
Functional Conversion of Acetyl-Coenzyme a Synthase to a Nickel Superoxide Dismutase via Rational Design of Coordination Microenvironment for the Nid-Site

Abstract
:1. Introduction
2. Results
2.1. Design of Nid-Site by Molecular Modeling
2.2. Mutagenesis, Expression, and Purification
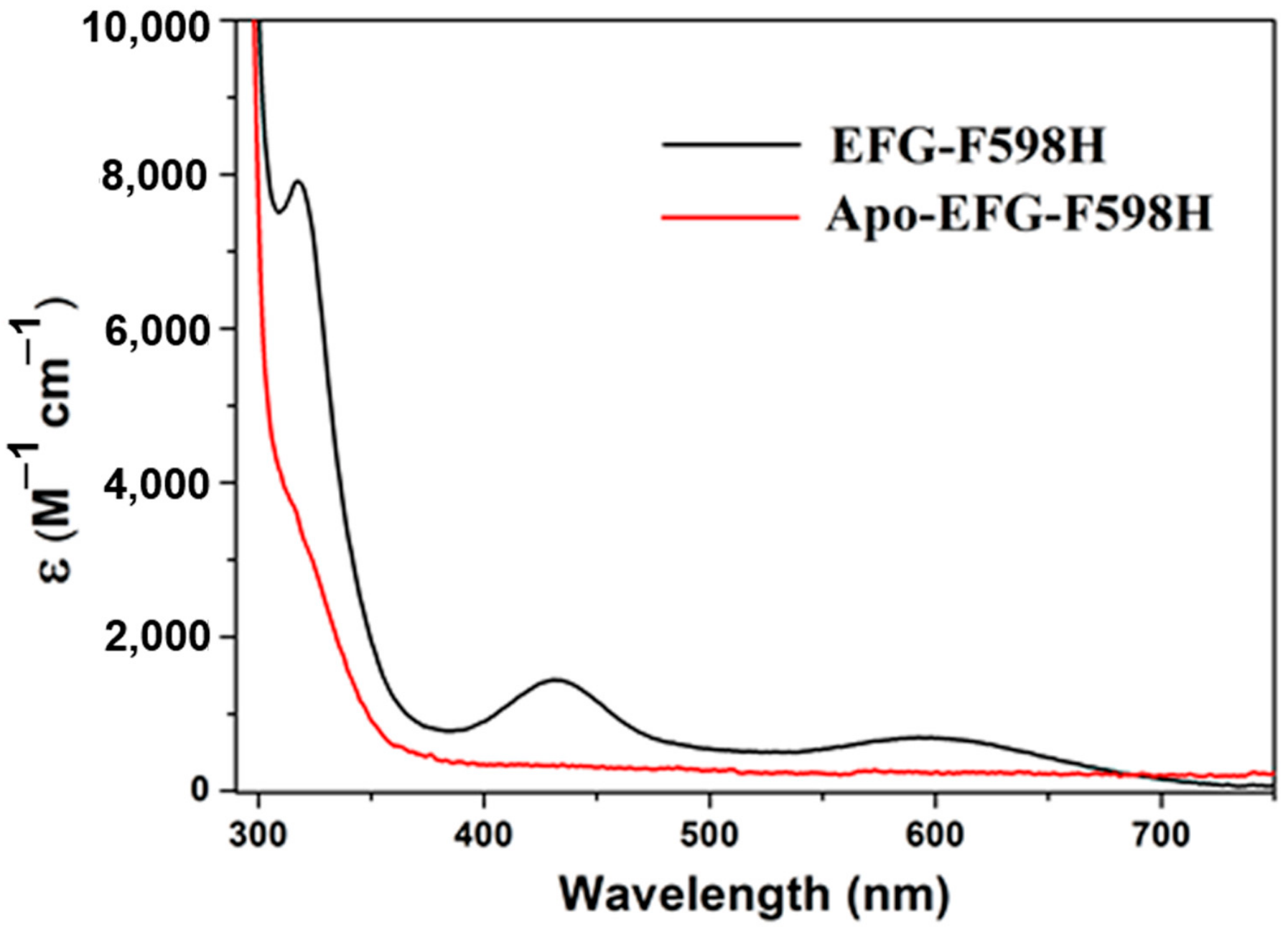
2.3. Nickel Binding Study by UV-Vis and ITC
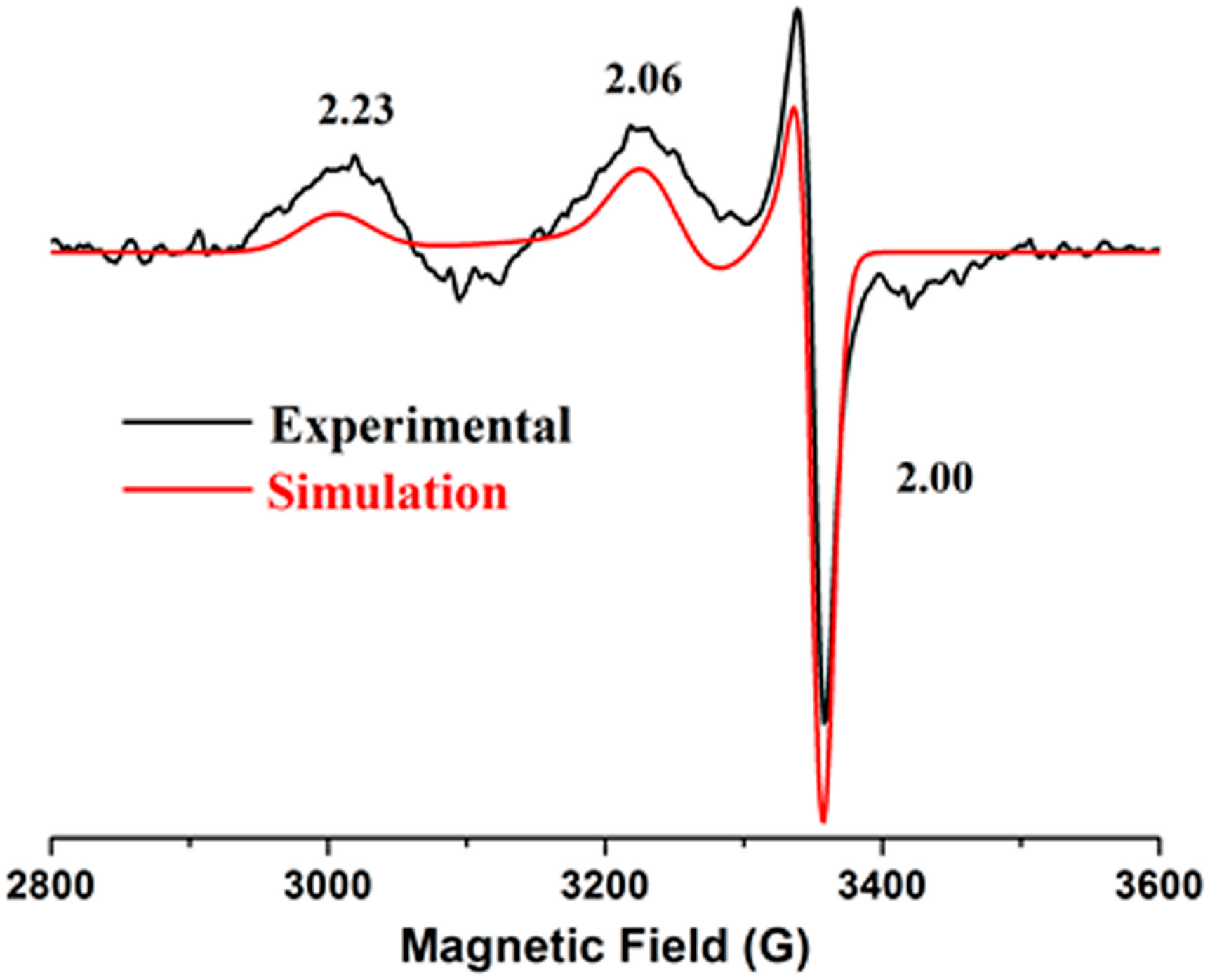
2.4. Electron Paramagnetic Resonance
2.5. Cyclic Voltammetry
2.6. SOD Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | No. of Ni Atoms per Protein | Em (mV vs. NHE) | SOD Activity (U/μmol) |
|---|---|---|---|
| F598H | 0.7 | 372 | 4360 |
| S594H | 0.8 | 374 | 4940 |
| S594H-GP | 0.8 | 375 | 4600 |
| EFG-F598H | 0.6 | 377 | 14,770 |
| YGP-F598H | 0.5 | 374 | 3420 |
2.7. Effect of pH on SOD Activity
2.8. Electro Spray Ionization-Mass Spectroscopy (ESI-MS)
2.9. Crystal Structure of F598H
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cloning, Expression, and Purification
4.3. Metal Reconstruction and Metal Analysis
4.4. Protein Characterization
4.5. Cyclic Voltammetry Assay
4.6. SOD Activity Assay
4.7. Molecular Dynamic Simulation
4.8. pH Titration Assay
4.9. Isothermal Titration Calorimetry (ITC) Assay
4.10. Electro Spray Ionization-Mass Spectroscopy (ESI-MS) Assay
4.11. Crystallography of F598H
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fontecilla-Camps, J.C.; Amara, P.; Cavazza, C.; Nicolet, Y.; Volbeda, A. Structure-function relationships of anaerobic gas-processing metalloenzymes. Nature 2009, 460, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Parker, M.J.; Stubbe, J. Choosing the Right Metal: Case Studies of Class I Ribonucleotide Reductases. J. Biol. Chem. 2014, 289, 28104–28111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Yeung, N.; Sieracki, N.; Marshall, N.M. Design of functional metalloproteins. Nature 2009, 460, 855–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrik, I.D.; Liu, J.; Lu, Y. Metalloenzyme design and engineering through strategic modifications of native protein scaffolds. Curr. Opin. Chem. Biol. 2014, 19, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.T.; Cangelosi, V.M.; Zastrow, M.L.; Tegoni, M.; Plegaria, J.S.; Tebo, A.G.; Mocny, C.S.; Ruckthong, L.; Qayyum, H.; Pecoraro, V.L. Protein Design: Toward Functional Metalloenzymes. Chem. Rev. 2014, 114, 3495–3578. [Google Scholar] [CrossRef] [Green Version]
- Bhagi-Damodaran, A.; Hosseinzadeh, P.; Mirts, E.; Reed, J.; Petrik, I.D.; Lu, Y. Design of Heteronuclear Metalloenzymes. In Peptide, Protein and Enzyme Design; Pecoraro, V.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 580, pp. 501–537. [Google Scholar]
- Nastri, F.; Chino, M.; Maglio, O.; Bhagi-Damodaran, A.; Lu, Y.; Lombardi, A. Design and engineering of artificial oxygen-activating metalloenzymes. Chem. Soc. Rev. 2016, 45, 5020–5054. [Google Scholar] [CrossRef]
- Lin, Y.W. Rational design of metalloenzymes: From single to multiple active sites. Coord. Chem. Rev. 2017, 336, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.F.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Kohler, V.; Lewis, J.C.; Ward, T.R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [Google Scholar] [CrossRef] [Green Version]
- Nastri, F.; D’Alonzo, D.; Leone, L.; Zambrano, G.; Pavone, V.; Lombardi, A. Engineering Metalloprotein Functions in Designed and Native Scaffolds. Trends Biochem. Sci. 2019, 44, 1022–1040. [Google Scholar] [CrossRef]
- Liu, Y.; Zhu, X.; Wang, F.; Ying, T.; Li, P.; Huang, Z.X.; Tan, X. Probing the role of the bridging C509 between the [Fe4S4] cubane and the [Ni(p)Ni(d)] centre in the A-cluster of acetyl-coenzyme A synthase. Chem. Commun. 2011, 47, 1291–1293. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, F.; Li, P.; Tan, X. Insights into the mechanistic role of the [Fe4S4] cubane in the A-cluster {[Fe4S4]-(SR)-[NipNid]} of acetyl-coenzyme A synthase. Chembiochem 2011, 12, 1417–1421. [Google Scholar] [CrossRef]
- Kung, Y.; Drennan, C.L. A role for nickel-iron cofactors in biological carbon monoxide and carbon dioxide utilization. Curr. Opin. Chem. Biol. 2011, 15, 276–283. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, P.A. Acetyl-coenzyme A synthase: The case for a Ni(p)(0)-based mechanism of catalysis. J. Biol. Inorg. Chem. 2004, 9, 516–524. [Google Scholar] [CrossRef]
- Wuerges, J.; Lee, J.W.; Yim, Y.I.; Yim, H.S.; Kang, S.O.; Djinovic Carugo, K. Crystal structure of nickel-containing superoxide dismutase reveals another type of active site. Proc. Natl. Acad. Sci. USA 2004, 101, 8569–8574. [Google Scholar] [CrossRef] [Green Version]
- Darnault, C.; Volbeda, A.; Kim, E.J.; Legrand, P.; Vernede, X.; Lindahl, P.A.; Fontecilla-Camps, J.C. Ni-Zn-[Fe4-S4] and Ni-Ni-[Fe4-S4] clusters in closed and open subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat. Struct. Biol. 2003, 10, 271–279. [Google Scholar] [CrossRef]
- Barondeau, D.P.; Kassmann, C.J.; Bruns, C.K.; Tainer, J.A.; Getzoff, E.D. Nickel superoxide dismutase structure and mechanism. Biochemistry 2004, 43, 8038–8047. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Q.; Wei, Y.; Lin, Y.W.; Li, W.; Su, J.H.; Wang, Z.; Tian, Y.; Huang, Z.X.; Tan, X. Functional conversion of nickel-containing metalloproteins via molecular design: From a truncated acetyl-coenzyme A synthase to a nickel superoxide dismutase. Chem. Commun. 2013, 49, 1452–1454. [Google Scholar] [CrossRef]
- Schmidt, M.; Zahn, S.; Carella, M.; Ohlenschlager, O.; Gorlach, M.; Kothe, E.; Weston, J. Solution structure of a functional biomimetic and mechanistic implications for nickel superoxide dismutases. ChemBioChem 2008, 9, 2135–2146. [Google Scholar] [CrossRef]
- Youn, H.D.; Kim, E.J.; Roe, J.H.; Hah, Y.C.; Kang, S.O. A novel nickel-containing superoxide dismutase from Streptomyces spp. Biochem. J. 1996, 318 Pt 3, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Herbst, R.W.; Guce, A.; Bryngelson, P.A.; Higgins, K.A.; Ryan, K.C.; Cabelli, D.E.; Garman, S.C.; Maroney, M.J. Role of conserved tyrosine residues in NiSOD catalysis: A case of convergent evolution. Biochemistry 2009, 48, 3354–3369. [Google Scholar] [CrossRef] [Green Version]
- Shearer, J.; Zhao, N. [Me4N](Ni(II)(BEAAM)): A synthetic model for nickel superoxide dismutase that contains Ni in a mixed amine/amide coordination environment. Inorg. Chem. 2006, 45, 9637–9639. [Google Scholar] [CrossRef] [PubMed]
- Gale, E.M.; Simmonett, A.C.; Telser, J.; Schaefer, H.F., 3rd; Harrop, T.C. Toward functional Ni-SOD biomimetics: Achieving a structural/electronic correlation with redox dynamics. Inorg. Chem. 2011, 50, 9216–9218. [Google Scholar] [CrossRef] [PubMed]
- Bryngelson, P.A.; Arobo, S.E.; Pinkham, J.L.; Cabelli, D.E.; Maroney, M.J. Expression, reconstitution, and mutation of recombinant Streptomycescoelicolor NiSOD. J. Am. Chem. Soc. 2004, 126, 460–461. [Google Scholar] [CrossRef]
- Mathrubootham, V.; Thomas, J.; Staples, R.; McCraken, J.; Shearer, J.; Hegg, E.L. Bisamidate and mixed amine/amidate NiN2S2 complexes as models for nickel-containing acetyl coenzyme A synthase and superoxide dismutase: An experimental and computational study. Inorg. Chem. 2010, 49, 5393–5406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakane, D.; Wasada-Tsutsui, Y.; Funahashi, Y.; Hatanaka, T.; Ozawa, T.; Masuda, H. A novel square-planar Ni(II) complex with an amino-carboxamido-dithiolato-type ligand as an active-site model of NiSOD. Inorg. Chem. 2014, 53, 6512–6523. [Google Scholar] [CrossRef]
- Tian, Y.; Mao, L.; Okajima, T.; Ohsaka, T. Electrochemistry and electrocatalytic activities of superoxide dismutases at gold electrodes modified with a self-assembled monolayer. Anal. Chem. 2004, 76, 4162–4168. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.B.; Lee, J.W.; Davidson, G.; Yim, Y.I.; Bose, K.; Sharma, M.L.; Kang, S.O.; Cabelli, D.E.; Maroney, M.J. Examination of the nickel site structure and reaction mechanism in Streptomyces seoulensis superoxide dismutase. Biochemistry 1999, 38, 3744–3752. [Google Scholar] [CrossRef]
- Leveque, V.J.; Vance, C.K.; Nick, H.S.; Silverman, D.N. Redox properties of human manganese superoxide dismutase and active-site mutants. Biochemistry 2001, 40, 10586–10591. [Google Scholar] [CrossRef]
- Fee, J.A.; DiCorleto, P.E. Observations on the oxidation-reduction properties of bovine erythrocyte superoxide dismutase. Biochemistry 1973, 12, 4893–4899. [Google Scholar] [CrossRef]
- Shearer, J. Use of a metallopeptide-based mimic provides evidence for a proton-coupled electron-transfer mechanism for superoxide reduction by nickel-containing superoxide dismutase. Angew. Chem. Int. Ed. 2013, 52, 2569–2572. [Google Scholar] [CrossRef] [Green Version]
- Tietze, D.; Tischler, M.; Voigt, S.; Imhof, D.; Ohlenschlager, O.; Gorlach, M.; Buntkowsky, G. Development of a functional cis-prolyl bond biomimetic and mechanistic implications for nickel superoxide dismutase. Chemistry 2010, 16, 7572–7578. [Google Scholar] [CrossRef]
- Vance, C.K.; Miller, A.F. Spectroscopic comparisons of the pH dependencies of Fe-substituted (Mn)superoxide dismutase and Fe-superoxide dismutase. Biochemistry 1998, 37, 5518–5527. [Google Scholar] [CrossRef]
- Yamakura, F.; Kobayashi, K.; Ue, H.; Konno, M. The pH-dependent changes of the enzymic activity and spectroscopic properties of iron-substituted manganese superoxide dismutase. A study on the metal-specific activity of Mn-containing superoxide dismutase. Eur. J. Biochem. 1995, 227, 700–706. [Google Scholar] [CrossRef]
- Neupane, K.P.; Shearer, J. The influence of amine/amide versus bisamide coordination in nickel superoxide dismutase. Inorg. Chem. 2006, 45, 10552–10566. [Google Scholar] [CrossRef]
- Shearer, J. Dioxygen and superoxide stability of metallopeptide based mimics of nickel containing superoxide dismutase: The influence of amine/amidate vs. bis-amidate ligation. J. Inorg. Biochem. 2013, 129, 145–149. [Google Scholar] [CrossRef]
- Hanss, J.; Kruger, H.J. First isolation and structural characterization of a nickel(III) complex containing aliphatic thiolate donors. Angew. Chem. Int. Ed. 1998, 37, 360–363. [Google Scholar] [CrossRef]
- Harrop, T.C.; Olmstead, M.M.; Mascharak, P.K. Novel folding of N,N′-naphthalenebis(o-mercaptobenzamide) in nickel(II) complexes: Monomeric and trimeric species with unexpected ‘butterfly’ and ‘slant chair’ structure. Inorg. Chim. Acta 2002, 338, 189–195. [Google Scholar] [CrossRef]
- Kruger, H.J.; Peng, G.; Holm, R.H. Low-potential nickel(III,II) complexes: New systems based on tetradentate amidate-thiolate ligands and the influence of ligand structure on potentials in relation to the nickel site in [NiFe]-hydrogenases. Inorg. Chem. 2002, 30, 734–742. [Google Scholar] [CrossRef]
- Neupane, K.P.; Gearty, K.; Francis, A.; Shearer, J. Probing variable axial ligation in nickel superoxide dismutase utilizing metallopeptide-based models: Insight into the superoxide disproportionation mechanism. J. Am. Chem. Soc. 2007, 129, 14605–14618. [Google Scholar] [CrossRef]
- Prabhakar, R.; Morokuma, K.; Musaev, D.G. A DFT study of the mechanism of Ni superoxide dismutase (NiSOD): Role of the active site cysteine-6 residue in the oxidative half-reaction. J. Comput. Chem. 2006, 27, 1438–1445. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laskowski, R.A.; Macarthur, M.W.; Moss, D.S.; Thornton, J.M. Procheck—A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]









| Sample | No. of Ni Atoms per Protein | Extinction Coefficient (M−1 cm−1) | EPR |
|---|---|---|---|
| 317, 430, 590 nm | |||
| F598H | 0.7 | 7760, 1495, 615 | g = 2.23, 2.06, 2.00 |
| S594H | 0.8 | 7065, 1565, 640 | g = 2.23, 2.06, 2.00 |
| S594H-GP | 0.8 | 7685, 1490, 615 | g = 2.23, 2.06, 2.00 |
| EFG-F598H | 0.6 | 7905, 1440, 695 | g = 2.23, 2.06, 2.00 |
| YGP-F598H | 0.5 | 7450, 1445, 590 | g = 2.23, 2.06, 2.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Zhou, Y.; Yuan, H.; Liu, Y.; Lin, Y.-W.; Su, J.; Tan, X. Functional Conversion of Acetyl-Coenzyme a Synthase to a Nickel Superoxide Dismutase via Rational Design of Coordination Microenvironment for the Nid-Site. Int. J. Mol. Sci. 2022, 23, 2652. https://doi.org/10.3390/ijms23052652
Wei Y, Zhou Y, Yuan H, Liu Y, Lin Y-W, Su J, Tan X. Functional Conversion of Acetyl-Coenzyme a Synthase to a Nickel Superoxide Dismutase via Rational Design of Coordination Microenvironment for the Nid-Site. International Journal of Molecular Sciences. 2022; 23(5):2652. https://doi.org/10.3390/ijms23052652
Chicago/Turabian StyleWei, Yaozhu, Yajun Zhou, Hong Yuan, Yi Liu, Ying-Wu Lin, Jihu Su, and Xiangshi Tan. 2022. "Functional Conversion of Acetyl-Coenzyme a Synthase to a Nickel Superoxide Dismutase via Rational Design of Coordination Microenvironment for the Nid-Site" International Journal of Molecular Sciences 23, no. 5: 2652. https://doi.org/10.3390/ijms23052652