
Prenylcysteine Oxidase 1 (PCYOX1), a New Player in Thrombosis

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Results
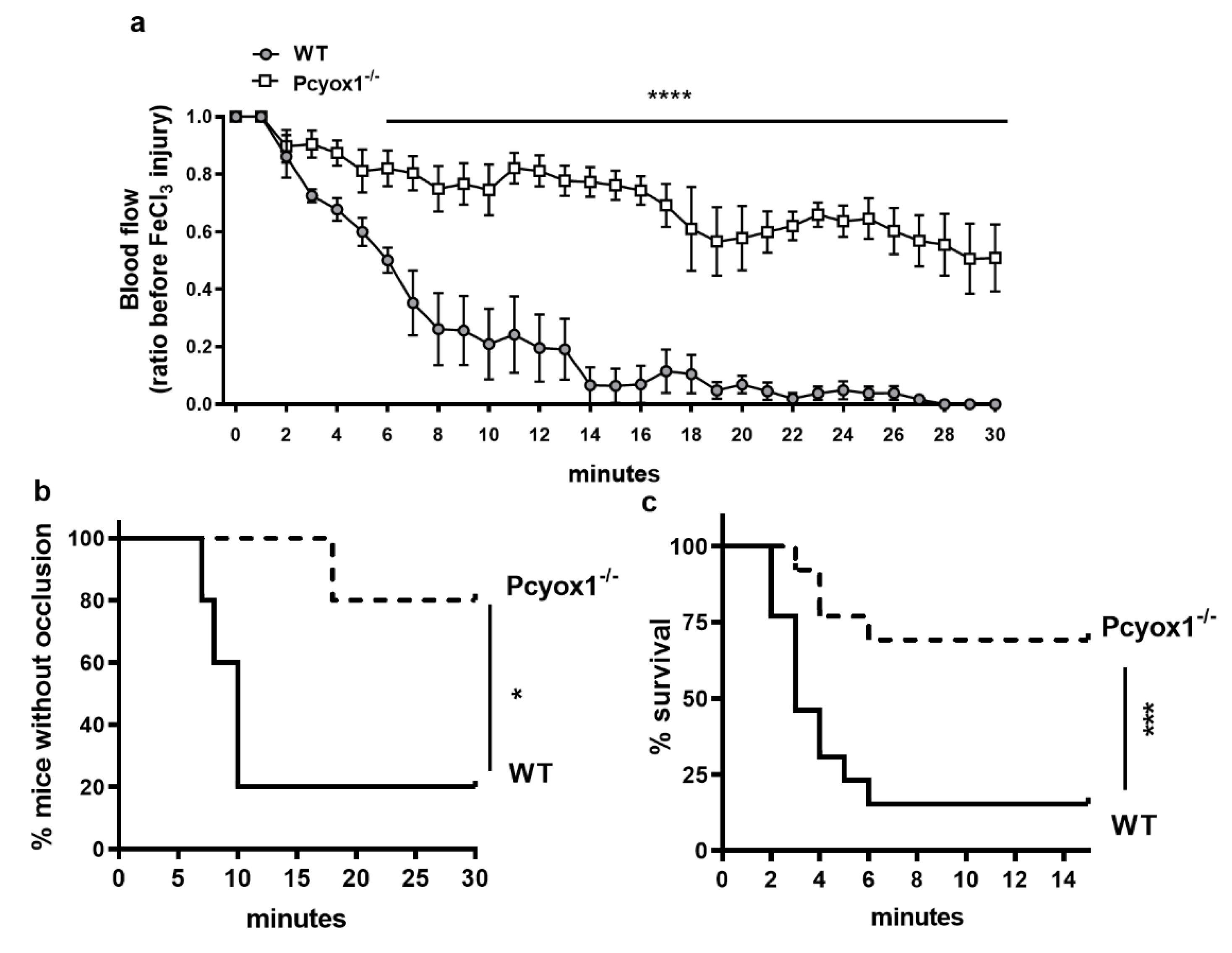
2.1. Pcyox1 Deficiency Affects Thrombosis In Vivo
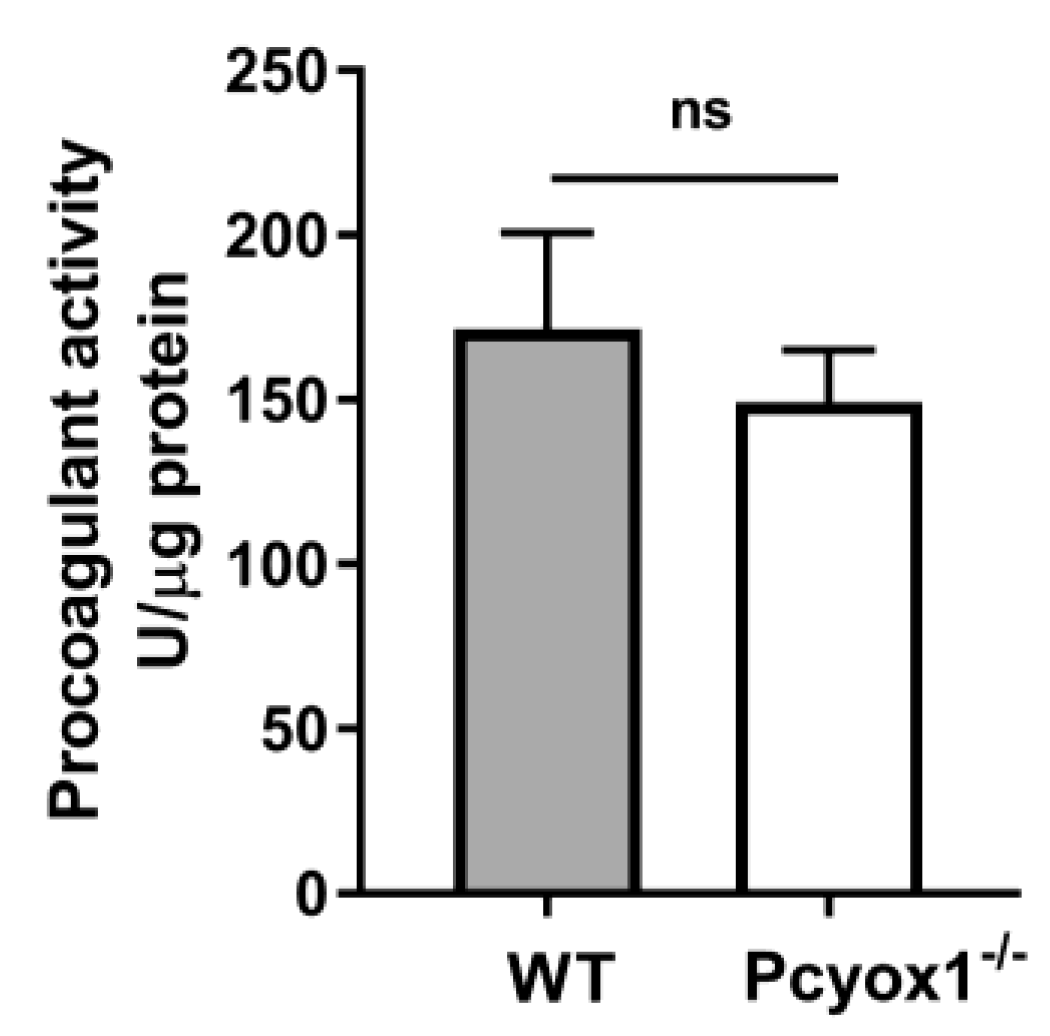
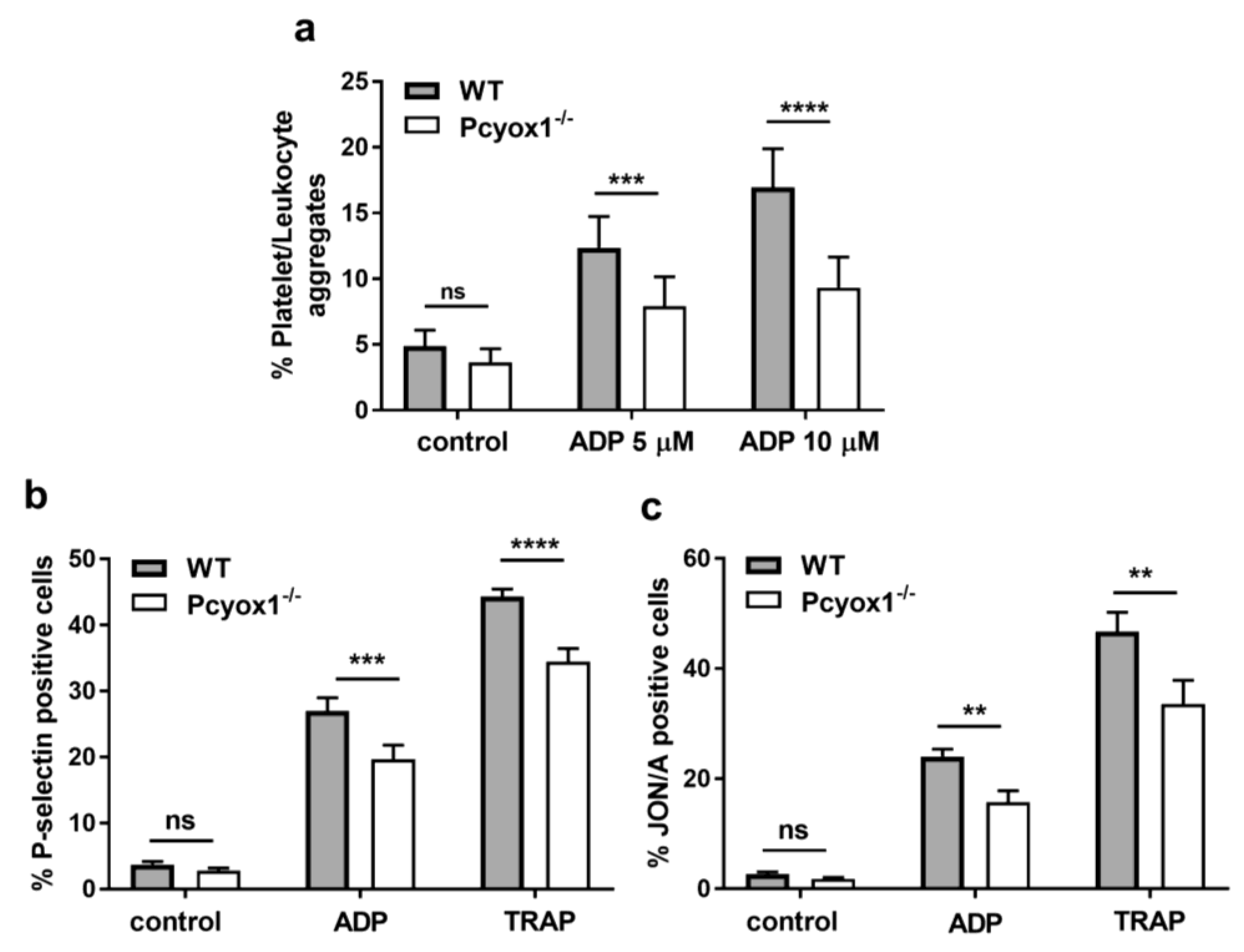
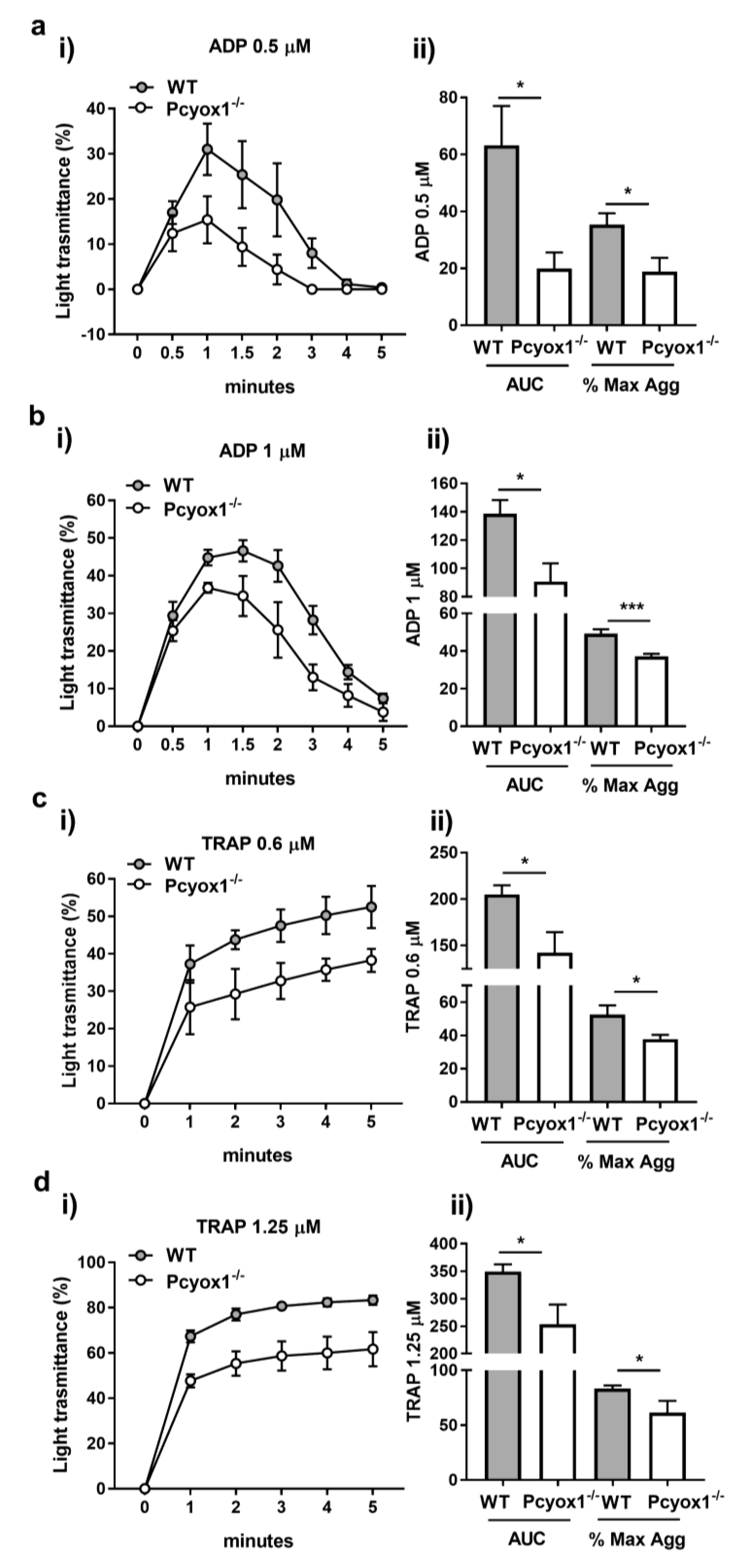
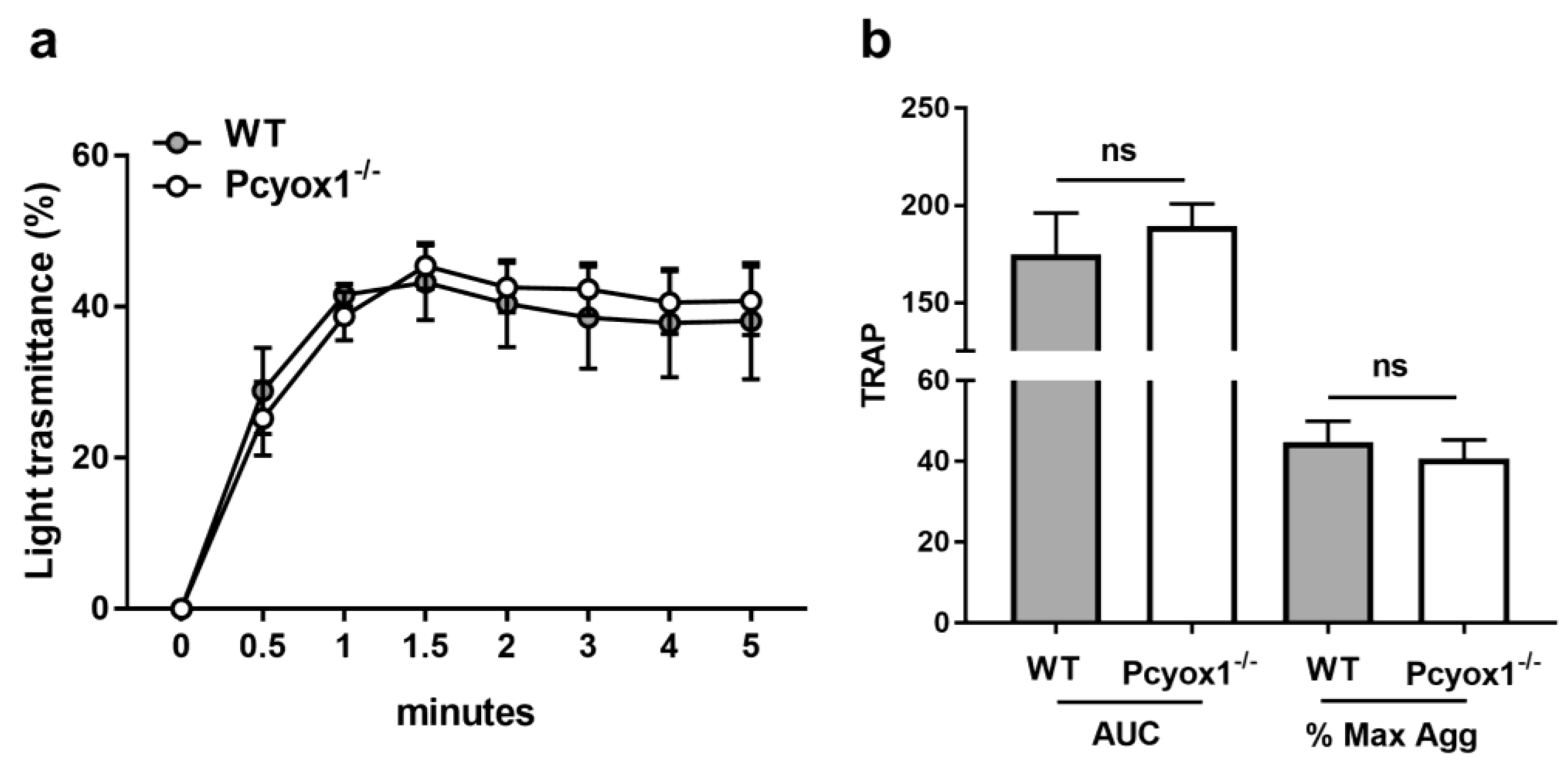
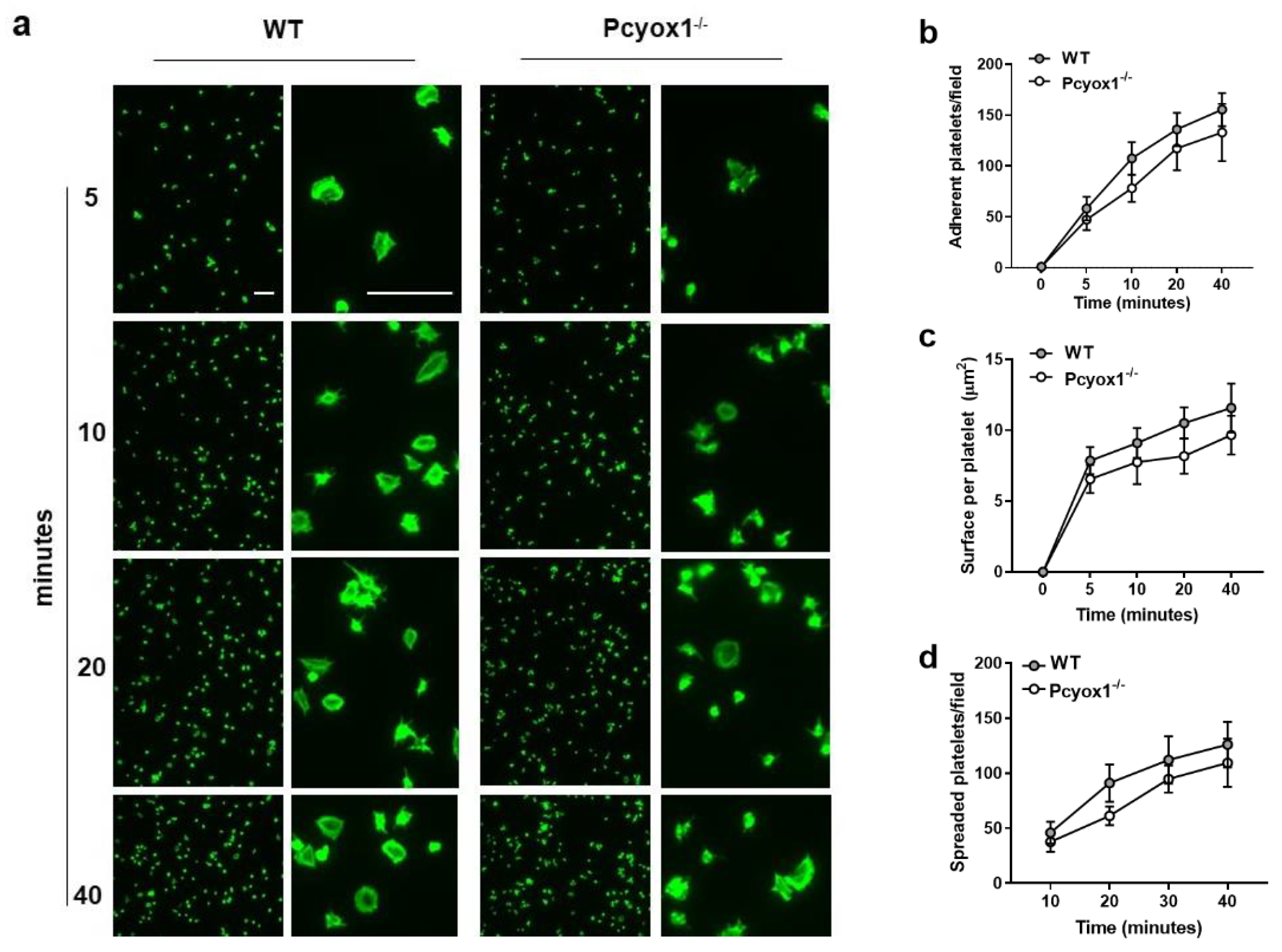
2.2. Impact of Pcyox1 Deletion on the Vascular Procoagulant Activity and Platelet Functions
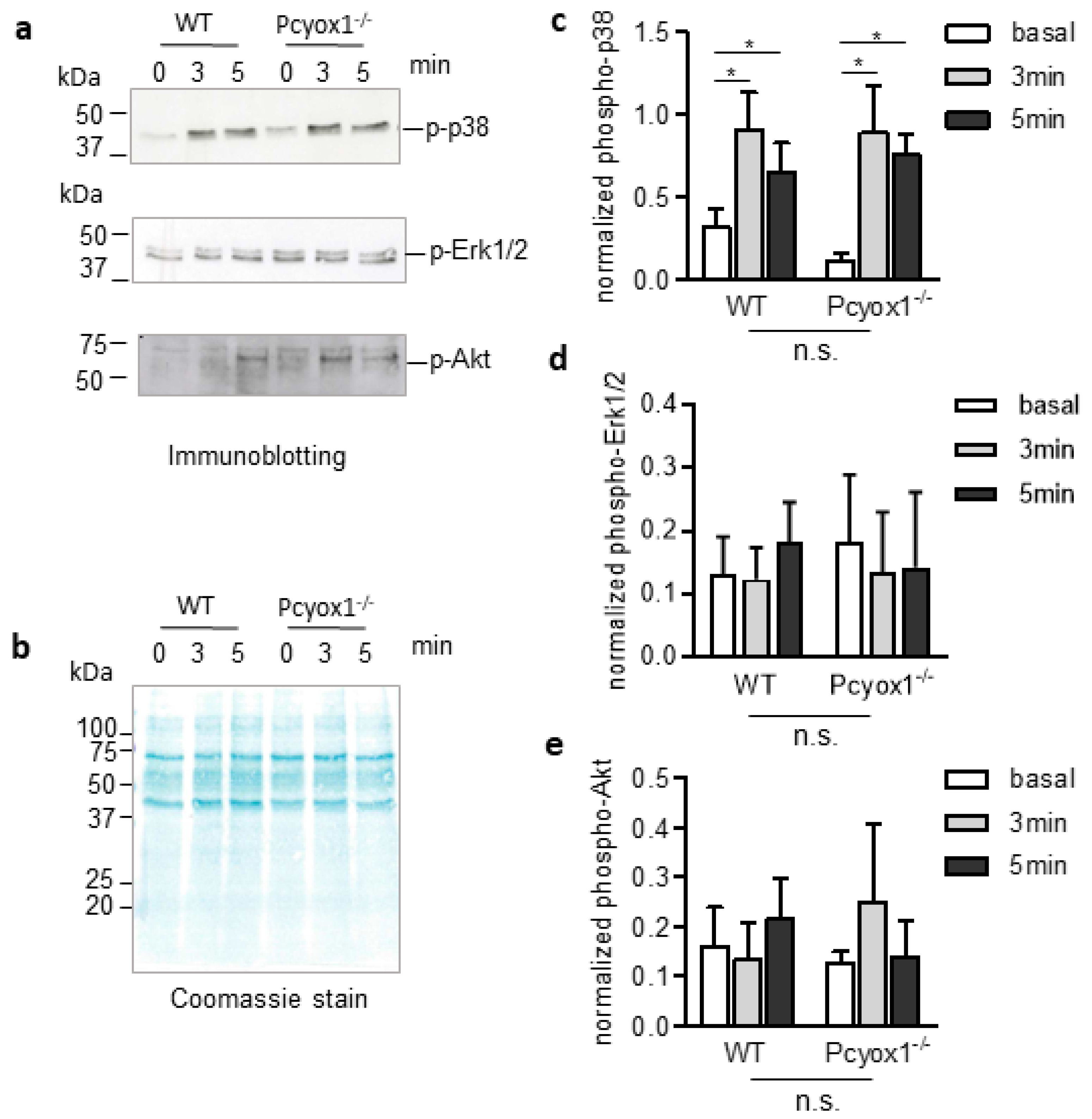
2.3. Role of PCYOX1 in Washed Platelets
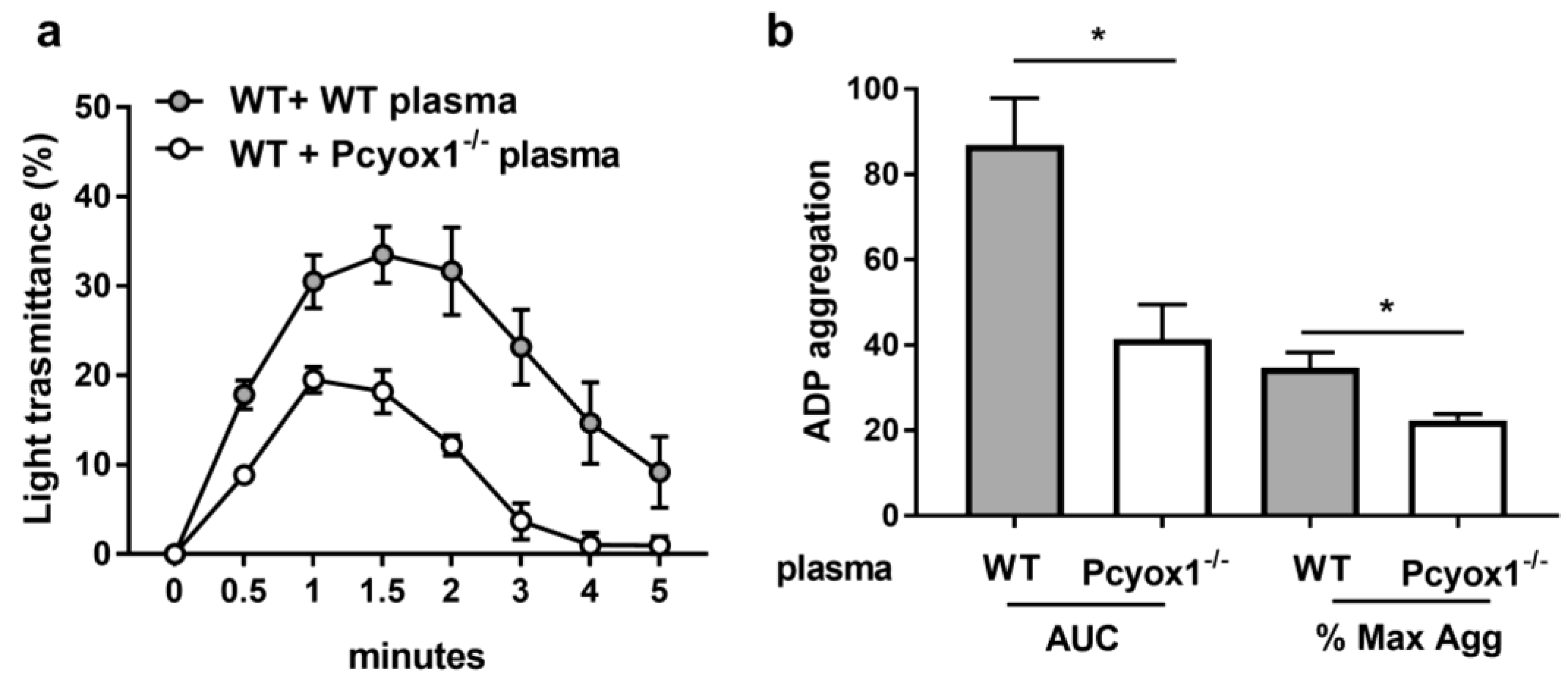
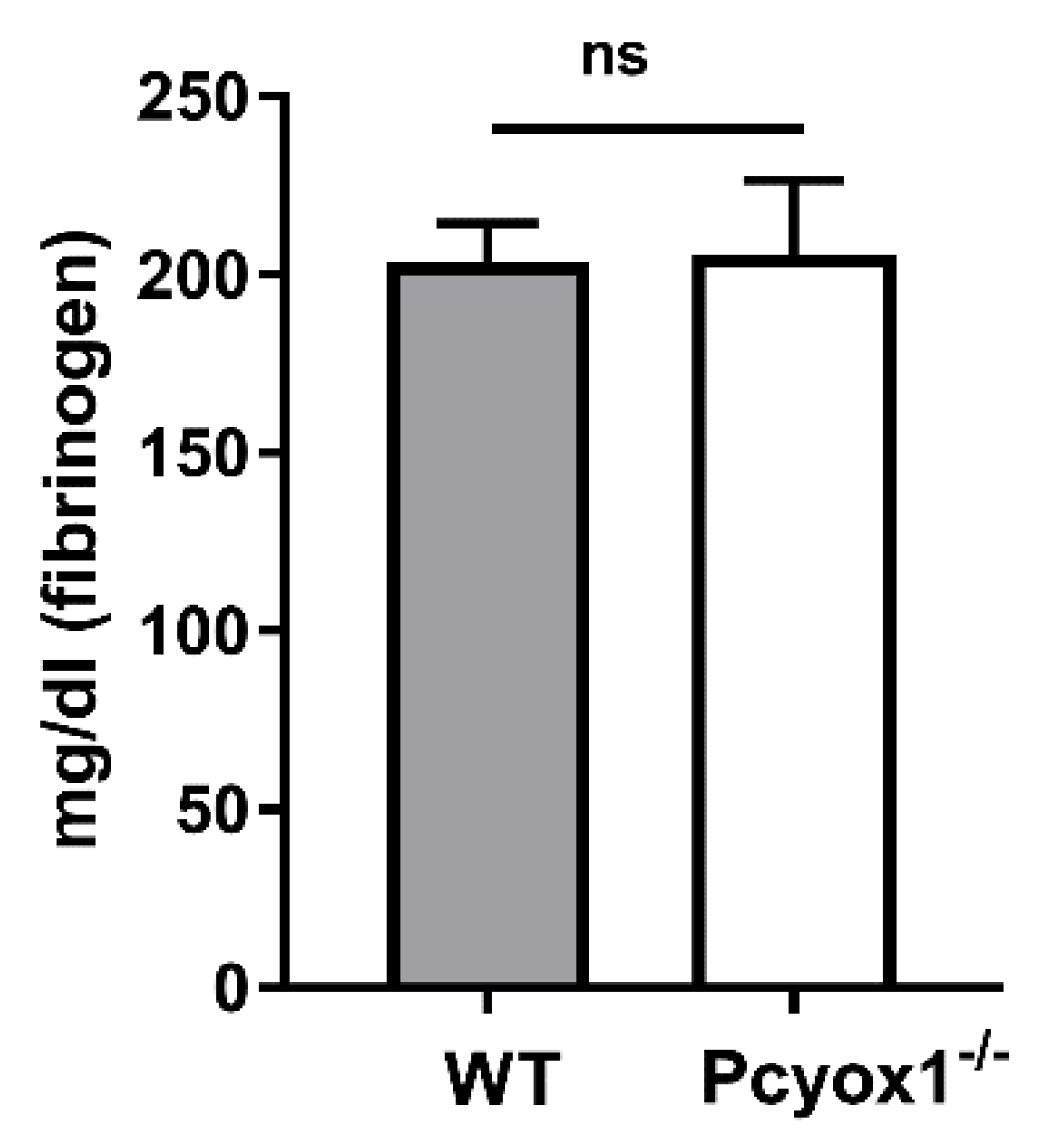
2.4. Role of Plasma Component in the Modulation of Platelet Activation in Pcyox1 Deficient Mice
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Blood Collection, Blood Cell Count and Plasma Preparation
4.3. Carotid Artery Thrombosis Model
4.4. Mouse Thromboembolism Model
4.5. Procoagulant Activity
4.6. Platelet-Rich Plasma Preparation
4.7. Platelet Flow Cytometry Analyses
4.8. Washed Platelet Preparation
4.9. Platelet Aggregation Studies
4.10. Western Blot
4.11. Platelet Adhesion
4.12. Functional Fibrinogen Assay
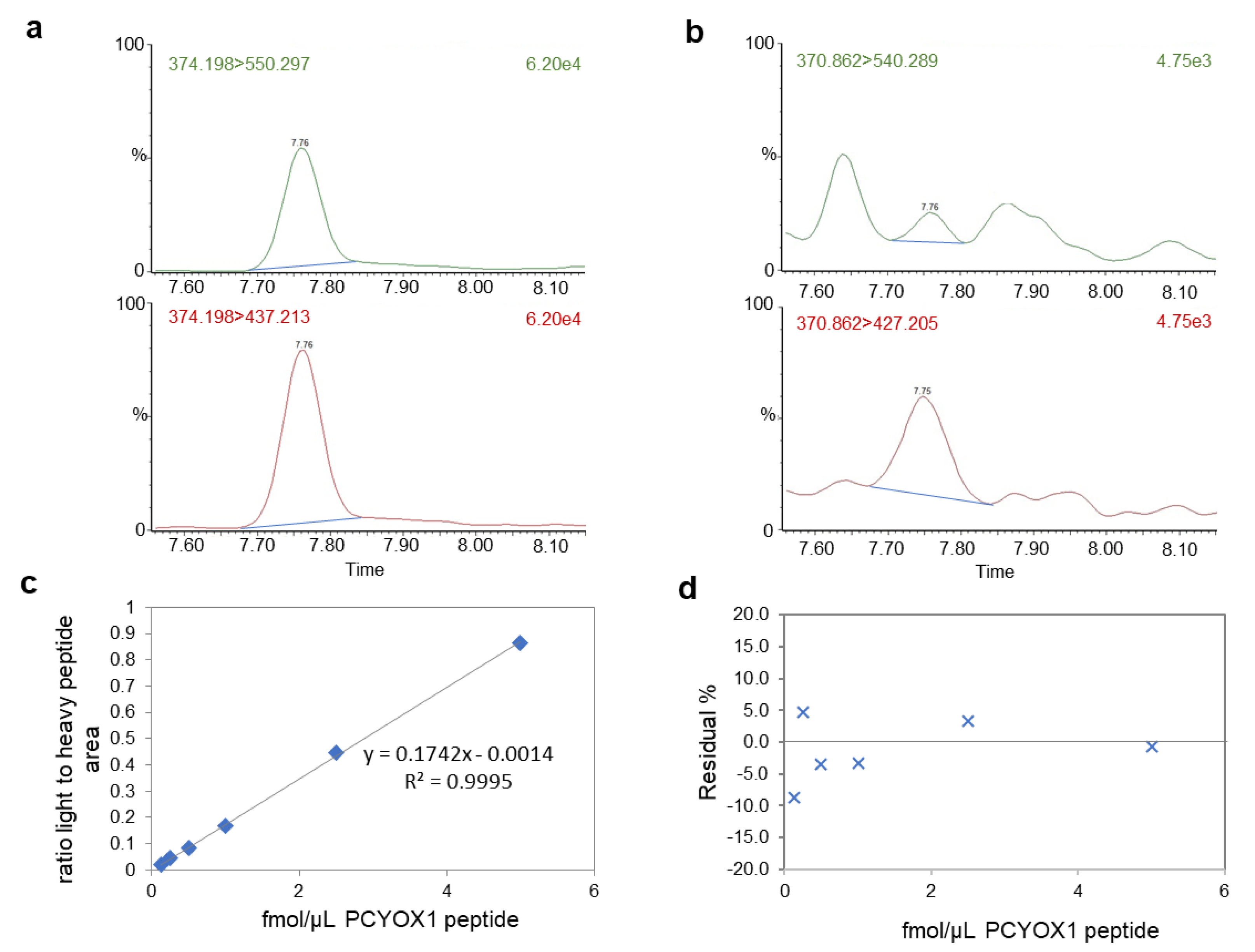
4.13. Mass Spectrometry Analysis
4.14. Statistical Analyses
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruggeri, Z.M. Platelets in atherothrombosis. Nat. Med. 2002, 8, 1227–1234. [Google Scholar] [CrossRef]
- Wang, L.; Tang, C. Targeting Platelet in Atherosclerosis Plaque Formation: Current Knowledge and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 9760. [Google Scholar] [CrossRef]
- Koupenova, M.; Kehrel, B.E.; Corkrey, H.A.; Freedman, J.E. Thrombosis and platelets: An update. Eur. Heart J. 2017, 38, 785–791. [Google Scholar] [CrossRef]
- Masselli, E.; Pozzi, G.; Vaccarezza, M.; Mirandola, P.; Galli, D.; Vitale, M.; Carubbi, C.; Gobbi, G. ROS in Platelet Biology: Functional Aspects and Methodological Insights. Int. J. Mol. Sci. 2020, 21, 4866. [Google Scholar] [CrossRef]
- Banfi, C.; Brioschi, M.; Barcella, S.; Wait, R.; Begum, S.; Galli, S.; Rizzi, A.; Tremoli, E. Proteomic analysis of human low-density lipoprotein reveals the presence of prenylcysteine lyase, a hydrogen peroxide-generating enzyme. Proteomics 2009, 9, 1344–1352. [Google Scholar] [CrossRef]
- Tschantz, W.R.; Digits, J.A.; Pyun, H.-J.; Coates, R.M.; Casey, P. Lysosomal Prenylcysteine Lyase Is a FAD-dependent Thioether Oxidase. J. Biol. Chem. 2001, 276, 2321–2324. [Google Scholar] [CrossRef] [Green Version]
- Banfi, C.; Baetta, R.; Barbieri, S.S.; Brioschi, M.; Guarino, A.; Ghilardi, S.; Sandrini, L.; Eligini, S.; Polvani, G.; Bergman, O.; et al. Prenylcysteine oxidase 1, an emerging player in atherosclerosis. Commun. Biol. 2021, 4, 1109. [Google Scholar] [CrossRef]
- Osada, J.; Herrera-Marcos, L.V.; Lou-Bonafonte, J.M.; Martinez-Gracia, M.V.; Arnal, C.; Navarro, M.A. Prenylcysteine oxidase 1 a pro-oxidant enzyme of low density lipoproteins. Front. Biosci. 2018, 23, 1020–1037. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Tschantz, W.R.; Casey, P. Isolation and Characterization of a Prenylcysteine Lyase from Bovine Brain. J. Biol. Chem. 1997, 272, 23354–23359. [Google Scholar] [CrossRef] [Green Version]
- Digits, J.A.; Pyun, H.-J.; Coates, R.M.; Casey, P. Stereospecificity and Kinetic Mechanism of Human Prenylcysteine Lyase, an Unusual Thioether Oxidase. J. Biol. Chem. 2002, 277, 41086–41093. [Google Scholar] [CrossRef] [Green Version]
- Pignatelli, P.; Pulcinelli, F.M.; Lenti, L.; Gazzaniga, P.P.; Violi, F. Hydrogen peroxide is involved in collagen-induced platelet activation. Blood 1998, 91, 484–490. [Google Scholar] [CrossRef] [Green Version]
- Dayal, S.; Wilson, K.M.; Motto, D.G.; Miller, F.J., Jr.; Chauhan, A.K.; Lentz, S.R. Hydrogen Peroxide Promotes Aging-Related Platelet Hyperactivation and Thrombosis. Circulation 2013, 127, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Korporaal, S.J.; Gorter, G.; van Rijn, H.J.; Akkerman, J.-W.N. Effect of Oxidation on the Platelet-Activating Properties of Low-Density Lipoprotein. Arter. Thromb. Vasc. Biol. 2005, 25, 867–872. [Google Scholar] [CrossRef]
- Banfi, C.; Camera, M.; Giandomenico, G.; Toschi, V.; Arpaia, M.; Mussoni, L.; Tremoli, E.; Colli, S. Vascular thrombogenicity induced by progressive LDL oxidation: Protection by antioxidants. Thromb. Haemost. 2003, 89, 544–553. [Google Scholar] [CrossRef]
- Colas, R.; Sassolas, A.; Guichardant, M.; Cugnet-Anceau, C.; Moret, M.; Moulin, P.; Lagarde, M.; Calzada, C. LDL from obese patients with the metabolic syndrome show increased lipid peroxidation and activate platelets. Diabetologia 2011, 54, 2931–2940. [Google Scholar] [CrossRef] [Green Version]
- Korporaal, S.J.; Van Eck, M.; Adelmeijer, J.; Ijsseldijk, M.; Out, R.; Lisman, T.; Lenting, P.J.; Van Berkel, T.J.; Akkerman, J.-W.N. Platelet Activation by Oxidized Low Density Lipoprotein Is Mediated by Cd36 and Scavenger Receptor-A. Arter. Thromb. Vasc. Biol. 2007, 27, 2476–2483. [Google Scholar] [CrossRef] [Green Version]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [Green Version]
- Jung, R.G.; Simard, T.; Labinaz, A.; Ramirez, F.D.; Di Santo, P.; Motazedian, P.; Rochman, R.; Gaudet, C.; Faraz, M.A.; Beanlands, R.S.; et al. Role of plasminogen activator inhibitor-1 in coronary pathophysiology. Thromb. Res. 2018, 164, 54–62. [Google Scholar] [CrossRef]
- Cerletti, C.; Tamburrelli, C.; Izzi, B.; Gianfagna, F.; de Gaetano, G. Platelet-leukocyte interactions in thrombosis. Thromb. Res. 2012, 129, 263–266. [Google Scholar] [CrossRef]
- Sandrini, L.; Ieraci, A.; Amadio, P.; Veglia, F.; Popoli, M.; Lee, F.S.; Tremoli, E.; Barbieri, S.S. Sub-Chronic Stress Exacerbates the Pro-Thrombotic Phenotype in BDNFVal/Met Mice: Gene-Environment Interaction in the Modulation of Arterial Thrombosis. Int. J. Mol. Sci. 2018, 19, 3235. [Google Scholar] [CrossRef] [Green Version]
- Sandrini, L.; Ieraci, A.; Amadio, P.; Zarà, M.; Mitro, N.; Lee, F.S.; Tremoli, E.; Barbieri, S.S. Physical Exercise Affects Adipose Tissue Profile and Prevents Arterial Thrombosis in BDNF Val66Met Mice. Cells 2019, 8, 875. [Google Scholar] [CrossRef] [Green Version]
- Amadio, P.; Rocca, B.; Pesce, M.; Machlus, K.R.; Ranelletti, F.O.; Gianellini, S.; Weksler, B.B.; Italiano, J.E., Jr.; Tremoli, E.; Barbieri, S.S.; et al. Abnormal megakaryopoiesis and platelet function in cyclooxygenase-2-deficient mice. Thromb. Haemost. 2015, 114, 1218–1229. [Google Scholar] [CrossRef]
- Sandrini, L.; Amadio, P.; Ieraci, A.; Malara, A.; Werba, J.P.; Soprano, P.M.; Balduini, A.; Zarà, M.; Bonomi, A.; Veglia, F.; et al. The α2-adrenergic receptor pathway modulating depression influences the risk of arterial thrombosis associated with BDNFVal66Met polymorphism. Biomed. Pharmacother. 2022, 146, 112557. [Google Scholar] [CrossRef]
- Sandrini, L.; Ieraci, A.; Amadio, P.; Popoli, M.; Tremoli, E.; Barbieri, S.S. Apocynin Prevents Abnormal Megakaryopoiesis and Platelet Activation Induced by Chronic Stress. Oxidative Med. Cell. Longev. 2017, 2017, 9258937. [Google Scholar] [CrossRef] [Green Version]
- Amadio, P.; Colombo, G.I.; Tarantino, E.; Gianellini, S.; Ieraci, A.; Brioschi, M.; Banfi, C.; Werba, J.P.; Parolari, A.; Lee, F.S.; et al. BDNFVal66met polymorphism: A potential bridge between depression and thrombosis. Eur. Hear. J. 2017, 38, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Torti, M.; Manganaro, D.; Visconte, C.; Zarà, M.; Canino, J.; Vismara, M.; Canobbio, I.; Guidetti, G.F. Stimulation of mTORC2 by integrin αIIbβ3 is required for PI3Kβ-dependent activation of Akt but is dispensable for platelet spreading on fibrinogen. Platelets 2020, 31, 521–529. [Google Scholar] [CrossRef]
- Baldassarre, D.; Amato, M.; Eligini, S.; Barbieri, S.S.; Mussoni, L.; Frigerio, B.; Kozakova, M.; Tremoli, E.; Sirtori, C.R.; Colli, S. Effect of n-3 fatty acids on carotid atherosclerosis and haemostasis in patients with combined hyperlipoproteinemia: A double-blind pilot study in primary prevention. Ann. Med. 2006, 38, 367–375. [Google Scholar] [CrossRef]
- Fernandez, A.E.M.; Regazzoni, L.; Brioschi, M.; Gianazza, E.; Agostoni, P.; Aldini, G.; Banfi, C. Pro-oxidant and pro-inflammatory effects of glycated albumin on cardiomyocytes. Free Radic. Biol. Med. 2019, 144, 245–255. [Google Scholar] [CrossRef]
- Amadio, P.; Tarantino, E.; Sandrini, L.; Tremoli, E.; Barbieri, S.S. Prostaglandin-endoperoxide synthase-2 deletion affects the natural trafficking of Annexin A2 in monocytes and favours venous thrombosis in mice. Thromb. Haemost. 2017, 117, 1486–1497. [Google Scholar] [CrossRef]
- Silva, J.C.; Denny, R.; Dorschel, C.; Gorenstein, M.V.; Li, G.-Z.; Richardson, K.; Wall, D.; Geromanos, S.J. Simultaneous Qualitative and Quantitative Analysis of theEscherichia coli Proteome. Mol. Cell. Proteom. 2006, 5, 589–607. [Google Scholar] [CrossRef] [Green Version]
- Brioschi, M.; Eligini, S.; Crisci, M.; Fiorelli, S.; Tremoli, E.; Colli, S.; Banfi, C. A mass spectrometry-based workflow for the proteomic analysis of in vitro cultured cell subsets isolated by means of laser capture microdissection. Anal. Bioanal. Chem. 2014, 406, 2817–2825. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cells | WT | Pcyox1−/− | p-Value |
|---|---|---|---|
| Erythrocytes (107/µL) | 15.47 ± 0.65 | 15.88 ± 0.99 | 0.650 |
| Leukocytes (106/µL) | 2.61 ± 0.97 | 2.81 ± 1.57 | 0.301 |
| Platelets (104/µL) | 71.9 ± 2.20 | 72.47 ± 2.61 | 0.707 |
| Peptide | Precursor m/z | Transition m/z | Collision Energy | Cone Voltage (V) |
|---|---|---|---|---|
| CPSIILHDR light | 370.86 | 540.29 | 15 | 35 |
| 427.20 * | 15 | 35 | ||
| CPSIILHDR heavy | 374.2 | 550.2971 | 15 | 35 |
| 437.2131 * | 15 | 35 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banfi, C.; Amadio, P.; Zarà, M.; Brioschi, M.; Sandrini, L.; Barbieri, S.S. Prenylcysteine Oxidase 1 (PCYOX1), a New Player in Thrombosis. Int. J. Mol. Sci. 2022, 23, 2831. https://doi.org/10.3390/ijms23052831
Banfi C, Amadio P, Zarà M, Brioschi M, Sandrini L, Barbieri SS. Prenylcysteine Oxidase 1 (PCYOX1), a New Player in Thrombosis. International Journal of Molecular Sciences. 2022; 23(5):2831. https://doi.org/10.3390/ijms23052831
Chicago/Turabian StyleBanfi, Cristina, Patrizia Amadio, Marta Zarà, Maura Brioschi, Leonardo Sandrini, and Silvia S. Barbieri. 2022. "Prenylcysteine Oxidase 1 (PCYOX1), a New Player in Thrombosis" International Journal of Molecular Sciences 23, no. 5: 2831. https://doi.org/10.3390/ijms23052831