopaminergic neurons. J. Neurosci. 1996, 16, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Nirenberg, M.J.; Chan, J.; Pohorille, A.; Vaughan, R.A.; Uhl, G.R.; Kuhar, M.J.; Pickel, V.M. The dopamine transporter: Comparative ultrastructure of dopaminergic axons in limbic and motor compartments of the nucleus accumbens. J. Neurosci. 1997, 17, 6899–6907. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Zhang, C.W.; Chen, G.Y.; Zhu, B.; Chai, C.; Xu, Q.H.; Tan, E.K.; Zhu, Q.; Lim, K.L.; Yao, S.Q. A sensitive two-photon probe to selectively detect monoamine oxidase B activity in Parkinson’s disease models. Nat. Commun. 2014, 5, 3276. [Google Scholar] [CrossRef]
- Nicotra, A.; Pierucci, F.; Parvez, H.; Senatori, O. Monoamine oxidase expression during development and aging. Neurotoxicology 2004, 25, 155–165. [Google Scholar] [CrossRef]
- Damier, P.; Kastner, A.; Agid, Y.; Hirsch, E.C. Does monoamine oxidase type B play a role in dopaminergic nerve cell death in Parkinson’s disease? Neurology 1996, 46, 1262–1269. [Google Scholar] [CrossRef]
- Chen, J.J.; Swope, D.M. Clinical pharmacology of rasagiline: A novel, second-generation propargylamine for the treatment of Parkinson disease. J. Clin. Pharmacol. 2005, 45, 878–894. [Google Scholar] [CrossRef]
- Fornai, F.; Chen, K.; Giorgi, F.S.; Gesi, M.; Alessandri, M.G.; Shih, J.C. Striatal dopamine metabolism in monoamine oxidase B-deficient mice: A brain dialysis study. J. Neurochem. 1999, 73, 2434–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, S.P.; Fairbrother, I.S.; Kelly, J.S.; Arbuthnott, G.W. Effects of selective monoamine oxidase inhibitors on the in vivo release and metabolism of dopamine in the rat striatum. J. Neurochem. 1990, 55, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Scarr, E.; Wingerchuk, D.M.; Juorio, A.V.; Paterson, I.A. The effects of monoamine oxidase B inhibition on dopamine metabolism in rats with nigro-striatal lesions. Neurochem. Res. 1994, 19, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Paterson, I.A.; Juorio, A.V.; Berry, M.D.; Zhu, M.Y. Inhibition of monoamine oxidase-B by (-)-deprenyl potentiates neuronal responses to dopamine agonists but does not inhibit dopamine catabolism in the rat striatum. J. Pharmacol. Exp. Ther. 1991, 258, 1019–1026. [Google Scholar]
- Sader-Mazbar, O.; Loboda, Y.; Rabey, M.J.; Finberg, J.P. Increased L-DOPA-derived dopamine following selective MAO-A or -B inhibition in rat striatum depleted of dopaminergic and serotonergic innervation. Br. J. Pharmacol. 2013, 170, 999–1013. [Google Scholar] [CrossRef] [Green Version]
- Lenders, J.W.; Eisenhofer, G.; Abeling, N.G.; Berger, W.; Murphy, D.L.; Konings, C.H.; Wagemakers, L.M.; Kopin, I.J.; Karoum, F.; van Gennip, A.H.; et al. Specific genetic deficiencies of the A and B isoenzymes of monoamine oxidase are characterized by distinct neurochemical and clinical phenotypes. J. Clin. Investig. 1996, 97, 1010–1019. [Google Scholar] [CrossRef] [Green Version]
- Fowler, C.J.; Benedetti, M.S. The metabolism of dopamine by both forms of monoamine oxidase in the rat brain and its inhibition by cimoxatone. J. Neurochem. 1983, 40, 1534–1541. [Google Scholar] [CrossRef]
- O’Carroll, A.M.; Fowler, C.J.; Phillips, J.P.; Tobbia, I.; Tipton, K.F. The deamination of dopamine by human brain monoamine oxidase. Specificity for the two enzyme forms in seven brain regions. Naunyn. Schmiedebergs. Arch. Pharmacol. 1983, 322, 198–202. [Google Scholar] [CrossRef]
- Davis, G.C.; Williams, A.C.; Markey, S.P.; Ebert, M.H.; Caine, E.D.; Reichert, C.M.; Kopin, I.J. Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979, 1, 249–254. [Google Scholar] [CrossRef]
- Jackson-Lewis, V.; Przedborski, S. Protocol for the MPTP mouse model of Parkinson’s disease. Nat. Protoc. 2007, 2, 141–151. [Google Scholar] [CrossRef]
- Uhl, G.R.; Javitch, J.A.; Snyder, S.H. Normal MPTP binding in parkinsonian substantial nigra: Evidence for extraneuronal toxin conversion in human brain. Lancet 1985, 1, 956–957. [Google Scholar] [CrossRef]
- Watanabe, Y.; Himeda, T.; Araki, T. Mechanisms of MPTP toxicity and their implications for therapy of Parkinson’s disease. Med. Sci. Monit. 2005, 11, RA17–RA23. [Google Scholar] [PubMed]
- Caccia, C.; Maj, R.; Calabresi, M.; Maestroni, S.; Faravelli, L.; Curatolo, L.; Salvati, P.; Fariello, R.G. Safinamide: From molecular targets to a new anti-Parkinson drug. Neurology 2006, 67 (7 Suppl. 2), S18–S23. [Google Scholar] [CrossRef]
- Sundstrom, E.; Luthman, J.; Goldstein, M.; Jonsson, G. Time course of MPTP-induced degeneration of the nigrostriatal dopamine system in C57 BL/6 mice. Brain Res Bull 1988, 21, 257–263. [Google Scholar] [CrossRef]
- Woo, J.; Min, J.O.; Kang, D.S.; Kim, Y.S.; Jung, G.H.; Park, H.J.; Kim, S.; An, H.; Kwon, J.; Kim, J.; et al. Control of motor coordination by astrocytic tonic GABA release through modulation of excitation/inhibition balance in cerebellum. Proc. Natl. Acad. Sci. USA 2018, 115, 5004–5009. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Yoon, B.E.; Berglund, K.; Oh, S.J.; Park, H.; Shin, H.S.; Augustine, G.J.; Lee, C.J. Channel-mediated tonic GABA release from glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef]
- Yoon, B.E.; Jo, S.; Woo, J.; Lee, J.H.; Kim, T.; Kim, D.; Lee, C.J. The amount of astrocytic GABA positively correlates with the degree of tonic inhibition in hippocampal CA1 and cerebellum. Mol. Brain. 2011, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Chun, H.; Im, H.; Kang, Y.J.; Kim, Y.; Shin, J.H.; Won, W.; Lim, J.; Ju, Y.; Park, Y.M.; Kim, S.; et al. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H2O2(-) production. Nat. Neurosci. 2020, 23, 1555–1566. [Google Scholar] [CrossRef]
- Ju, Y.H.; Bhalla, M.; Hyeon, S.J.; Oh, J.E.; Yoo, S.; Chae, U.; Kwon, J.; Koh, W.; Lim, J.; Park, Y.M.; et al. Astrocytic urea cycle detoxifies Aβ-derived ammonia while impairing memory in Alzheimer’s disease. bioRxiv 2021, arXiv:15.464517. [Google Scholar] [CrossRef]
- Chamoli, M.; Chinta, S.J.; Andersen, J.K. An inducible MAO-B mouse model of Parkinson’s disease: A tool towards better understanding basic disease mechanisms and developing novel therapeutics. J. Neural. Transm. 2018, 125, 1651–1658. [Google Scholar] [CrossRef]
- Tan, Y.Y.; Jenner, P.; Chen, S.D. Monoamine Oxidase-B Inhibitors for the Treatment of Parkinson’s Disease: Past, Present, and Future. J. Parkinsons Dis. 2022, 12, 477–493. [Google Scholar] [CrossRef] [PubMed]
- Binde, C.D.; Tvete, I.F.; Gasemyr, J.I.; Natvig, B.; Klemp, M. Comparative effectiveness of dopamine agonists and monoamine oxidase type-B inhibitors for Parkinson’s disease: A multiple treatment comparison meta-analysis. Eur. J. Clin. Pharmacol. 2020, 76, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Ostadkarampour, M.; Putnins, E.E. Monoamine Oxidase Inhibitors: A Review of Their Anti-Inflammatory Therapeutic Potential and Mechanisms of Action. Front Pharmacol. 2021, 12, 676239. [Google Scholar] [CrossRef] [PubMed]
- Pisano, C.A.; Brugnoli, A.; Novello, S.; Caccia, C.; Keywood, C.; Melloni, E.; Vailati, S.; Padoani, G.; Morari, M. Safinamide inhibits in vivo glutamate release in a rat model of Parkinson’s disease. Neuropharmacology 2020, 167, 108006. [Google Scholar] [CrossRef] [PubMed]
- Teo, K.C.; Ho, S.L. Monoamine oxidase-B (MAO-B) inhibitors: Implications for disease-modification in Parkinson’s disease. Transl. Neurodegener. 2013, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finberg, J.P.; Gillman, K. Selective inhibitors of monoamine oxidase type B and the “cheese effect”. Int. Rev. Neurobiol. 2011, 100, 169–190. [Google Scholar]
- Knoll, J.; Ecseri, Z.; Kelemen, K.; Nievel, J.; Knoll, B. Phenylisopropylmethylpropinylamine (E-250), a new spectrum psychic energizer. Arch. Int. Pharmacodyn. Ther. 1965, 155, 154–164. [Google Scholar]
- Riederer, P.; Youdim, M.B. Monoamine oxidase activity and monoamine metabolism in brains of parkinsonian patients treated with l-deprenyl. J. Neurochem. 1986, 46, 1359–1365. [Google Scholar] [CrossRef]
- Riederer, P.; Lachenmayer, L. Selegiline’s neuroprotective capacity revisited. J. Neural Transm. 2003, 110, 1273–1278. [Google Scholar] [CrossRef]
- Palhagen, S.; Heinonen, E.; Hagglund, J.; Kaugesaar, T.; Maki-Ikola, O.; Palm, R.; Swedish Parkinson Study, G. Selegiline slows the progression of the symptoms of Parkinson disease. Neurology 2006, 66, 1200–1206. [Google Scholar] [CrossRef]
- De la Cruz, C.P.; Revilla, E.; Steffen, V.; Rodriguez-Gomez, J.A.; Cano, J.; Machado, A. Protection of the aged substantia nigra of the rat against oxidative damage by (-)-deprenyl. Br. J. Pharmacol. 1996, 117, 1756–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munirathinam, S.; Lakshmana, M.K.; Raju, T.R. (-) deprenyl attenuates aluminium induced neurotoxicity in primary cortical cultures. Neurodegeneration 1996, 5, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, W.; Takahashi, T.; Naoi, M. (-)-Deprenyl protects human dopaminergic neuroblastoma SH-SY5Y cells from apoptosis induced by peroxynitrite and nitric oxide. J. Neurochem. 1998, 70, 2510–2515. [Google Scholar] [CrossRef] [PubMed]
- Tatton, W.G.; Chalmers-Redman, R.M.; Ju, W.J.; Mammen, M.; Carlile, G.W.; Pong, A.W.; Tatton, N.A. Propargylamines induce antiapoptotic new protein synthesis in serum- and nerve growth factor (NGF)-withdrawn, NGF-differentiated PC-12 cells. J. Pharmacol. Exp. Ther. 2002, 301, 753–764. [Google Scholar] [CrossRef]
- Fowler, J.S.; Logan, J.; Volkow, N.D.; Shumay, E.; McCall-Perez, F.; Jayne, M.; Wang, G.J.; Alexoff, D.L.; Apelskog-Torres, K.; Hubbard, B.; et al. Evidence that formulations of the selective MAO-B inhibitor, selegiline, which bypass first-pass metabolism, also inhibit MAO-A in the human brain. Neuropsychopharmacology 2015, 40, 650–657. [Google Scholar] [CrossRef]
- Maurer, H.H.; Kraemer, T. Toxicological detection of selegiline and its metabolites in urine using fluorescence polarization immunoassay (FPIA) and gas chromatography-mass spectrometry (GC-MS) and differentiation by enantioselective GC-MS of the intake of selegiline from abuse of methamphetamine or amphetamine. Arch. Toxicol. 1992, 66, 675–678. [Google Scholar]
- Finberg, J.P.; Youdim, M.B. Pharmacological properties of the anti-Parkinson drug rasagiline; modification of endogenous brain amines, reserpine reversal, serotonergic and dopaminergic behaviours. Neuropharmacology 2002, 43, 1110–1118. [Google Scholar] [CrossRef]
- Kamakura, K.; Mochizuki, H.; Kaida, K.; Hirata, A.; Kanzaki, M.; Masaki, T.; Nakamura, R.; Motoyoshi, K. Therapeutic factors causing hallucination in Parkinson’s disease patients, especially those given selegiline. Parkinsonism Relat. Disord. 2004, 10, 235–242. [Google Scholar] [CrossRef]
- Sadeghian, M.; Mullali, G.; Pocock, J.M.; Piers, T.; Roach, A.; Smith, K.J. Neuroprotection by safinamide in the 6-hydroxydopamine model of Parkinson’s disease. Neuropathol. Appl. Neurobiol. 2016, 42, 423–435. [Google Scholar] [CrossRef]
- Perez-Lloret, S.; Rascol, O. The safety and efficacy of safinamide mesylate for the treatment of Parkinson’s disease. Expert. Rev. Neurother. 2016, 16, 245–258. [Google Scholar] [CrossRef]
- Marzo, A.; Dal Bo, L.; Monti, N.C.; Crivelli, F.; Ismaili, S.; Caccia, C.; Cattaneo, C.; Fariello, R.G. Pharmacokinetics and pharmacodynamics of safinamide, a neuroprotectant with antiparkinsonian and anticonvulsant activity. Pharmacol. Res. 2004, 50, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Bulaklak, K.; Gersbach, C.A. The once and future gene therapy. Nat. Commun. 2020, 11, 5820. [Google Scholar] [CrossRef]
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid. Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.H.; Ye, S.; Lim, S.; Jo, A.; Lee, H.; Hong, F.; Lee, S.E.; Oh, S.J.; Kim, N.R.; Kim, K.; et al. CRISPR-Cas9 Gene Editing Protects from the A53T-SNCA Overexpression-Induced Pathology of Parkinson’s Disease In Vivo. CRISPR J. 2022, 5, 95–108. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nam, M.-H.; Sa, M.; Ju, Y.H.; Park, M.G.; Lee, C.J. Revisiting the Role of Astrocytic MAOB in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 4453. https://doi.org/10.3390/ijms23084453
Nam M-H, Sa M, Ju YH, Park MG, Lee CJ. Revisiting the Role of Astrocytic MAOB in Parkinson’s Disease. International Journal of Molecular Sciences. 2022; 23(8):4453. https://doi.org/10.3390/ijms23084453
Chicago/Turabian StyleNam, Min-Ho, Moonsun Sa, Yeon Ha Ju, Mingu Gordon Park, and C. Justin Lee. 2022. "Revisiting the Role of Astrocytic MAOB in Parkinson’s Disease" International Journal of Molecular Sciences 23, no. 8: 4453. https://doi.org/10.3390/ijms23084453