
Metabolic Syndrome and β-Oxidation of Long-Chain Fatty Acids in the Brain, Heart, and Kidney Mitochondria

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Origin and Features of Metabolic Syndrome
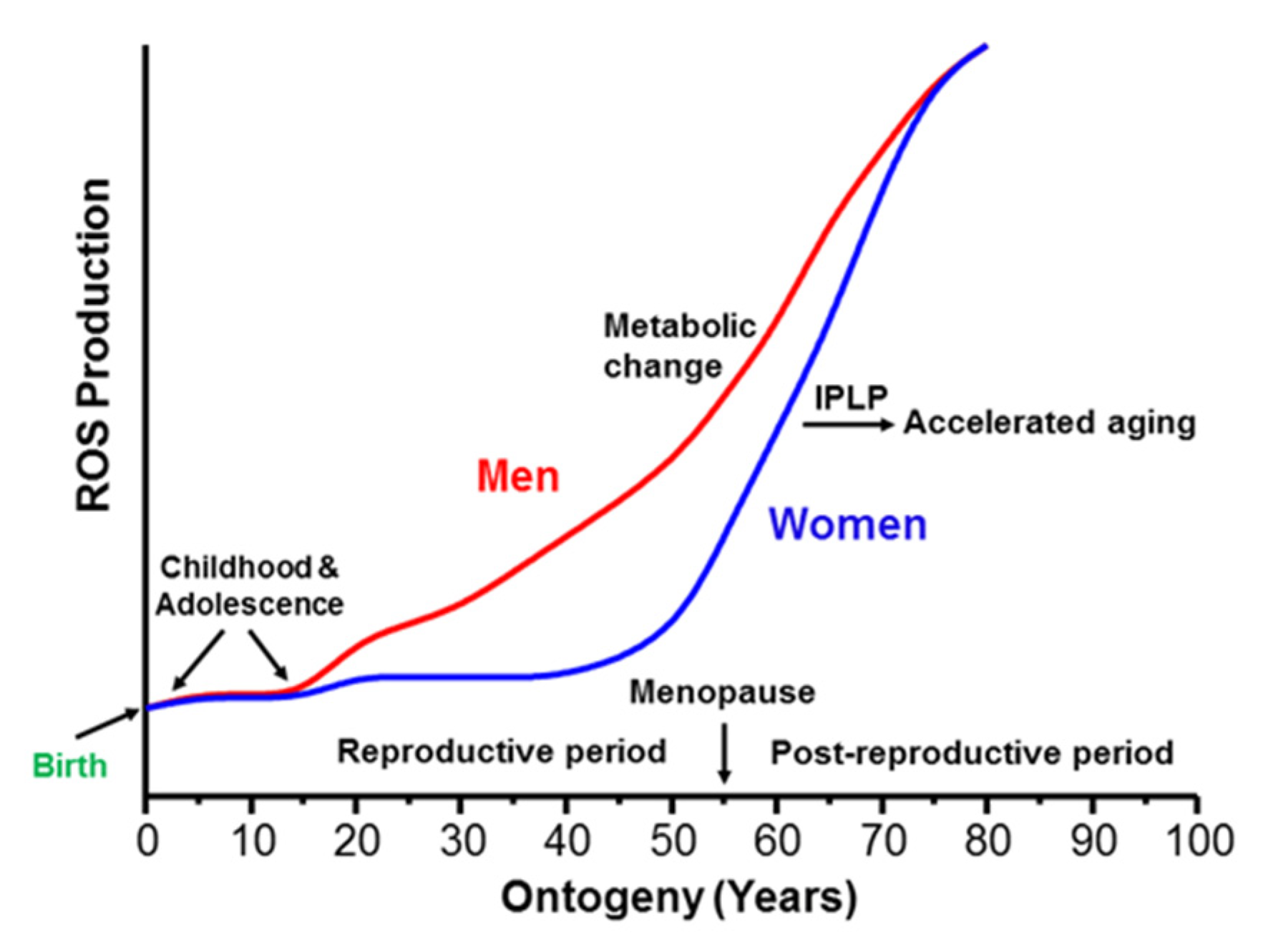
2.1. Metabolic Syndrome Represents the Beginning of the Last Stage of Human Postembryonic Ontogenesis
2.2. Gender Differences in Energy Metabolism Affect the Rate of Appearance of the Metabolic Syndrome
3. Sex-Specific Differences in the Rates of Aging and Longevity
3.1. Sex-Specific Differences in the Rate of Fat Utilization for Production of Energy
3.2. Sex Differences in Substrate Utilization during Physical Activities
3.3. Gender Differences in the Transition from Reproductive to the Postreproductive Stage
4. β-Oxidation of Fatty Acids Is the Primary Source of Energy in Humans
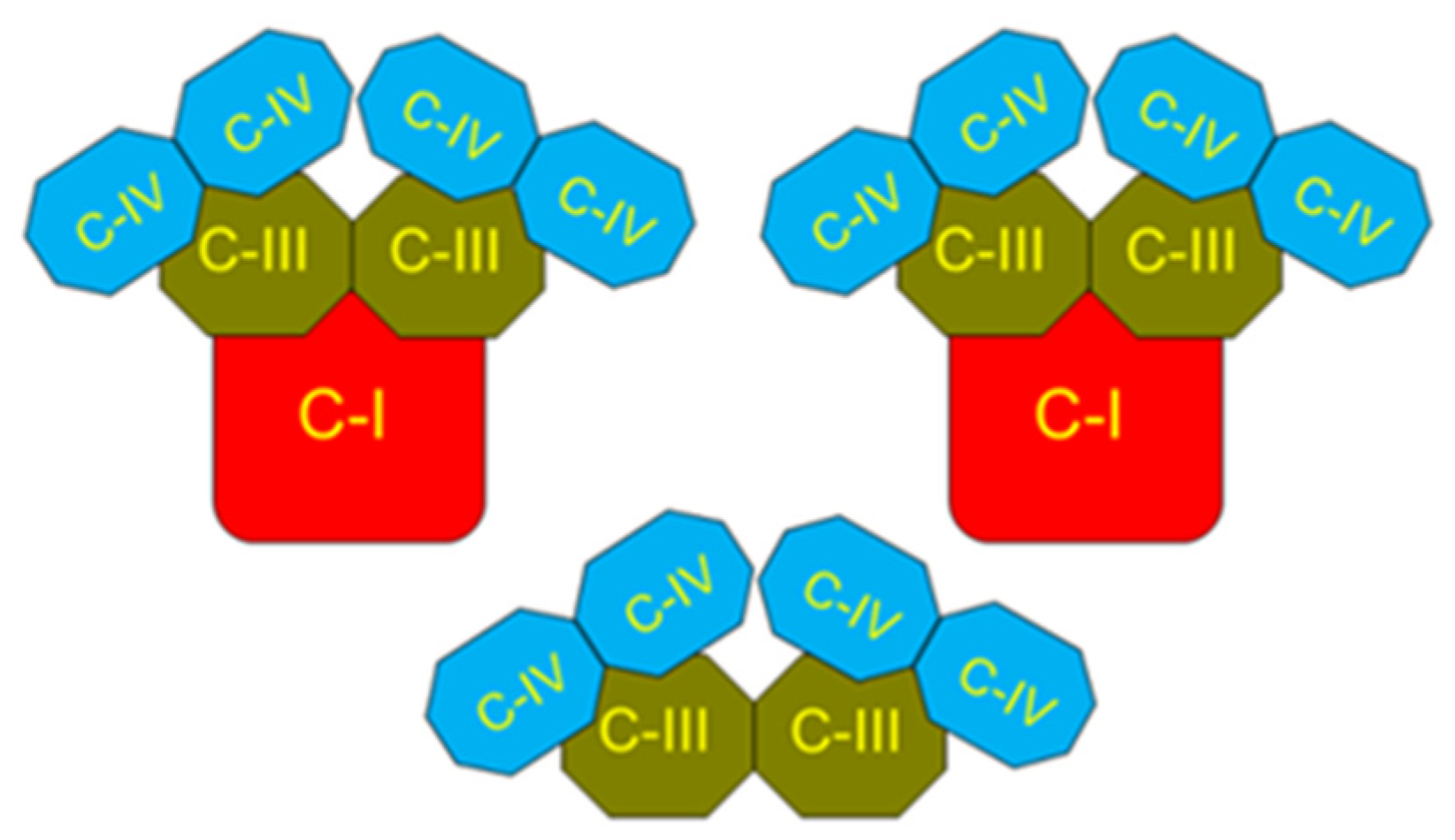
5. Properties of β-Oxidation of Long-Chain Fatty Acids and Generation of Superoxide Radicals by the Kidney, Brain, and Heart Mitochondria
5.1. Intrinsic Inhibition of Succinate Dehydrogenase (Complex II) as a Protection against Excessive Production of ROS
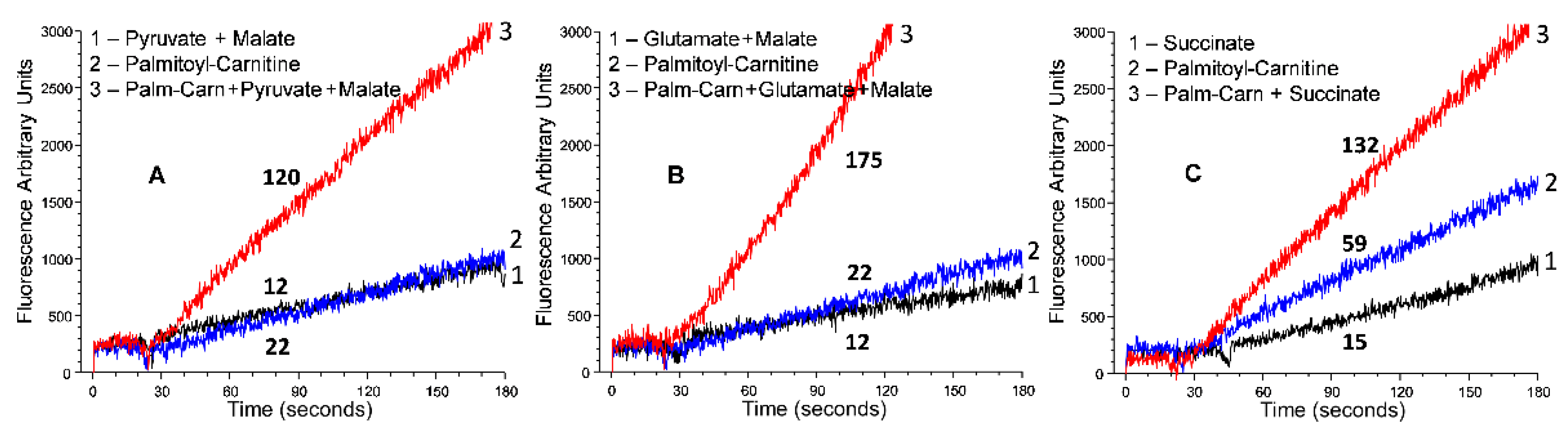
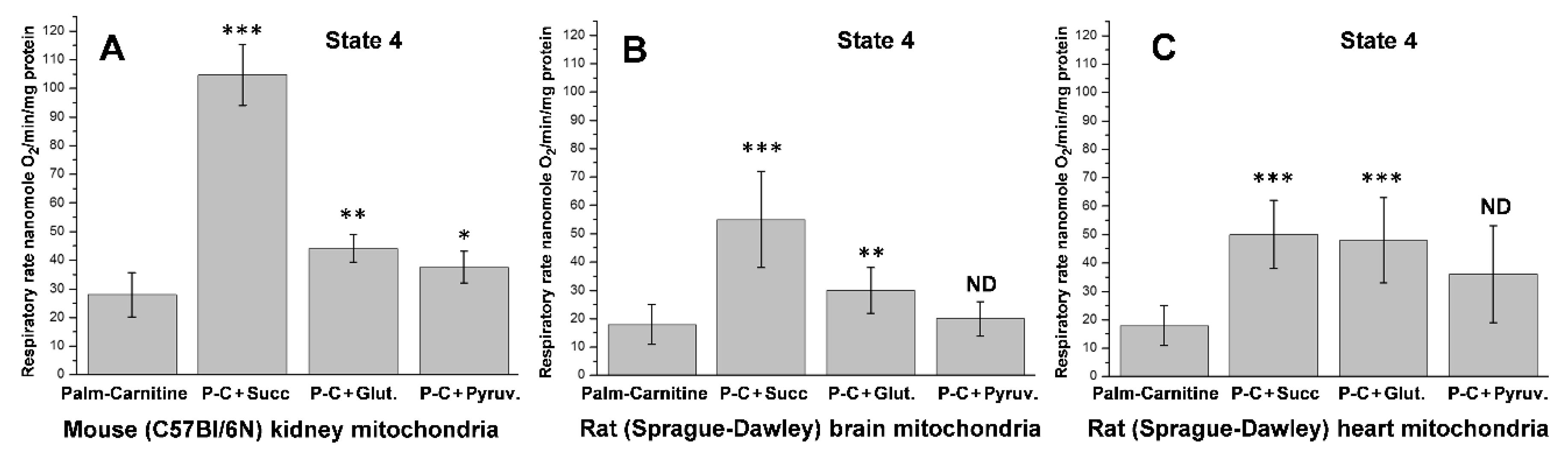
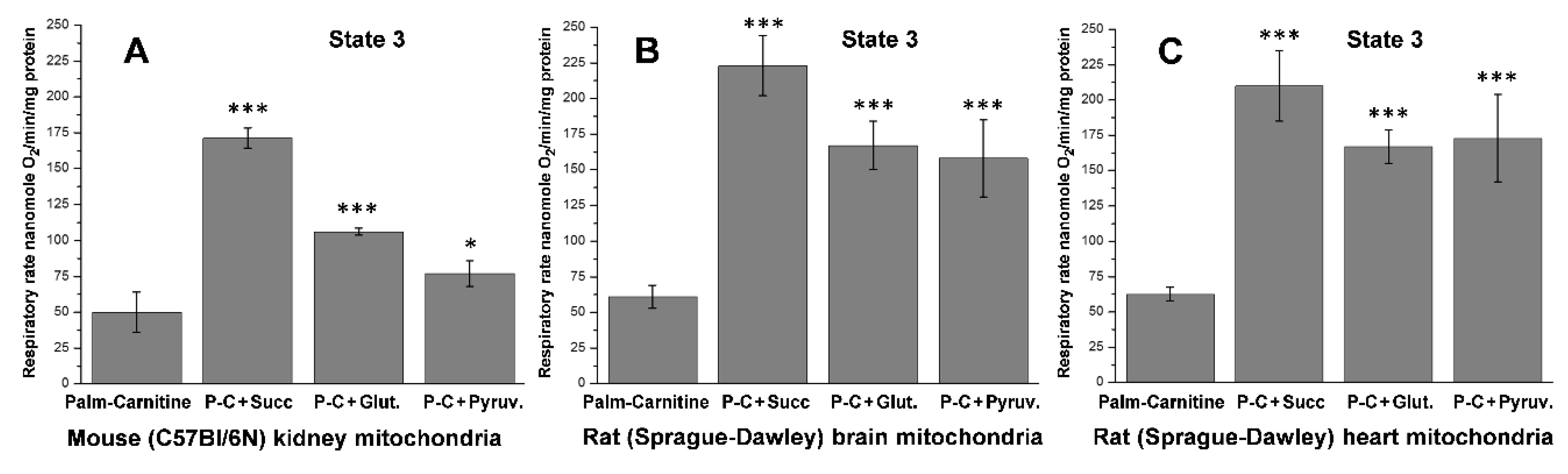
5.2. β-Oxidation of Long-Chain Fatty Acid by the Isolated Kidney, Brain, and Heart Mitochondria
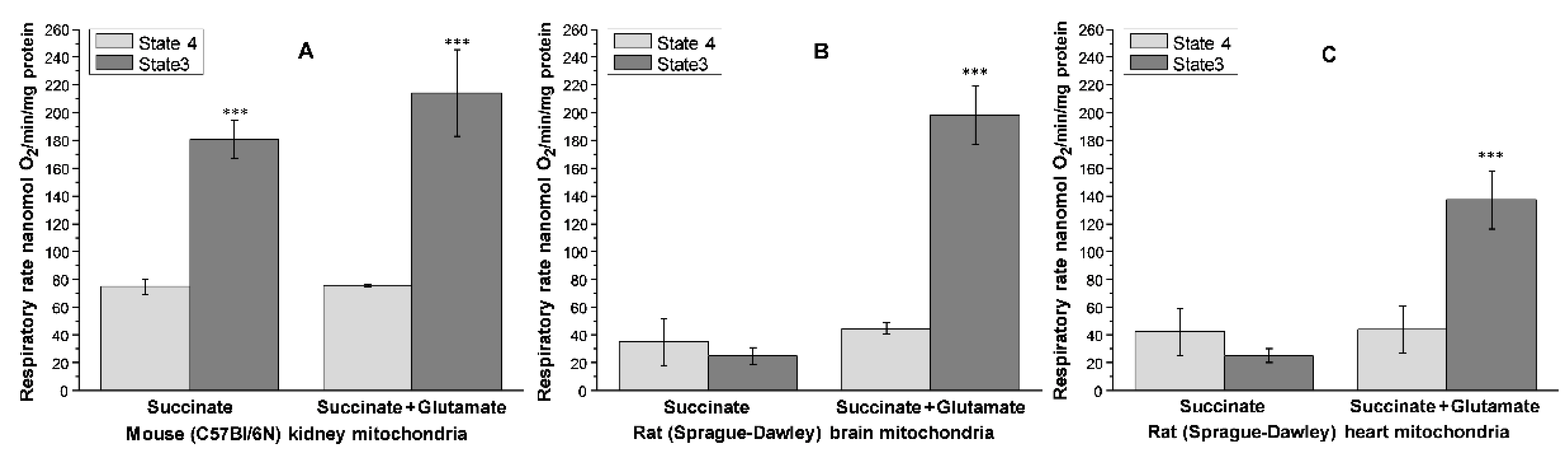
5.3. Generation of Superoxide Radicals by the Kidney, Brain, and Heart Mitochondria
5.4. Oxidative Stress, Promoted by Metabolic Syndrome, Is the Primary Damaging Factor for the Heart and Brain but Not for the Kidney
6. The Mechanisms of Kidney Failure under Conditions of the Metabolic Syndrome
- Normally, both glucose, and Na+, as symport members, are reciprocal at physiological levels.
- The concentration of Na+ in the glomerular filtrate exceeds glucose. Therefore, for the complete Na+ reabsorption, kidneys produce glucose via gluconeogenesis. Since 95.5% of the symport of glucose and Na2+ occurs in the proximal S1 and S3 sections of the tubule [89], glucose synthesis must occur in the podocytes.
- The concentration of glucose in the glomerular filtrate dramatically exceeds that of sodium. In the absence of a reciprocal amount of Na+, the untransported glucose becomes excreted with urine. Because glucose is a valuable metabolite for the body, we suggest that one of the mechanisms to diminish glycosuria may be converting glucose into lactic acid in the distal parts of tubules. Lactate is also a valuable metabolite reabsorbed back into the blood. We suggest that a high concentration of lactate in the medullar part of the kidneys shows high glycolytic activity not to produce ATP but rather to save valuable substrate for other organs, such as the liver and the central nervous system, and diminish the level of hyperglycemia.
- Both concentrations of glucose and sodium in the glomerular filtrate are unphysiologically high. Under this condition, the kidneys must work extremely hard, which results in hypoxia and finally causes kidney dysfunction.
6.1. β-Oxidation of Long-Chain Fatty Acids in the Presence of Supporting Substrates Provides the Highest Rates of ATP Production
6.2. Mechanism of Kidney Hypoxia Development at Persisting Hyperglycemia
6.3. Mechanisms of Oxidative Stress under Conditions of Persistent Hyperglycemia
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Bertermann, H.; Gronow, G.; Schirmer, A.; Weiss, C. Contribution of long chain fatty acids to the energy supply of the rat kidney cortex. Pflug. Arch. 1975, 356, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Wirthensohn, G.; Guder, W.G. Triacylglycerol metabolism in isolated rat kidney cortex tubules. Biochem. J. 1980, 186, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonfeld, P.; Reiser, G. Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem. Int. 2017, 109, 68–77. [Google Scholar] [CrossRef]
- Julius, U.; Leonhardt, W.; Schulze, J.; Schollberg, K.; Hanefeld, M.; Haller, H. Hyperinsulinemia in patients with low fractional catabolic rate of triglycerides. Acta Diabetol. Lat. 1981, 18, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L. A comprehensive definition for metabolic syndrome. Dis. Models Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, S.M. Metabolic syndrome: A multiplex cardiovascular risk factor. J. Clin. Endocrinol. Metabol. 2007, 92, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Koyama, K.; Chen, G.; Lee, Y.; Unger, R.H. Tissue triglycerides, insulin resistance, and insulin production: Implications for hyperinsulinemia of obesity. Am. J. Physiol. 1997, 273, E708–E713. [Google Scholar] [CrossRef]
- Reaven, G.M. The metabolic syndrome: Is this diagnosis necessary? Am. J. Clin. Nutrit. 2006, 83, 1237–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchard, G. Genetics and the metabolicard syndrome. Intern. J. Obes. 1995, 19, S52–S59. [Google Scholar]
- Poulsen, P.; Vaag, A.; Kyvik, K.; Beck-Nielsen, H. Genetic versus environmental aetiology of the metabolic syndrome among male and female twins. Diabetologia 2001, 44, 537–543. [Google Scholar] [CrossRef] [Green Version]
- Edwardson, C.L.; Gorely, T.; Davies, M.J.; Gray, L.J.; Khunti, K.; Wilmot, E.G.; Yates, T.; Biddle, S.J. Association of sedentary behaviour with metabolic syndrome: A meta-analysis. PLoS ONE 2012, 7, e34916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Ren, M.; Liu, D.; Wang, C.; Yang, C.; Yan, L. Alcohol consumption and risk of metabolic syndrome: A meta-analysis of prospective studies. Clin. Nutr. 2014, 33, 596–602. [Google Scholar] [CrossRef]
- Lee, H.K.; Cho, Y.M.; Kwak, S.H.; Lim, S.; Park, K.S.; Shim, E.B. Mitochondrial dysfunction and metabolic syndrome-looking for environmental factors. Biochim. Biophys. Acta 2010, 1800, 282–289. [Google Scholar] [CrossRef]
- Kim, J.T.; Lee, H.K. Metabolic syndrome and the environmental pollutants from mitochondrial perspectives. Rev. Endocr. Metab. Disord. 2014, 15, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S.; Giles, W.H.; Dietz, W.H. Prevalence of the metabolic syndrome among US adults: Findings from the third National Health and Nutrition Examination Survey. JAMA 2002, 287, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Paik, J.K.; Kang, R.; Kim, S.Y. Increased oxidative stress in normal-weight postmenopausal women with metabolic syndrome compared with metabolically healthy overweight/obese individuals. Metabolism 2013, 62, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Kloting, N.; Bluher, M. Adipocyte dysfunction, inflammation and metabolic syndrome. Rev. Endocr. Metab. Disord. 2014, 15, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Voulgari, C.; Tentolouris, N.; Dilaveris, P.; Tousoulis, D.; Katsilambros, N.; Stefanadis, C. Increased heart failure risk in normal-weight people with metabolic syndrome compared with metabolically healthy obese individuals. J. Am. Coll. Cardiol. 2011, 58, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Stančáková, A.; Laakso, M. Genetics of metabolic syndrome. Rev. Endocr. Metab. Disord. 2014, 15, 243–252. [Google Scholar] [CrossRef]
- Panov, A.V.; Dikalov, S.I. Mitochondrial Metabolism and the Age-Associated Cardiovascular Diseases. EC Cardiol. 2018, 5, 750–769. [Google Scholar]
- Panov, A.V. A New Look at the Causes of Heart Failure at Old Age. EC Cardiol. 2020, 7, 01–07. [Google Scholar]
- Harman, D. Free radical theory of aging: Consequences of mitochondrial aging. Age 1983, 6, 86–94. [Google Scholar] [CrossRef]
- Morris, C.W. Academic Press Dictionary of Science and Technology; Gulf Professional Publishing: Oxford, UK, 1992. [Google Scholar]
- Nair, K.S.; Bigelow, M.L.; Asmann, Y.W.; Chow, L.S.; Coenen-Schimke, J.M.; Klaus, K.A.; Guo, Z.; Sreekumar, R.; Irving, B.A. Asian Indians have enhanced skeletal muscle mitochondrial capacity to produce ATP in association with severe insulin resistance. Diabetes 2008, 57, 1166–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neel, J.V. Diabetes mellitus: A “thrifty” genotype rendered detrimental by “progress”? Am. J. Hum. Genet. 1962, 14, 353–362. [Google Scholar] [PubMed]
- Joffe, B.; Zimmet, P. The thrifty genotype in type 2 diabetes: An unfinished symphony moving to its finale? Endocrine 1998, 9, 139–141. [Google Scholar] [CrossRef]
- Carulli, L.; Rondinella, S.; Lombardini, S.; Canedi, I.; Loria, L.; Carulli, N. Review article: Diabetes, genetics and ethnicity. Aliment. Pharmacol. Ther. 2005, 22, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Golubenko, M.V.; Salakhov, R.R.; Makeeva, O.A.; Goncharova, I.A.; Kashtalap, V.V.; Barbarash, O.A.; Puzyrev, V.P. Mitochondrial DNA polymorphism association with myocardial infarction and prognostic signs for atherosclerosis. J. Mol. Biol. 2015, 49, 968–976. [Google Scholar] [CrossRef]
- Baltakhinova, M.E.; Klimova, T.M.; Zakharova, R.N.; Ammosova, E.P. Territorial differences in the incidence of 2 type diabetes in the Republic of Sakha (Yakutia). Siberian Med. Rev. 2019, 6, 80–87. [Google Scholar] [CrossRef]
- Liou, C.W.; Chen, J.B.; Tiao, M.M.; Weng, S.W.; Huang, T.L.; Chuang, J.H.; Chen, S.D.; Chuang, Y.C.; Lee, W.C.; Lin, T.K. Mitochondrial DNA coding and control region variants as genetic risk factors for type 2 diabetes. Diabetes 2012, 61, 2642–2651. [Google Scholar] [CrossRef] [Green Version]
- Puzyrev, V.P.; Kucher, A.N. Evolutionary ontogenetic aspects of pathogenetics of chronic human diseases. Russ. J. Genet. 2011, 47, 1573–1585. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.W.; Zhu, S.; Palaniappan, L.; Heshka, S.; Carnethon, M.R.; Heymsfield, S.B. The metabolic syndrome: Prevalence and associated risk factor findings in the US population from the Third National Health and Nutrition Examination Survey, 1988-1994. Arch. Intern. Med. 2003, 163, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Carr, M.C. The emergence of the metabolic syndrome with menopause. J. Clin. Endocrinol. Metabol. 2003, 88, 2404–2411. [Google Scholar] [CrossRef]
- Kim, J.H.; Im, J.A.; Lee, D.C. The relationship between leukocyte mitochondrial DNA contents and metabolic syndrome in postmenopausal women. Menopause 2012, 19, 582–587. [Google Scholar] [CrossRef]
- Baños, G.; Carbó, R.; Pérez-Torres, I. Arachidonic Acid, Metabolic Syndrome and Alternative Therapeutic Agents. In Arachidonic Acid: Dietary Sources and General Functions; Dumancas, G.G., Murdianti, B.S., Lucas, E.A., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2012; ISBN 978-1-62257-481-0. [Google Scholar]
- Barton, N.H.; Charlesworth, B. Why sex and recombination? Science 1998, 281, 1986–1990. [Google Scholar] [CrossRef]
- Marinou, K.; Adiels, M.; Hodson, L.; Frayn, K.N.; Karpe, F.; Fielding, B.A. Young women partition fatty acids towards ketone body production rather than VLDL-TAG synthesis, compared with young men. Br. J. Nutr. 2011, 105, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Hodson, L.; Banerjee, R.; Rial, B.; Arlt, W.; Adiels, M.; Boren, J.; Marinou, K.; Fisher, C.; Mostad, I.L.; Stratton, I.M. Menopausal Status and Abdominal Obesity Are Significant Determinants of Hepatic Lipid Metabolism in Women. J. Am. Heart. Assoc. 2015, 4, e002258. [Google Scholar] [CrossRef] [Green Version]
- Hoekstra, R.F. Evolutionary biology: Why sex is good. Nature 2005, 434, 571–573. [Google Scholar] [CrossRef]
- Goddard, M.R.; Godfray, H.C.; Burt, A. Sex increases the efficacy of natural selection in experimental yeast populations. Nature 2005, 434, 636–640. [Google Scholar] [CrossRef]
- de Visser, J.A.; Elena, S.F. The evolution of sex: Empirical insights into the roles of epistasis and drift. Nat. Rev. Genet. 2007, 8, 139–149. [Google Scholar] [CrossRef]
- Vina, J.; Borras, C.; Gambini, J.; Sastre, J.; Pallardo, F.V. Why females live longer than males? Importance of the upregulation of longevity-associated genes by oestrogenic compounds. FEBS Lett. 2005, 579, 2541–2545. [Google Scholar] [CrossRef] [Green Version]
- Ballard, J.W.; Melvin, R.G.; Miller, J.T.; Katewa, S.D. Sex differences in survival and mitochondrial bioenergetics during aging in Drosophila. Aging Cell. 2007, 6, 699–708. [Google Scholar] [CrossRef]
- Sengupta, P. The Laboratory Rat: Relating Its Age with Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar] [PubMed]
- Demeter, E.; Sarter, M.; Lustig, C. Rats and humans paying attention: Cross-species task development for translational research. Neuropsychology 2008, 22, 787–799. [Google Scholar] [CrossRef] [Green Version]
- Zucker, I.; Beery, A.K. Males still dominate animal studies. Nature 2010, 465, 690. [Google Scholar] [CrossRef]
- Vijay, V.; Han, T.; Moland, C.L.; Kwekel, J.C.; Fuscoe, J.C.; Desai, V.G. Sexual dimorphism in the expression of mitochondria-related genes in rat heart at different ages. PLoS ONE 2015, 10, e0117047. [Google Scholar] [CrossRef]
- Olivetti, G.; Giordano, G.; Corradi, D.; Melissari, M.; Lagrasta, C.; Gambert, S.R.; Anversa, P. Gender differences and aging: Effects on the human heart. J. Am. Coll. Cardiol. 1995, 26, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Blaak, E. Gender differences in fat metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 499–502. [Google Scholar] [CrossRef] [Green Version]
- Lemieux, S.; Prud’homme, D.; Bouchard, C.; Tremblay, A.; Després, J.P. Sex differences in the relation of visceral adipose tissue accumulation to total body fatness. Am. J. Clin. Nutr. 1993, 58, 463–467. [Google Scholar] [CrossRef]
- Bjorntorp, P. The regulation of adipose tissue distribution in humans. Int. J. Obes. Relat. Metab. Disord. 1996, 20, 291–302. [Google Scholar] [PubMed]
- Tipton, K.D. Gender differences in protein metabolism. Curr. Opin. Clin. Nutr. Metab. Care 2001, 4, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Rennie, C.; Tarnopolsky, M.A. Substrate utilization during endurance exercise in men and women after endurance training. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E898–E907. [Google Scholar] [CrossRef] [Green Version]
- Tarnopolsky, M.A. Gender differences in substrate metabolism during endurance exercise. Can. J. Appl. Physiol. 2000, 25, 312–327. [Google Scholar] [CrossRef] [PubMed]
- Lamont, L.S.; McCullough, A.J.; Kalhan, S.C. Gender differences in the regulation of amino acid metabolism. J. Appl. Physiol. 2003, 95, 1259–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panov, A.V. Synergistic oxidation of fatty acids, glucose and amino acids metabolites by isolated rat heart mitochondria. EC Cardiol. 2018, 5, 198–208. [Google Scholar]
- Joyner, M.J. Physiological limits to endurance exercise performance: Influence of sex. J. Physiol. 2017, 595, 2949–2954. [Google Scholar] [CrossRef] [Green Version]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty Acids in Energy Metabolism of the Central Nervous System. Review Article. BioMed Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef] [Green Version]
- Panov, A.; Darenskaya, M.A.; Dikalov, S.I.; Kolesnikov, S.I. Chapter 3. Metabolic syndrome as the first stage of eldership; the beginning of real aging. In Update in Geriatrics; Amornyotin, S., Ed.; InTech: London, UK, 2021; pp. 1–31. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Piquereau, J.; Garnier, A.; Mericskay, M.; Lemaire, C.; Crozatier, B. Gender issues in cardiovascular diseases. Focus on energy metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165722. [Google Scholar] [CrossRef]
- Legouis, D.; Faivre, A.; Cippa, P.E.; de Seigneux, S. Renal gluconeogenesis: An underestimated role of the kidney in systemic glucose metabolism. Nephrol. Dial. Transplant. 2020, gfaa302. [Google Scholar] [CrossRef]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Hemendinger, R.; Greenamyre, J.T.; Rosenfeld, J. Species- and tissue-specific relationships between mitochondrial permeability transition and generation of ROS in brain and liver mitochondria of rats and mice. Am. J. Physiol. Cell Physiol. 2007, 292, C708–C718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Perevoshchikova, I.V.; Quinlan, C.L.; Orr, A.L.; Gerencser, A.A.; Brand, M.D. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2013, 61C, 298–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [Green Version]
- Schagger, H. respiratory chain supercomplexes. IUBMB Life 2001, 52, 119–128. [Google Scholar] [CrossRef]
- Panov, A. Perhydroxyl radical (HO2) as inducer of the isoprostane lipid peroxidation in mitochondria. Mol. Biol. 2018, 52, 295–305. [Google Scholar] [CrossRef]
- Sumegi, B.; Srere, P.A. Complex I Binds Several Mitochondrial NAD-coupled Dehydrogenases. J. Biol. Chem. 1984, 259, 15040–15045. [Google Scholar] [CrossRef] [PubMed]
- Srere, P.A. Complexes of sequential metabolic enzymes. Annu. Rev. Biochem. 1987, 56, 89–124. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mohsen, A.W.; Mihalik, S.J.; Goetzman, E.S.; Vockley, J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J. Biol. Chem. 2010, 285, 29834–29841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley, C.J.; Perham, R.N. Purification of 2-oxo acid dehydrogenase multyenzyme complexes from ox heart by a new method. Biochem. J. 1980, 191, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Rolfe, D.F.S.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panov, A.; Orynbayeva, Z. Determination of mitochondrial metabolic phenotype through investigation of the intrinsic inhibition of succinate dehydrogenase. Analyt. Biochem. 2018, 552, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Panov, A.V.; Mayorov, V.I.; Dikalov, S.I. Metabolic properties of murine kidney mitochondria. bioRxiv 2022. [Google Scholar] [CrossRef]
- Schonfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in the energy metabolism—The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef] [Green Version]
- Brown, G.C.; Brand, M.D. On the nature of the mitochondrial proton leak. Biochim. Biophys. Acta 1991, 1059, 55–62. [Google Scholar] [CrossRef]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Kalra, S.; Unnikrishnan, A.G.; Baruah, M.P.; Sahay, R.; Bantwal, G. Metabolic and Energy Imbalance in Dysglycemia-Based Chronic Disease. Diabetes Metab. Syndr. Obes. 2021, 14, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.; Chacko, B.; Ballinger, S.W.; Bailey, S.M.; Zhang, J.; Darley-Usmar, V. Convergent mechanisms for dysregulation of mitochondrial quality control in metabolic disease: Implications for mitochondrial therapeutics. Biochem. Soc. Trans. 2013, 41, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Same, R.V.; Feldman, D.I.; Shah, N.; Martin SSAl Rifai, M.; Blaha, M.J.; Graham, G.; Ahmed, H. Relationship Between Sedentary Behavior and Cardiovascular Risk. Curr. Cardiol. Rep. 2016, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Clapp, W.L. Renal Anatomy. In Silva’s Diagnostic Renal Pathology; Zhou, X.J., Laszik, Z., Nadasdy, T., D’Agati, V.D., Silva, F.G., Eds.; Cambridge University Press: New York, NY, USA, 2009. [Google Scholar]
- Lobanova, M.V. Sugar Diabetes. Woyennaya Med. 2018, 2, 139–144. [Google Scholar]
- Poulsen, S.B.; Fenton, R.A.; Rieg, T. Sodium-glucose cotransport. Curr. Opin. Nephrol. Hypertens. 2015, 24, 463–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, E.M.; Loo, D.D.F.; Hirayama, B.A. Biology of human sodium-glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaller, W.; Rittinger, M. Quantitative morphology of the rat kidney. Int. J. Biochem. 1981, 12, 17–22. [Google Scholar] [CrossRef]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, F. Intrarenal oxygen in diabetes and a possible link to diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 2006, 33, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Hansell, P.; Welch, W.J.; Blantz, R.C.; Palm, F. Determinants of kidney oxygen consumption and their relationship to tissue oxygen tension in diabetes and hypertension. Clin. Exp. Pharmacol. Physiol. 2013, 40, 123–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, A.; Miracle, C.M.; Suarez, J.M.; Lortie, M.; Satriano, J.; Thomson, S.C.; Munger, K.A.; Blantz, R.C. Oxygen consumption in the kidney: Effects of nitric oxide synthase isoforms and angiotensin II. Kidney Int. 2005, 68, 723–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, F.; Nordquist, N. Renal oxidative stress, oxygenation, and hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1229–R1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, P.L. Sodium and potassium ion pump in kidney tubules. Physiol. Rev. 1980, 60, 864–917. [Google Scholar] [CrossRef]
- Katz, A.I. Renal Na-K-ATPase: Its role in tubular sodium and potassium transport. Am. J. Physiol. 1982, 242, F207–F219. [Google Scholar] [CrossRef] [PubMed]
- Mandel, L.; Balaban, R.S. Stoichiometry and coupling of active transport to oxidative metabolism in epithelial tissues. Am. Physiol. 1981, 240, F357–F371. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.S.; Noh, M.R.; Kim, J.; Padanilam, B.J. Defective Mitochondrial Fatty Acid Oxidation and Lipotoxicity in Kidney Diseases. Front. Med. 2020, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Soltoff, S.P. ATP and the regulation of renal cell function. Annu. Rev. Physiol. 1986, 48, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Gregoire, F. Oxidative metabolism of the normal rat glomerulus. Kidney. Int. 1975, 7, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.I.; Balaban, R.S.; Barrett, L.; Mandel, L.J. Mitochondrial respiratory capacity and Na+- and K+-dependent adenosine triphosphatase-mediated ion transport in the intact renal cell. J. Biol. Chem. 1981, 256, 10319–10328. [Google Scholar] [CrossRef] [PubMed]
- Chretien, D.; Benit, P.; Ha, H.H.; Keipert, S.; El-Khoury, R.; Chang, Y.T.; Jastroch, M.; Jacobs, H.T.; Rustin, P.; Rak, M. Mitochondria are physiologically maintained at close to 50 degrees C. PLoS Biol. 2018, 16, e2003992. [Google Scholar] [CrossRef] [Green Version]
- Gabbai, F.B. Effects of nitric oxide synthase blockers on renal function. Nephrol. Dial. Transplant. 2001, 16, 10–13. [Google Scholar] [CrossRef] [Green Version]
- Blantz, R.C.; Weir, M.R. Are the oxygen costs of kidney function highly regulated? Curr. Opin. Nephrol. Hypertens. 2004, 13, 67–71. [Google Scholar] [CrossRef]
- Nangaku, M.; Fujita, T. Activation of the renin-angiotensin system and chronic hypoxia of the kidney. Hypertens. Res. 2008, 31, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Leipziger, J.; Praetorius, H. Renal Autocrine and Paracrine Signaling: A Story of Self-protection. Physiol. Rev. 2020, 100, 1229–1289. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laycock, S.K.; Vogel, T.; Forfia, P.R.; Tuzman, J.; Xu, X.; Ochoa, M.; Thompson, C.A.; Nasjletti, A.; Hintze, T.H. Role of nitric oxide in the control of renal oxygen consumption and the regulation of chemical work in the kidney. Circ. Res. 1998, 82, 1263–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remuzzi, A.; Conti, S.; Ene-Iordache, B.; Tomasoni, S.; Rizzo, P.; Benigni, A.; Remuzzi, G. Role of ultrastructural determinants of glomerular permeability in ultrafiltration function loss. JCI Insight 2020, 5, e137249. [Google Scholar] [CrossRef]
- Friederich-Persson, M.; Welch, W.J.; Luo, Z.; Palm, F.; Nordquist, L. Angiotensin II reduces transport-dependent oxygen consumption but increases transport-independent oxygen consumption in immortalized mouse proximal tubular cells. Adv. Exp. Med. Biol. 2014, 812, 157–163. [Google Scholar] [CrossRef] [Green Version]
- Palm, F.; Cederberg, J.; Hansell, P.; Liss, P.; Carlsson, P.O. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia 2003, 46, 1153–1160. [Google Scholar] [CrossRef] [Green Version]
- Rolo, A.P.; Palmeira, C.M. Diabetes and mitochondrial function: Role of hyperglycemia and oxidative stress. Toxicol. Appl. Pharmacol. 2006, 212, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Coughlan, M.T.; Cooper, M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes 2008, 57, 1446–1454. [Google Scholar] [CrossRef] [Green Version]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef]
- Munro, D.; Treberg, J.R. A radical shift in perspective: Mitochondria as regulators of reactive oxygen species. J. Exp. Biol. 2017, 220, 1170–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Barja, G. Updating the mitochondrial free radical theory of aging: An integrated view, key aspects, and confounding concepts. Antioxid. Redox Signal 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Vusse, G.J.; Glatz, J.F.C.; Van Nieuwenhoven, F.A.; Reneman, R.S.; Bassingthwaighte, J.B. Transport of long-chain fatty acids across the muscular endothelium. Adv. Exp. Med. Biol. 1998, 441, 181–191. [Google Scholar]
- Xiong, J.; Kawagishi, H.; Yan, Y.; Liu, J.; Wells, Q.S.; Edmunds, L.R.; Fergusson, M.M.; Yu, Z.; Rovira, I.I.; Brittain, E.L.; et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol. Cell 2018, 69, 689–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, A.; Yucel, N.; Kim, B.; Arany, Z. Local Mitochondrial ATP Production Regulates Endothelial Fatty Acid Uptake and Transport. Cell Metab. 2020, 32, 309–319. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panov, A.; Mayorov, V.I.; Dikalov, S. Metabolic Syndrome and β-Oxidation of Long-Chain Fatty Acids in the Brain, Heart, and Kidney Mitochondria. Int. J. Mol. Sci. 2022, 23, 4047. https://doi.org/10.3390/ijms23074047
Panov A, Mayorov VI, Dikalov S. Metabolic Syndrome and β-Oxidation of Long-Chain Fatty Acids in the Brain, Heart, and Kidney Mitochondria. International Journal of Molecular Sciences. 2022; 23(7):4047. https://doi.org/10.3390/ijms23074047
Chicago/Turabian StylePanov, Alexander, Vladimir I. Mayorov, and Sergey Dikalov. 2022. "Metabolic Syndrome and β-Oxidation of Long-Chain Fatty Acids in the Brain, Heart, and Kidney Mitochondria" International Journal of Molecular Sciences 23, no. 7: 4047. https://doi.org/10.3390/ijms23074047