
Using ELP Repeats as a Scaffold for De Novo Construction of Gadolinium-Binding Domains within Multifunctional Recombinant Proteins for Targeted Delivery of Gadolinium to Tumour Cells

 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Protein Modelling
2.1.1. Construction of the Basic Metal Binding Domain Containing the 4MBS-Domain
2.1.2. Construction of the RGD-Containing Region (3RGD-Domain)
2.1.3. Linking the Designed Domains
2.2. Engineering DNA Constructs
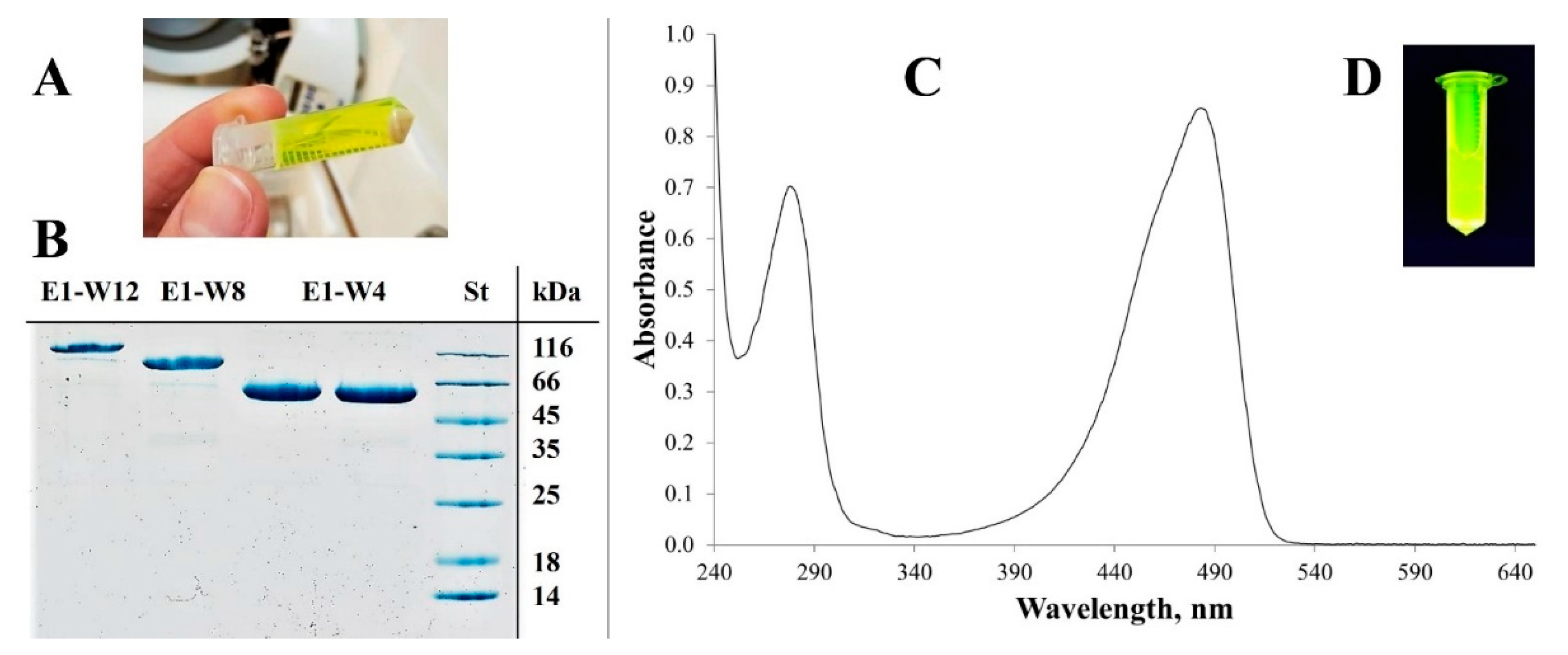
2.3. Protein Purification
2.4. Size Measurement
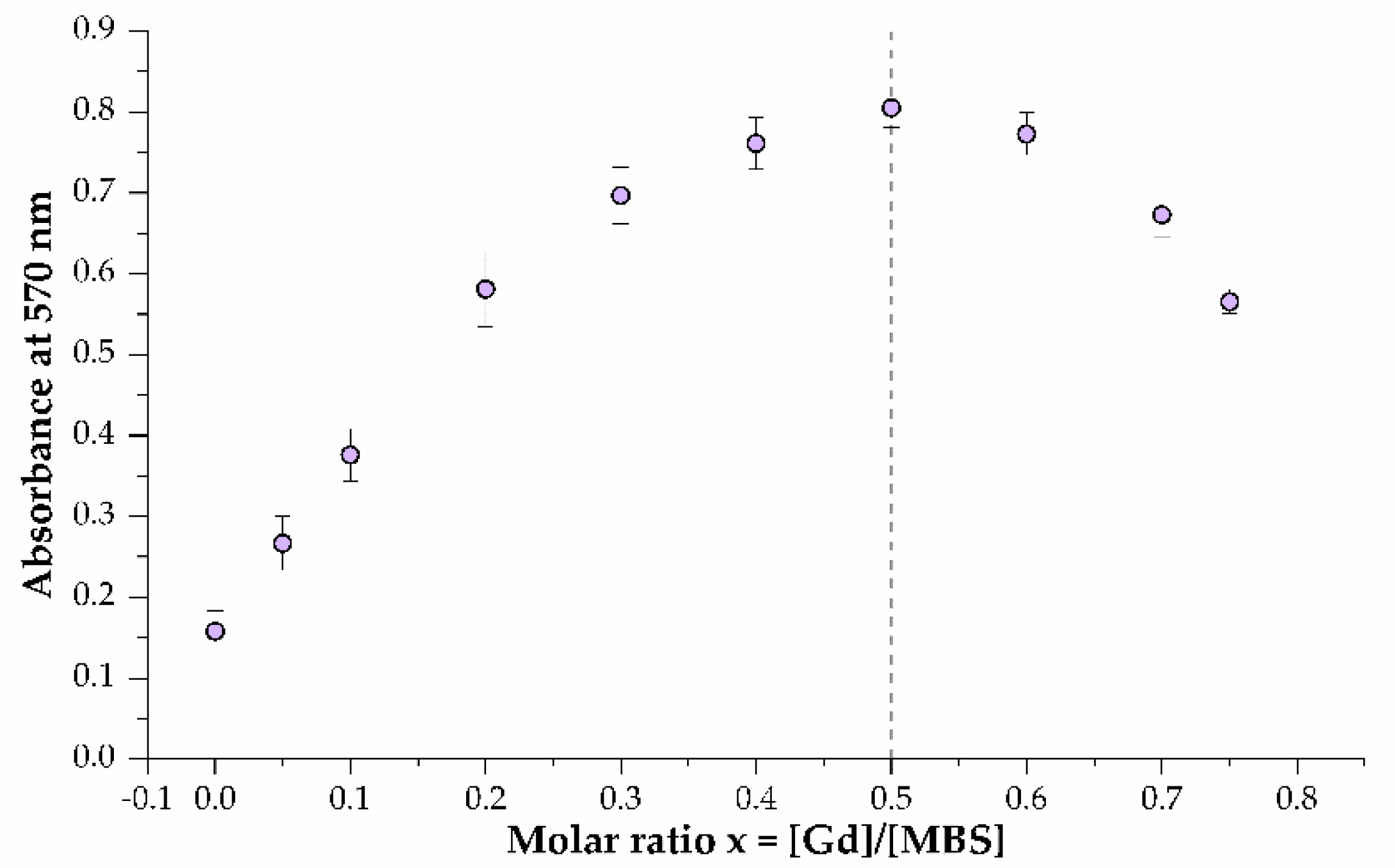
2.5. Gd3+-Binding Affinity (mKd) and Binding Stoichiometry Determination
2.6. Relaxivity and MRI
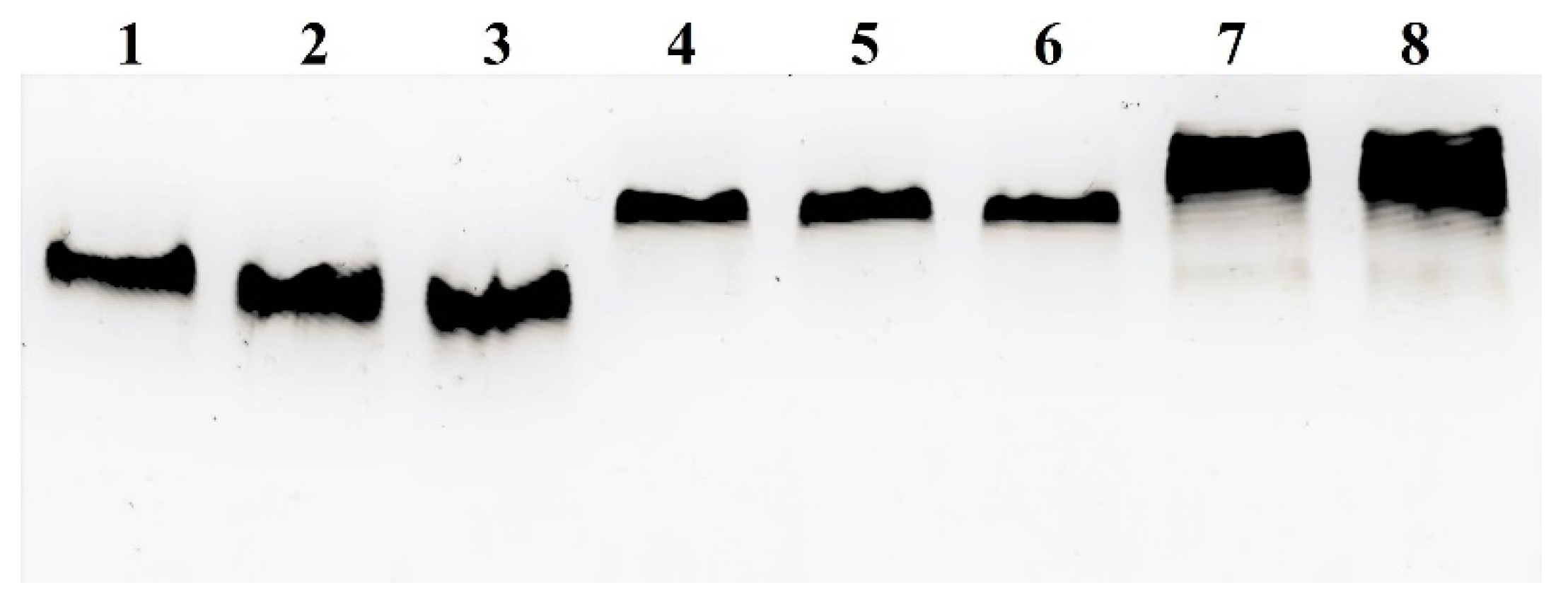
2.7. In Vitro Assessment of the Stability of MBS-Gd3+ Complexes in the Presence of Serum
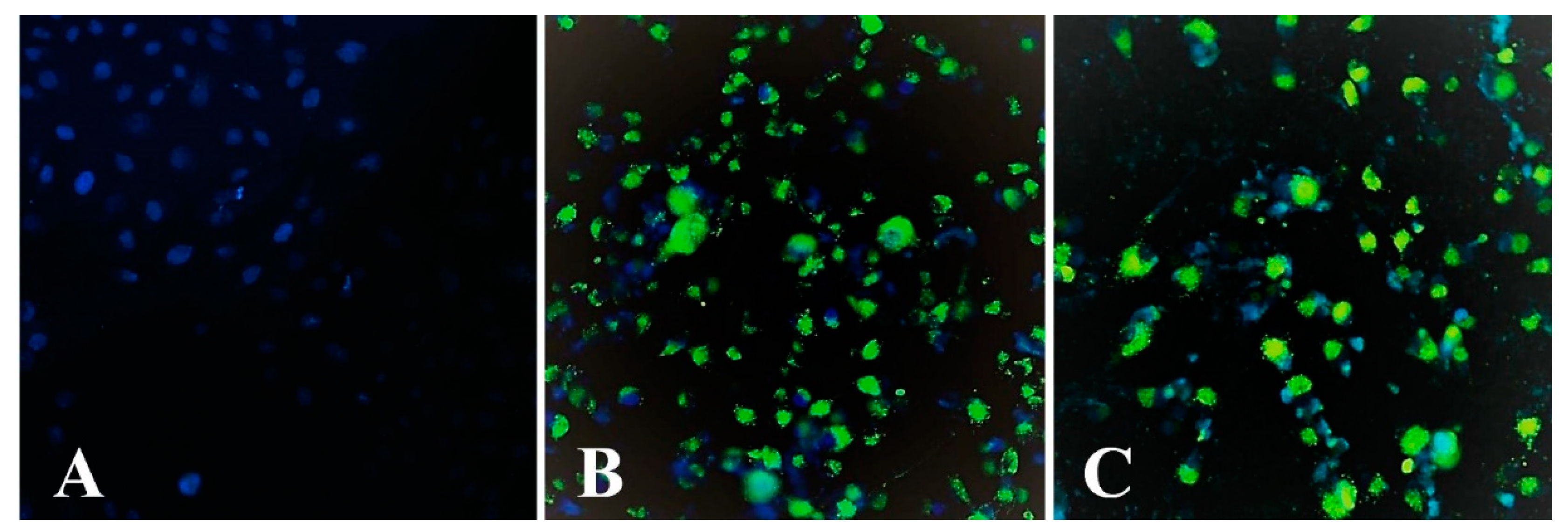
2.8. Assay of MBS-Protein Uptake by Tumour Cells Using Fluorescence Microscopy
2.9. Flow Cytometry
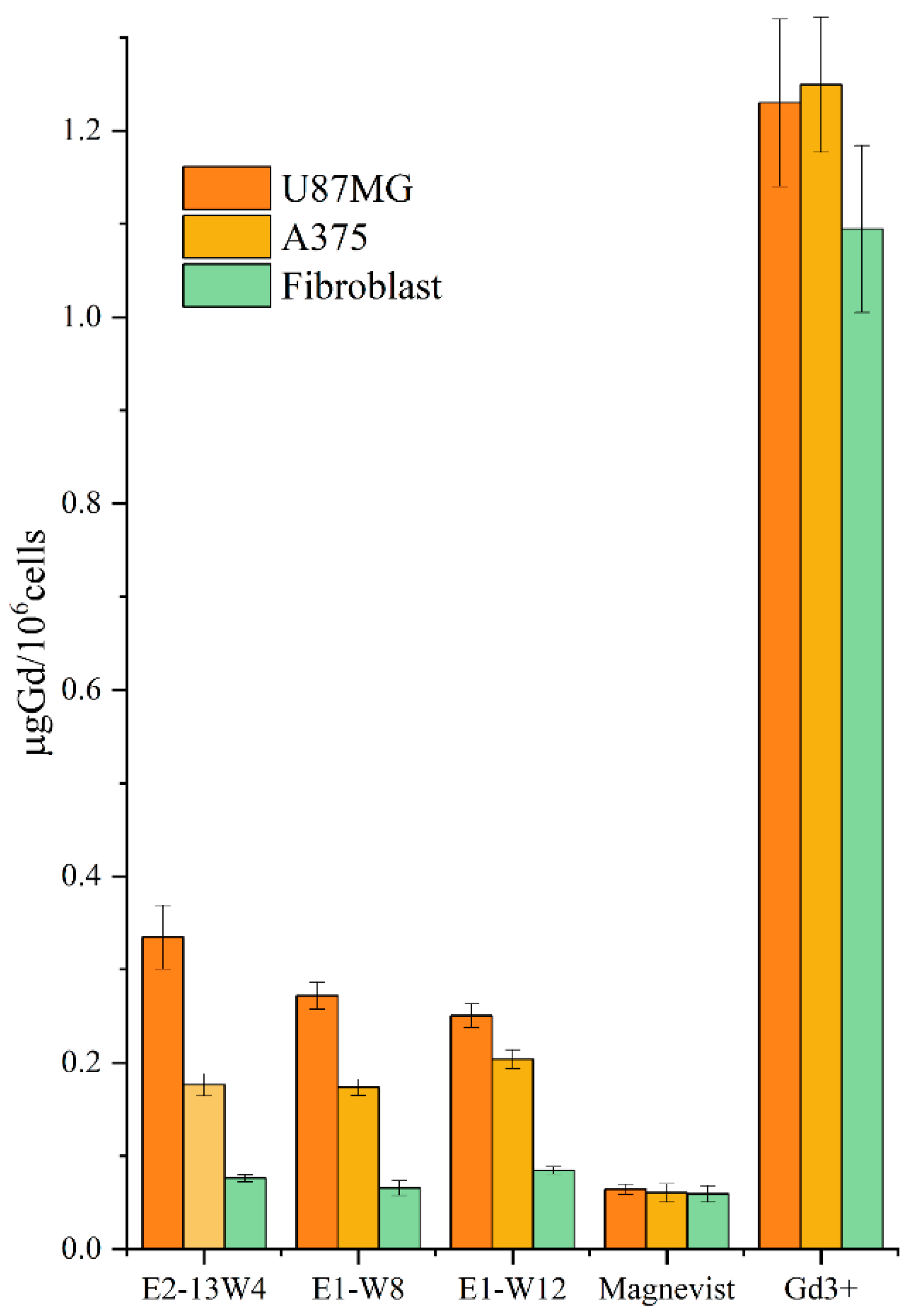
2.10. In Vitro Accumulation of Gd3+ Carried by the MBS-Protein in the Cells
2.11. Ex Vivo Imaging
3. Materials and Methods
3.1. Materials
3.1.1. Genetic Constructions
3.1.2. Protein Purification
3.2. Software
3.3. Cell Cultures
3.4. Methods
Genetic Engineering Manipulations
3.5. Protein Purification
3.5.1. Culture Growth
3.5.2. Cell Lysis and Clearing the Lysate
3.5.3. Ion-Exchange Chromatography
3.5.4. Metal-Chelate Chromatography
3.5.5. Metal Ion Elimination
3.5.6. Desalting the Protein
3.6. Size Measurement
3.7. Gd3+-Binding Affinity Determination
3.8. Binding Stoichiometry Determination
3.9. Relaxivity Measurement
3.10. MRI of Phantoms
3.11. Assessment of Stability of MBS-Proteins in Complexes with Gd3+ in the Presence of Serum In Vitro
3.12. Assay of MBS-Proteins Uptake by Tumour Cells by Fluorescence Microscopy
3.13. Flow Cytometry
3.14. In Vitro Accumulation of Gd3+ in Cultured Cells
3.15. Ex Vivo Imaging
3.15.1. Labelling MBS-Proteins with Cy7 Fluorescent Dye
3.15.2. Animal Experiment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lux, F.; Sancey, L.; Bianchi, A.; Crémillieux, Y.; Roux, S.; Tillement, O. Gadolinium-Based Nanoparticles for Theranostic MRI-Radiosensitization. Nanomedicine 2015, 10, 1801–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathmanathan, A.U.; van As, N.J.; Kerkmeijer, L.G.W.; Christodouleas, J.; Lawton, C.A.F.; Vesprini, D.; van der Heide, U.A.; Frank, S.J.; Nill, S.; Oelfke, U.; et al. Magnetic Resonance Imaging-Guided Adaptive Radiation Therapy: A “Game Changer” for Prostate Treatment? Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 361–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancey, L.; Lux, F.; Kotb, S.; Roux, S.; Dufort, S.; Bianchi, A.; Crémillieux, Y.; Fries, P.; Coll, J.-L.; Rodriguez-Lafrasse, C.; et al. The Use of Theranostic Gadolinium-Based Nanoprobes to Improve Radiotherapy Efficacy. Br. J. Radiol. 2014, 87, 20140134. [Google Scholar] [CrossRef] [PubMed]
- Taupin, F.; Flaender, M.; Delorme, R.; Brochard, T.; Mayol, J.-F.; Arnaud, J.; Perriat, P.; Sancey, L.; Lux, F.; Barth, R.F.; et al. Gadolinium Nanoparticles and Contrast Agent as Radiation Sensitizers. Phys. Med. Biol. 2015, 60, 4449–4464. [Google Scholar] [CrossRef] [PubMed]
- Brachman, D.G.; Pugh, S.L.; Ashby, L.S.; Thomas, T.A.; Dunbar, E.M.; Narayan, S.; Robins, H.I.; Bovi, J.A.; Rockhill, J.K.; Won, M.; et al. Phase 1/2 Trials of Temozolomide, Motexafin Gadolinium, and 60-Gy Fractionated Radiation for Newly Diagnosed Supratentorial Glioblastoma Multiforme: Final Results of RTOG 0513. Int. J. Radiat. Oncol. Biol. Phys. 2015, 91, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damme, N.M.; Fernandez, D.P.; Wang, L.-M.; Wu, Q.; Kirk, R.A.; Towner, R.A.; McNally, J.S.; Hoffman, J.M.; Morton, K.A. Analysis of Retention of Gadolinium by Brain, Bone, and Blood Following Linear Gadolinium-Based Contrast Agent Administration in Rats with Experimental Sepsis. Magn. Reson. Med. 2020, 83, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Verry, C.; Sancey, L.; Dufort, S.; Le Duc, G.; Mendoza, C.; Lux, F.; Grand, S.; Arnaud, J.; Quesada, J.L.; Villa, J.; et al. Treatment of Multiple Brain Metastases Using Gadolinium Nanoparticles and Radiotherapy: NANO-RAD, a Phase I Study Protocol. BMJ Open 2019, 9, e023591. [Google Scholar] [CrossRef]
- Detappe, A.; Kunjachan, S.; Rottmann, J.; Robar, J.; Tsiamas, P.; Korideck, H.; Tillement, O.; Berbeco, R. AGuIX Nanoparticles as a Promising Platform for Image-Guided Radiation Therapy. Cancer Nanotechnol. 2015, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; van der Meel, R.; Chen, X.; Lammers, T. The EPR Effect and beyond: Strategies to Improve Tumor Targeting and Cancer Nanomedicine Treatment Efficacy. Theranostics 2020, 10, 7921–7924. [Google Scholar] [CrossRef]
- 1Benachour, H.; Sève, A.; Bastogne, T.; Frochot, C.; Vanderesse, R.; Jasniewski, J.; Miladi, I.; Billotey, C.; Tillement, O.; Lux, F.; et al. Multifunctional Peptide-Conjugated Hybrid Silica Nanoparticles for Photodynamic Therapy and MRI. Theranostics 2012, 2, 889–904. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Cai, R.; Zhao, T.; Wu, L.; Zhang, L.; Jin, J.; Xu, L.; Li, P.; Li, T.; Zhang, M.; et al. Hyaluronic Acid-Functionalized Gadolinium Oxide Nanoparticles for Magnetic Resonance Imaging-Guided Radiotherapy of Tumors. Nanoscale Res. Lett. 2020, 15, 94. [Google Scholar] [CrossRef] [PubMed]
- Lacerda, S. Targeted Contrast Agents for Molecular MRI. Inorganics 2018, 6, 129. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Peng, J.; Li, J.; Huang, L.; Yang, J.; Huang, K.; Li, H.; Jiang, N.; Zheng, S.; Zhang, X.; et al. Tumor-Targeted and Clearable Human Protein-Based MRI Nanoprobes. Nano Lett. 2017, 17, 4096–4100. [Google Scholar] [CrossRef] [PubMed]
- Milech, N.; Watt, P. The Construction of “Phylomer” Peptide Libraries as a Rich Source of Potent Inhibitors of Protein/Protein Interactions. Methods Mol. Biol. 2012, 899, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Watt, P.M.; Milech, N.; Stone, S.R. Structure-Diverse Phylomer Libraries as a Rich Source of Bioactive Hits from Phenotypic and Target Directed Screens against Intracellular Proteins. Curr. Opin. Chem. Biol. 2017, 38, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Yang, H.; Qiao, J.; Pu, F.; Jiang, J.; Hubbard, K.; Hekmatyar, K.; Langley, J.; Salarian, M.; Long, R.C.; et al. Protein MRI Contrast Agent with Unprecedented Metal Selectivity and Sensitivity for Liver Cancer Imaging. Proc. Natl. Acad. Sci. USA 2015, 112, 6607–6612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Li, S.; Yang, J.; Ye, Y.; Zou, J.; Wang, L.; Long, R.; Zurkiya, O.; Zhao, T.; Johnson, J.; et al. Protein-Based MRI Contrast Agents for Molecular Imaging of Prostate Cancer. Mol. Imaging Biol. 2011, 13, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Salarian, M.; Turaga, R.C.; Xue, S.; Nezafati, M.; Hekmatyar, K.; Qiao, J.; Zhang, Y.; Tan, S.; Ibhagui, O.Y.; Hai, Y.; et al. Early Detection and Staging of Chronic Liver Diseases with a Protein MRI Contrast Agent. Nat. Commun. 2019, 10, 4777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grum, D.; Franke, S.; Kraff, O.; Heider, D.; Schramm, A.; Hoffmann, D.; Bayer, P. Design of a Modular Protein-Based MRI Contrast Agent for Targeted Application. PLoS ONE 2013, 8, e65346. [Google Scholar] [CrossRef] [Green Version]
- Patil, R.; Galstyan, A.; Grodzinski, Z.B.; Shatalova, E.S.; Wagner, S.; Israel, L.L.; Ding, H.; Black, K.L.; Ljubimova, J.Y.; Holler, E. Single- and Multi-Arm Gadolinium MRI Contrast Agents for Targeted Imaging of Glioblastoma. Int. J. Nanomed. 2020, 15, 3057–3070. [Google Scholar] [CrossRef]
- Thumshirn, G.; Hersel, U.; Goodman, S.L.; Kessler, H. Multimeric Cyclic RGD Peptides as Potential Tools for Tumor Targeting: Solid-Phase Peptide Synthesis and Chemoselective Oxime Ligation. Chemistry 2003, 9, 2717–2725. [Google Scholar] [CrossRef] [PubMed]
- Liolios, C.; Sachpekidis, C.; Kolocouris, A.; Dimitrakopoulou-Strauss, A.; Bouziotis, P. PET Diagnostic Molecules Utilizing Multimeric Cyclic RGD Peptide Analogs for Imaging Integrin Avβ3 Receptors. Molecules 2021, 26, 1792. [Google Scholar] [CrossRef] [PubMed]
- Erazo-Oliveras, A.; Najjar, K.; Dayani, L.; Wang, T.-Y.; Johnson, G.A.; Pellois, J.-P. Protein Delivery into Live Cells by Incubation with an Endosomolytic Agent. Nat. Methods 2014, 11, 861–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åmand, H.L.; Nordén, B.; Fant, K. Functionalization with C-Terminal Cysteine Enhances Transfection Efficiency of Cell-Penetrating Peptides through Dimer Formation. Biochem. Biophys. Res. Commun. 2012, 418, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Quiroz, F.G.; Chilkoti, A. Sequence Heuristics to Encode Phase Behaviour in Intrinsically Disordered Protein Polymers. Nat. Mater. 2015, 14, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Muiznieks, L.D.; Keeley, F.W. Proline Periodicity Modulates the Self-Assembly Properties of Elastin-like Polypeptides. J. Biol. Chem. 2010, 285, 39779–39789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holehouse, A.S.; Pappu, R.V. Protein Polymers: Encoding Phase Transitions. Nat. Mater. 2015, 14, 1083–1084. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, F.; Chen, X. Integrin Alpha(v)Beta(3)-Targeted Cancer Therapy. Drug Dev. Res. 2008, 69, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Vannini, A.; Leoni, V.; Barboni, C.; Sanapo, M.; Zaghini, A.; Malatesta, P.; Campadelli-Fiume, G.; Gianni, T. Avβ3-Integrin Regulates PD-L1 Expression and Is Involved in Cancer Immune Evasion. Proc. Natl. Acad. Sci. USA 2019, 116, 20141–20150. [Google Scholar] [CrossRef] [Green Version]
- Smart, J.A.; Oleksak, J.E.; Hartsough, E.J. Cell Adhesion Molecules in Plasticity and Metastasis. Mol. Cancer Res. 2021, 19, 25–37. [Google Scholar] [CrossRef]
- Su, C.-Y.; Li, J.-Q.; Zhang, L.-L.; Wang, H.; Wang, F.-H.; Tao, Y.-W.; Wang, Y.-Q.; Guo, Q.-R.; Li, J.-J.; Liu, Y.; et al. The Biological Functions and Clinical Applications of Integrins in Cancers. Front. Pharmacol. 2020, 11, 579068. [Google Scholar] [CrossRef] [PubMed]
- Koutsioumpa, M.; Papadimitriou, E. Cell Surface Nucleolin as a Target for Anti-Cancer Therapies. Recent Pat. Anti-Cancer Drug Discov. 2014, 9, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Hovanessian, A.G.; Soundaramourty, C.; El Khoury, D.; Nondier, I.; Svab, J.; Krust, B. Surface Expressed Nucleolin Is Constantly Induced in Tumor Cells to Mediate Calcium-Dependent Ligand Internalization. PLoS ONE 2010, 5, e15787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, B.; Belbekhouche, S.; Habert, D.; Houppe, C.; Vallée, B.; Bourgoin-Voillard, S.; Cohen, J.L.; Cascone, I.; Courty, J. Cell Surface Nucleolin as Active Bait for Nanomedicine in Cancer Therapy: A Promising Option. Nanotechnology 2021, 32, 322001. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-P.; Wang, X.; Xie, X.-L.; Zhang, G.-P.; Lv, F.-J.; Weng, W.-T.; Qiu, F.; Li, Z.-F.; Lin, J.-S.; Diao, Y. Cell Surface Expression of Nucleolin Mediates the Antiangiogenic and Antitumor Activities of Kallistatin. Oncotarget 2018, 9, 2220–2235. [Google Scholar] [CrossRef] [Green Version]
- Jia, W.; Yao, Z.; Zhao, J.; Guan, Q.; Gao, L. New Perspectives of Physiological and Pathological Functions of Nucleolin (NCL). Life Sci. 2017, 186, 1–10. [Google Scholar] [CrossRef]
- Berger, C.M.; Gaume, X.; Bouvet, P. The Roles of Nucleolin Subcellular Localization in Cancer. Biochimie 2015, 113, 78–85. [Google Scholar] [CrossRef]
- Shi, H.; Huang, Y.; Zhou, H.; Song, X.; Yuan, S.; Fu, Y.; Luo, Y. Nucleolin Is a Receptor That Mediates Antiangiogenic and Antitumor Activity of Endostatin. Blood 2007, 110, 2899–2906. [Google Scholar] [CrossRef]
- Zhuo, W.; Luo, C.; Wang, X.; Song, X.; Fu, Y.; Luo, Y. Endostatin Inhibits Tumour Lymphangiogenesis and Lymphatic Metastasis via Cell Surface Nucleolin on Lymphangiogenic Endothelial Cells. J. Pathol. 2010, 222, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Nakamura, T.; Shimizu, Y. Calcium Binding by Gadolinium-Based MR Contrast Agents. Radiat. Med. 2005, 23, 545–549. [Google Scholar]
- Sutresno, A.; Haryanto, F.; Viridi, S.; Arif, I. Influence Blocking by Gadolinium in Calcium Diffusion on Synapse Model: A Monte Carlo Simulation Study. J. Biomed. Phys. Eng. 2020, 10, 251–260. [Google Scholar] [CrossRef]
- Hayakawa, K.; Maeda, M.; Mitsumori, M.; Torizuka, T.; Okuno, Y.; Tanaka, F.; Matsui, A.; Misaki, T. Electrolyte Disturbances Caused by Intravenous Contrast Media. Radiat. Med. 1992, 10, 171–175. [Google Scholar] [PubMed]
- Bourne, G.W.; Trifaró, J.M. The Gadolinium Ion: A Potent Blocker of Calcium Channels and Catecholamine Release from Cultured Chromaffin Cells. Neuroscience 1982, 7, 1615–1622. [Google Scholar] [CrossRef]
- Martin, L.J. Development of Lanthanide-Binding Tags (LBTs) as Powerful and Versatile Peptides for Use in Studies of Proteins and Protein Interactions. Ph.D. Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA, 2008. [Google Scholar]
- Kanayama, Y.; Tsuji, T.; Enomoto, S.; Amano, R. Multitracer Screening: Brain Delivery of Trace Elements by Eight Different Administration Methods. Biometals 2005, 18, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Siew, E.L.; Farris, A.F.; Rashid, N.; Chan, K.M.; Rajab, N.F. In Vitro Toxicological Assessment of Gadolinium (III) Chloride in V79-4 Fibroblasts. Genes Environ. 2020, 42, 22. [Google Scholar] [CrossRef]
- Caravan, P.; Greenwood, J.M.; Welch, J.T.; Franklin, S.J. Gadolinium-Binding Helix-Turn-Helix Peptides: DNA-Dependent MRI Contrast Agents. Chem. Commun. 2003, 20, 2574–2575. [Google Scholar] [CrossRef] [PubMed]
- Chambre, L.; Martín-Moldes, Z.; Parker, R.N.; Kaplan, D.L. Bioengineered Elastin- and Silk-Biomaterials for Drug and Gene Delivery. Adv. Drug Deliv. Rev. 2020, 160, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved Protein Structure Prediction Using Predicted Interresidue Orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Lucie, S.; Elisabeth, G.; Stéphanie, F.; Guy, S.; Amandine, H.; Corinne, A.-R.; Didier, B.; Catherine, S.; Alexeï, G.; Pascal, D.; et al. Clustering and Internalization of Integrin Avβ3 With a Tetrameric RGD-Synthetic Peptide. Mol. Ther. 2009, 17, 837–843. [Google Scholar] [CrossRef]
- Arnold, M.; Hirschfeld-Warneken, V.C.; Lohmüller, T.; Heil, P.; Blümmel, J.; Cavalcanti-Adam, E.A.; López-García, M.; Walther, P.; Kessler, H.; Geiger, B.; et al. Induction of Cell Polarization and Migration by a Gradient of Nanoscale Variations in Adhesive Ligand Spacing. Nano Lett. 2008, 8, 2063–2069. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Giannone, G.; Critchley, D.R.; Fukumoto, E.; Sheetz, M.P. Two-Piconewton Slip Bond between Fibronectin and the Cytoskeleton Depends on Talin. Nature 2003, 424, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Schvartzman, M.; Palma, M.; Sable, J.; Abramson, J.; Hu, X.; Sheetz, M.P.; Wind, S.J. Nanolithographic Control of the Spatial Organization of Cellular Adhesion Receptors at the Single-Molecule Level. Nano Lett. 2011, 11, 1306–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, S.; Akiyama, S.K.; Yamada, K.M. Synergistic Roles for Receptor Occupancy and Aggregation in Integrin Transmembrane Function. Science 1995, 267, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.-H.; Furukawa, T.; Waki, A.; Akaji, K.; Coll, J.-L.; Saga, T.; Fujibayashi, Y. Effect of Multimerization of a Linear Arg-Gly-Asp Peptide on Integrin Binding Affinity and Specificity. Biol. Pharm. Bull. 2010, 33, 370–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cost, G.J.; Cozzarelli, N.R. Directed Assembly of DNA Molecules via Simultaneous Ligation and Digestion. Biotechniques 2007, 42, 86–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, S.W.; Yang, J.-S.; Kim, I.; Yang, J.; Min, B.E.; Kim, S.; Jung, G.Y. Predictive Design of MRNA Translation Initiation Region to Control Prokaryotic Translation Efficiency. Metab. Eng. 2013, 15, 67–74. [Google Scholar] [CrossRef]
- Karimi, Z.; Nezafat, N.; Negahdaripour, M.; Berenjian, A.; Hemmati, S.; Ghasemi, Y. The Effect of Rare Codons Following the ATG Start Codon on Expression of Human Granulocyte-Colony Stimulating Factor in Escherichia Coli. Protein. Expr. Purif. 2015, 114, 108–114. [Google Scholar] [CrossRef]
- Cubitt, A.B.; Woollenweber, L.A.; Heim, R. Understanding Structure-Function Relationships in the Aequorea Victoria Green Fluorescent Protein. Methods Cell Biol. 1999, 58, 19–30. [Google Scholar] [CrossRef]
- Shave, S.; Pham, N.T.; Śmieja, C.B.; Auer, M. Quantitative Microdialysis: Experimental Protocol and Software for Small Molecule Protein Affinity Determination and for Exclusion of Compounds with Poor Physicochemical Properties. Methods Protoc. 2020, 3, 55. [Google Scholar] [CrossRef]
- Pollard, T.D. A Guide to Simple and Informative Binding Assays. Mol. Biol. Cell 2010, 21, 4061–4067. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Yang, L.; He, L. Overview of the Detection Methods for Equilibrium Dissociation Constant KD of Drug-Receptor Interaction. J. Pharm. Anal. 2018, 8, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Crouch, T.H.; Klee, C.B. Positive Cooperative Binding of Calcium to Bovine Brain Calmodulin. Biochemistry 1980, 19, 3692–3698. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y. Determination of Binding Stoichiometry by the Continuous Variation Method: The Job Plot. Methods Enzymol. 1982, 87, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Belleza, O.J.V.; Villaraza, A.J.L. Ion Charge Density Governs Selectivity in the Formation of Metal–Xylenol Orange (M–XO) Complexes. Inorg. Chem. Commun. 2014, 47, 87–92. [Google Scholar] [CrossRef]
- Yang, J.J.; Yang, J.; Wei, L.; Zurkiya, O.; Yang, W.; Li, S.; Zou, J.; Zhou, Y.; Maniccia, A.L.W.; Mao, H.; et al. Rational Design of Protein-Based MRI Contrast Agents. J. Am. Chem. Soc. 2008, 130, 9260–9267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldshmit, Y.; Trangle, S.S.; Kloog, Y.; Pinkas-Kramarski, R. Interfering with the Interaction between ErbB1, Nucleolin and Ras as a Potential Treatment for Glioblastoma. Oncotarget 2014, 5, 8602–8613. [Google Scholar] [CrossRef] [Green Version]
- di Leandro, L.; Giansanti, F.; Mei, S.; Ponziani, S.; Colasante, M.; Ardini, M.; Angelucci, F.; Pitari, G.; d’Angelo, M.; Cimini, A.; et al. Aptamer-Driven Toxin Gene Delivery in U87 Model Glioblastoma Cells. Front. Pharmacol. 2021, 12, 588306. [Google Scholar] [CrossRef]
- Balça-Silva, J.; do Carmo, A.; Tão, H.; Rebelo, O.; Barbosa, M.; Moura-Neto, V.; Sarmento-Ribeiro, A.B.; Lopes, M.C.; Moreira, J.N. Nucleolin Is Expressed in Patient-Derived Samples and Glioblastoma Cells, Enabling Improved Intracellular Drug Delivery and Cytotoxicity. Exp. Cell Res. 2018, 370, 68–77. [Google Scholar] [CrossRef]
- Shaw, S.K.; Schreiber, C.L.; Roland, F.M.; Battles, P.M.; Brennan, S.P.; Padanilam, S.J.; Smith, B.D. High Expression of Integrin Avβ3 Enables Uptake of Targeted Fluorescent Probes into Ovarian Cancer Cells and Tumors. Bioorg. Med. Chem. 2018, 26, 2085–2091. [Google Scholar] [CrossRef] [Green Version]
- Cluzel, C.; Saltel, F.; Lussi, J.; Paulhe, F.; Imhof, B.A.; Wehrle-Haller, B. The Mechanisms and Dynamics of (Alpha)v(Beta)3 Integrin Clustering in Living Cells. J. Cell Biol. 2005, 171, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Pan, D.; Yan, Q.; Song, Y. Activation Mechanisms of AVβ3 Integrin by Binding to Fibronectin: A Computational Study. Protein Sci. 2017, 26, 1124–1137. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Wang, R.; Zhang, Y.; Han, X.; Ampah, K.K.; Liu, W.; Zeng, X. Identification of Nucleolin as a Lipid-Raft-Dependent Β1-Integrin-Interacting Protein in A375 Cell Migration. Mol. Cells 2013, 36, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sader, M.; Courty, J.; Destouches, D. Nanoparticles Functionalized with Ligands of Cell Surface Nucleolin for Cancer Therapy and Diagnosis. J. Nanomed. Nanotechnol. 2015, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yang, C.; Wang, R.; Jiao, Y.; Ampah, K.K.; Wang, X.; Zeng, X. C-Abl Kinase Is a Regulator of Avβ3 Integrin Mediated Melanoma A375 Cell Migration. PLoS ONE 2013, 8, e66108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, J.; Wang, R.; Zeng, X. Lipid Rafts Regulate the Lamellipodia Formation of Melanoma A375 Cells via Actin Cytoskeleton-Mediated Recruitment of Β1 and Β3 Integrin. Oncol. Lett. 2018, 16, 6540–6546. [Google Scholar] [CrossRef] [Green Version]
- de Campos, T.P.R.; Dalmazio, I.; Augusti, R.; Almeida, I.G. Gd-GLU toward NMR Imaging: Synthesis, Characterization and Breast Cell Uptake Assay. Braz. J. Pharm. Sci. 2020, 56, e18122. [Google Scholar] [CrossRef] [Green Version]
- Shikata, F.; Tokumitsu, H.; Ichikawa, H.; Fukumori, Y. In Vitro Cellular Accumulation of Gadolinium Incorporated into Chitosan Nanoparticles Designed for Neutron-Capture Therapy of Cancer. Eur. J. Pharm. Biopharm. 2002, 53, 57–63. [Google Scholar] [CrossRef]
- Peters, T.; Grunewald, C.; Blaickner, M.; Ziegner, M.; Schütz, C.; Iffland, D.; Hampel, G.; Nawroth, T.; Langguth, P. Cellular Uptake and in Vitro Antitumor Efficacy of Composite Liposomes for Neutron Capture Therapy. Radiat. Oncol. 2015, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, E.; Amanlou, M.; Ebrahimi, S.E.S.; Hamedani, M.P.; Mahrooz, A.; Mehravi, B.; Emami, B.A.; Aghasadeghi, M.R.; Bitarafan-Rajabi, A.; Akbar, H.R.P.A.; et al. Cellular Uptake, Imaging and Pathotoxicological Studies of a Novel Gd[III]–DO3A-Butrol Nano-Formulation. RSC Adv. 2014, 4, 45984–45994. [Google Scholar] [CrossRef]
- Tang, A.M.; Ananta, J.S.; Zhao, H.; Cisneros, B.T.; Lam, E.Y.; Wong, S.T.; Wilson, L.J.; Wong, K.K. Cellular Uptake and Imaging Studies of Gadolinium-Loaded Single-Walled Carbon Nanotubes as MRI Contrast Agents. Contrast Media Mol. Imaging 2011, 6, 93–99. [Google Scholar] [CrossRef]
- Hall, A.J.; Robertson, A.G.; Hill, L.R.; Rendina, L.M. Synthesis and Tumour Cell Uptake Studies of Gadolinium(III)-Phosphonium Complexes. Sci. Rep. 2021, 11, 598. [Google Scholar] [CrossRef]
- Venanzi, F.; Shifrin, V.; Sherman, M.Y.; Gabai, V.; Kisilev, O.; Komissarov, A.; Grudinin, M.; Shartakova, M.; Romanovskaya-Romanko, E.; Kudryavets, Y.; et al. Broad-Spectrum Anti-Tumor and Anti-Metastatic DNA Vaccine Based on P62-Encoding Vector. Oncotarget 2013, 4, 1829–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jirtle, R.; Clifton, K.H. The Effect of Tumor Size and Host Anemia on Tumor Cell Survival after Irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1978, 4, 395–400. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suazo, K.F.G.; Villaraza, A.J.L. Gd–XO: A Colourimetric Probe for the Complexation of Gd3+ with DO3A-Type Ligands. Anal. Methods 2015, 7, 8967–8969. [Google Scholar] [CrossRef]
- Tamura, H.; Yanagawa, I.; Hikichi, T.; Matsumoto, K.; Takahashi, S.; Sakamoto, K. T1 Measurements with Clinical MR Units. Tohoku J. Exp. Med. 1995, 175, 249–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanson, G. Bioconjugate Techniques, 3rd ed.; Academic Press: Amsterdam, The Netherlands, 2013; pp. 1–1146. [Google Scholar] [CrossRef]
- Fleischmann, T.; Jirkof, P.; Henke, J.; Arras, M.; Cesarovic, N. Injection Anaesthesia with Fentanyl-Midazolam-Medetomidine in Adult Female Mice: Importance of Antagonization and Perioperative Care. Lab. Anim. 2016, 50, 264–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E2-13W4 | E1-W8 | E1-W12 | EmGFP | BSA | |
|---|---|---|---|---|---|
| MW, kDa | 52.2 | 68.4 | 84.6 | 26.9 | 69.3 |
| Theoretical pI | 5.9 | 6.01 | 6.03 | 5.71 | 5.82 |
| Ext. coefficient at 280 nm (1 g/L) | 0.6 | 0.5 | 0.5 | 0.8 | 0.7 |
| Estimated half-life: | |||||
| -Lysate of mammalian reticulocytes, in vitro | 30 h | 30 h | 30 h | 30 h | |
| -Yeast, within the cells | 20 h | 20 h | >20 h | >20 h | |
| -Escherichia coli, within the cells | 10 h | 10 h | >10 h | >10 h | |
| The instability index | 31.07 (stable) | 33.55 (stable) | 31.07 (stable) | 27.73 (stable) | 40.28 (unstable) |
| Aliphatic index | 55.85 | 53.44 | 52.07 | 77.82 | 77.46 |
| Grand average of hydropathicity (GRAVY) | −0.665 | −0.679 | −0.690 | −0.491 | −0.429 |
| Estimated charge at pH 7.00 | −6.2 | −5.2 | −5.1 | −6.6 | −12.2 |
| Symbol | Function | Source (Amino Acid) | Sequence (Amino Acid) |
|---|---|---|---|
| M | Metal binding site | Calmodulin (human) (D21–L33) | DKDGDGTITTKEL |
| ELP | Forms the secondary structure | Elastin (human) | [VPGSG] |
| Immunoglobulin heavy chain junction region | [VPGYG] | ||
| L | Linker | Small antibody fragment linker (single-chain Fv fragment) | [G4S]2[G3S]1, [SG]5GS |
| F3 | Ligand of tumour receptors | Non-histone chromosomal protein HMG-17 (human) | KDEPQRRSARLSAKPA PPKPEPKPKKAPAKK |
| RGD | Ligand of tumour receptors | Fibronectin (human) | AVTGRGD |
| Motif | Backbone Vector | ||||
|---|---|---|---|---|---|
| Plasmid | MBS | F3 | RGD | ELP | |
| pB7 | 2 | - | - | 6 | pJET1.2 |
| pFLB77 | 4 | 1 | - | 14 | pJET1.2 |
| pRGD1(1) | - | - | 1 | 1 | pRSET-EmGFP |
| Plasmid | RGD | F3 | ELP | MBS | EmGFP |
|---|---|---|---|---|---|
| pE2-13W4 | 3 | 1 | 5 | 4 | 1 |
| pE1-W8 | 3 | 2 | 10 | 8 | 1 |
| pE1-W12 | 3 | 3 | 15 | 12 | 1 |
| Sample Name | MW, kDa | “Monomer”, d., nm | % Mass, d., nm |
|---|---|---|---|
| E2-13W4 | 52.2 | 9.2 | 99.87 |
| E1-W8 | 68.4 | 10.7 | 99.98 |
| E1-W12 | 84.6 | 11.3 | 99.99 |
| BSA | 69.3 | 4.7 | 100.00 |
| Protein | mKd (±Standard Deviation), µM |
|---|---|
| E2-13W4 | 0.21 ± 0.03 |
| E1-W8 | 0.17 ± 0.02 |
| E1-W12 | 0.19 ± 0.04 |
| E2-13W4-Gd4 | E1-W8-Gd8 | E1-W12-Gd12 | Magnevist® | |
|---|---|---|---|---|
| r1, mM−1 s−1 | 6.84 | 6.61 | 6.66 | 4.43 |
| Functional Elements of the Samples in the Cell Incubation Medium | Results | |||||
|---|---|---|---|---|---|---|
| Sample Name | [Protein], µM | [RGD] *, µM | [F3] **, µM | [Gd], µM | U87/Fb Ratio | A375/Fb Ratio |
| E2-13W4-Gd4 | 2.50 | 7.50 | 2.50 | 10 | 4.4 | 2.3 |
| E1-W8-Gd8 | 1.25 | 3.75 | 2.50 | 10 | 4.1 | 2.6 |
| E1-W12-Gd12 | 0.85 | 2.55 | 2.55 | 10 | 3.0 | 2.4 |
| Gd(NO3)3 | 10 | - | - | 10 | 1.1 | 1.1 |
| Magnevist® | 10 | - | - | 10 | 1.1 | 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pozdniakova, N.V.; Ryabaya, O.V.; Semkina, A.S.; Skribitsky, V.A.; Shevelev, A.B. Using ELP Repeats as a Scaffold for De Novo Construction of Gadolinium-Binding Domains within Multifunctional Recombinant Proteins for Targeted Delivery of Gadolinium to Tumour Cells. Int. J. Mol. Sci. 2022, 23, 3297. https://doi.org/10.3390/ijms23063297
Pozdniakova NV, Ryabaya OV, Semkina AS, Skribitsky VA, Shevelev AB. Using ELP Repeats as a Scaffold for De Novo Construction of Gadolinium-Binding Domains within Multifunctional Recombinant Proteins for Targeted Delivery of Gadolinium to Tumour Cells. International Journal of Molecular Sciences. 2022; 23(6):3297. https://doi.org/10.3390/ijms23063297
Chicago/Turabian StylePozdniakova, Natalia V., Oxana V. Ryabaya, Alevtina S. Semkina, Vsevolod A. Skribitsky, and Alexei B. Shevelev. 2022. "Using ELP Repeats as a Scaffold for De Novo Construction of Gadolinium-Binding Domains within Multifunctional Recombinant Proteins for Targeted Delivery of Gadolinium to Tumour Cells" International Journal of Molecular Sciences 23, no. 6: 3297. https://doi.org/10.3390/ijms23063297