
Ferritinophagy and α-Synuclein: Pharmacological Targeting of Autophagy to Restore Iron Regulation in Parkinson’s Disease


 ,
,
Abstract
:1. Introduction
2. Roles of Iron, Calcium and α-Synuclein in Nigral Neurons
3. Synaptic Role of α-Synuclein
4. Iron and Calcium Regulation of α-Synuclein
4.1. Aggregation Induction of α-Synuclein
4.2. Iron-Induced Catecholamine Oxidation and Redox Damage
5. Iron Entry, Regulation and Cellular Metabolism
6. Iron Metabolism and Ferritin
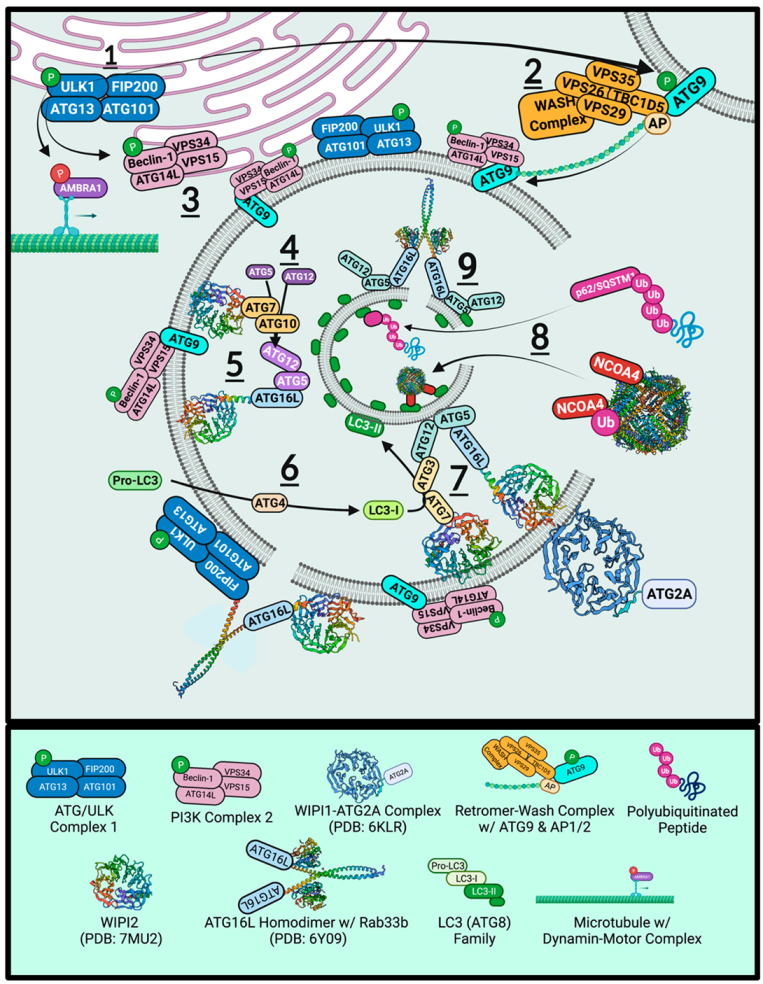
7. Autophagy and Ferritinophagy
7.1. ULK1
7.2. Retromer
7.3. ATG9
7.4. Beclin-1 and Vps34
7.5. WIPIs and ATGs
7.6. ATG2 Tethering and ATG16L1 Constriction
7.7. ATG8
7.8. NCOA4 and LC3-Interacting Region
7.9. p62/Sequestosome-1
7.10. SNARE Proteins
8. α-Synuclein Aggregates Inhibit Ferritinophagy
9. Therapeutic Targets for Ferritinophagy
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAAD | Aromatic amino acid decarboxylase |
| AD | Alzheimer’s disease |
| AKT1 | RAC-alpha serine/threonine-protein kinase |
| ALS | Amyotrophic lateral sclerosis |
| AMBRA1 | Autophagy and beclin-1 regulator 1 |
| AMP | Adenosine monophosphate |
| AMPK | AMP protein kinase |
| AP1/2 | Adaptor protein ½ |
| APP | Amyloid precursor protein |
| Arp2/3 | Actin-related proteins 2/3 |
| ATG9 | Autophagy-related protein 9 |
| ATG13 | Autophagy-related protein 13 |
| ATG101 | Autophagy-related protein 101 |
| BARA | β-α Repeated autophagy |
| BCL-2 | Apoptosis regulator B-cell lymphoma 2 Bcl-2 |
| BCL-XL | B-cell lymphoma-extra large |
| CALB1 | Calbindin-D28K |
| Cav1.2 | Voltage-dependent L-type calcium channel subunit α-1C |
| Cav1.3 | Calcium channel, voltage-dependent, L-type, α-1D subunit |
| CCD | Coiled-coil domain |
| CI-MPR | Cation-independent-mannose-6-phosphate receptor |
| CMA | Chaperone-mediated autophagy |
| COX-1 | Cyclooxygenase-1 |
| CSF | Cerebral spinal fluid |
| CSPα | Cysteine-string protein-α |
| DAT | Dopamine transporter |
| DLB | Dementia with Lewy bodies |
| DMT1 | Divalent metal transporter 1 |
| ECD | Evolutionarily conserved domain |
| ER | Endoplasmic reticulum |
| FAK | Focal adhesion kinase |
| Fe-S | Iron-sulfur cluster |
| FIP200 | FAK family kinase-interacting protein of 200 kDa |
| FPN1 | Ferroportin |
| Ft | Ferritin |
| FtH1 | Ferritin heavy chain |
| FtL | Ferritin light chain |
| FTLD | Frontotemporal lobar dementia |
| FtMt | Mitochondrial ferritin |
| GA | Ginkgolic acid |
| GABA | γ-Aminobutyric acid |
| GABARAP | γ-Aminobutyric acid receptor-associated protein |
| GABARAPL1 | γ-Aminobutyric acid receptor-associated protein-like 1 |
| GABARAPL2 | γ-Aminobutyric acid receptor-associated protein-like 2 |
| GABRR3 | γ-Aminobutyric acid receptor subunit rho-3 |
| HD | Huntington’s disease |
| HERC2 | E3 ubiquitin protein ligase 2 |
| HOPS | Homotypic fusion and vacuole protein sorting |
| HP1 | Hydrophobic pocket 1 |
| HRB | HIV-1 Rev-binding-protein |
| Hsc70 | Heat shock cognate 70 kDa |
| Hsp | Heat shock protein |
| Htt | Huntingtin |
| IRE | Iron-responsive element |
| IRP1 | Iron regulatory protein 1 |
| IMM | Inner-mitochondrial membrane |
| KO | Knockout |
| LAMP2a | Lysosomal-associated membrane protein 2a |
| LB | Lewy body |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3 |
| LIP | Labile iron pool |
| LIR | LC3 interacting region |
| LN | Lewy neurites |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MRI | Magnetic resonance imaging |
| MSA | Multiple system atrophy |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | mTOR complex 1 |
| Munc-13 | Mammalian uncoordinated 13 |
| Munc-18 | Mammalian uncoordinated 18 |
| MVB | Multi-vesicular body |
| MWW | Weighted-average molecular weight |
| NAC | Non-amyloid component |
| NBIA5 | Neurodegeneration with brain iron accumulation 5 |
| NCOA4 | Nuclear co-activator 4 |
| NLRP3 | NOD-like receptor protein 3 |
| NOD | Nucleotide binding domain |
| Nrf2 | Nuclear factor erythroid 2–related factor 2 |
| 6-OHDA | 6-Hydroxydopamine |
| PAWR | PRKC-apoptosis-WT1-regulator |
| PCBP2 | Poly-r(C)-binding protein 2 |
| PD | Parkinson’s disease |
| PE | Phosphatidylethanolamine |
| PI3KC3 | Phosphatidylinositol 3-kinase catalytic subunit type 3 |
| PI3P | Phosphatidylinositol 3-phosphate |
| PNN | Pedunculopontine nucleus |
| Rab1a | RAS-related protein in brain 1a |
| Rab7 | RAS-related protein Rab-7a |
| RB1CC1 | RB1-inducible coiled-coil protein 1 |
| RPE | Retinal pigmented epithelial cells |
| SENP | Sentrin-specific protease |
| SGT | Small glutamate-rich tetratricopeptide repeat-containing protein |
| SMV | Small single membrane vesicles |
| SNAP25 | Synaptosomal-associated protein of 25 kDa |
| SNARE | Soluble N-ethylmaleimide sensitive attachment factor receptor |
| SNpc | Substantia nigra pars compacta |
| SNpr | Substantia nigra pars reticulata |
| SQSTM1 | Sequestosome-1 (p62) |
| STEAP1 | Six transmembrane epithelial antigen of the prostate-1 |
| STX17 | Syntaxin-17 |
| SUMO1 | Small-ubiquitin-like modifier 1 |
| SYNGAP | Synaptic Ras-like GTPase-activating protein 1 |
| TBC1D5 | TBC1 domain family member 5 |
| TFEB | Transcription factor EB |
| TFR1 | Transferrin receptor 1 |
| TFR2 | Transferrin receptor 2 |
| TH | Tyrosine hydroxylase |
| TGN | trans-Golgi network |
| TLR4 | Toll-like receptor 4 |
| TOM20 | Translocase of outer membrane of 20 kDa |
| TOMM20 | Mitochondrial import receptor subunit TOM20 homolog |
| t-SNARE | Target SNARE |
| UBA | Ubiquitin-associated |
| ULK1 | Unc-51 like autophagy activating kinase |
| Unc-51 | Serine/threonine-protein kinase unc-51 |
| UPS | Ubiquitin/proteasome system |
| UVRAG | UV radiation resistance-associated gene protein |
| VAMP2 | Vesicle-associated membrane protein 2 (also known as synaptobrevin) |
| VGCC | Voltage-gated calcium channel |
| VMAT2 | Vesicular-monoamine transporter |
| VPS26 | Vacuolar protein sorting-associated protein 26A |
| VPS29 | Vacuolar protein sorting-associated protein 29 |
| VPS35 | VPS35 endosomal protein-sorting factor-like |
| v-SNARE | Vesicular SNARE |
| WASH | Wiskott–Aldrich syndrome protein and SCAR homologue |
| WIPI2 | WD repeat domain phosphoinositide-interacting protein 2 |
References
- Parkinson, J. An essay on the shaking palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, I.; Vachal, E.; Tomita, T. Dense core vesicles around the Lewy body in incidental Parkinson’s disease: An electron microscopic study. Acta Neuropathol. 1977, 39, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Choong, C.J.; Baba, K. Parkinson’s disease and iron. J. Neural Transm 2020, 127, 181–187. [Google Scholar] [CrossRef]
- Singh, P.K.; Muqit, M.M.K. Parkinson’s: A Disease of Aberrant Vesicle Trafficking. Annu. Rev. Cell Dev. Biol. 2020, 36, 237–264. [Google Scholar] [CrossRef]
- Abeyawardhane, D.L.; Lucas, H.R. Iron Redox Chemistry and Implications in the Parkinson’s Disease Brain. Oxid. Med. Cell Longev. 2019, 2019, 4609702. [Google Scholar] [CrossRef] [Green Version]
- Dexter, D.T.; Carayon, A.; Javoy-Agid, F.; Agid, Y.; Wells, F.R.; Daniel, S.E.; Lees, A.J.; Jenner, P.; Marsden, C.D. Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 1991, 114, 1953–1975. [Google Scholar] [CrossRef]
- Zecca, L.; Shima, T.; Stroppolo, A.; Goj, C.; Battiston, G.A.; Gerbasi, R.; Sarna, T.; Swartz, H.M. Interaction of neuromelanin and iron in substantia nigra and other areas of human brain. Neuroscience 1996, 73, 407–415. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Kaji, S.; Maki, T.; Ishimoto, T.; Yamakado, H.; Takahashi, R. Insights into the pathogenesis of multiple system atrophy: Focus on glial cytoplasmic inclusions. Transl. Neurodegener. 2020, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Calo, L.; Wegrzynowicz, M.; Santivanez-Perez, J.; Grazia Spillantini, M. Synaptic failure and alpha-synuclein. Mov. Disord. 2016, 31, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Di Marco Vieira, B.; Radford, R.A.W.; Hayashi, J.; Eaton, E.D.; Greenaway, B.; Jambas, M.; Petcu, E.B.; Chung, R.S.; Pountney, D.L. Extracellular Alpha-Synuclein Promotes a Neuroinhibitory Secretory Phenotype in Astrocytes. Life 2020, 10, 183. [Google Scholar] [CrossRef] [PubMed]
- Moons, R.; Konijnenberg, A.; Mensch, C.; Van Elzen, R.; Johannessen, C.; Maudsley, S.; Lambeir, A.M.; Sobott, F. Metal ions shape alpha-synuclein. Sci. Rep. 2020, 10, 16293. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Day, J.R.; Connor, J.R.; Beard, J.L. H and L ferritin subunit mRNA expression differs in brains of control and iron-deficient rats. J. Nutr. 2002, 132, 2769–2774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, T.A.; Zhang, D.L.; Jeong, S.Y. Brain iron homeostasis, the choroid plexus, and localization of iron transport proteins. Metab. Brain Dis. 2009, 24, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Meyron-Holtz, E.G.; Cohen, L.A.; Fahoum, L.; Haimovich, Y.; Lifshitz, L.; Magid-Gold, I.; Stuemler, T.; Truman-Rosentsvit, M. Ferritin polarization and iron transport across monolayer epithelial barriers in mammals. Front. Pharmacol. 2014, 5, 194. [Google Scholar] [CrossRef] [Green Version]
- Truman-Rosentsvit, M.; Berenbaum, D.; Spektor, L.; Cohen, L.A.; Belizowsky-Moshe, S.; Lifshitz, L.; Ma, J.; Li, W.; Kesselman, E.; Abutbul-Ionita, I.; et al. Ferritin is secreted via 2 distinct nonclassical vesicular pathways. Blood 2018, 131, 342–352. [Google Scholar] [CrossRef]
- Everett, J.; Brooks, J.; Lermyte, F.; O’Connor, P.B.; Sadler, P.J.; Dobson, J.; Collingwood, J.F.; Telling, N.D. Iron stored in ferritin is chemically reduced in the presence of aggregating Abeta(1-42). Sci. Rep. 2020, 10, 10332. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Health and Disease. Pharmaceuticals 2018, 11, 114. [Google Scholar] [CrossRef] [Green Version]
- Tang, M.; Chen, Z.; Wu, D.; Chen, L. Ferritinophagy/ferroptosis: Iron-related newcomers in human diseases. J. Cell Physiol. 2018, 233, 9179–9190. [Google Scholar] [CrossRef]
- Quiles Del Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, G.K.; Butcher, L.L. Pars compacta of the substantia nigra modulates motor activity but is not involved importantly in regulating food and water intake. Naunyn. Schmiedebergs Arch. Pharmacol. 1980, 313, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Reimao, S.; Carvalho, M.; Nunes, R.G.; Abreu, D.; Guedes, L.C.; Bouca, R.; Lobo, P.P.; Godinho, C.; Coelho, M.; et al. Substantia Nigra Neuromelanin as an Imaging Biomarker of Disease Progression in Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.L.; Sinton, C.M.; Sonsalla, P.K.; German, D.C. Midbrain dopaminergic neurons in the mouse that contain calbindin-D28k exhibit reduced vulnerability to MPTP-induced neurodegeneration. Neurodegeneration 1996, 5, 313–318. [Google Scholar] [CrossRef] [PubMed]
- de Berker, A.O.; Rutledge, R.B. A role for the human substantia nigra in reinforcement learning. J. Neurosci. 2014, 34, 12947–12949. [Google Scholar] [CrossRef] [Green Version]
- Galtieri, D.J.; Estep, C.M.; Wokosin, D.L.; Traynelis, S.; Surmeier, D.J. Pedunculopontine glutamatergic neurons control spike patterning in substantia nigra dopaminergic neurons. Elife 2017, 6, e30352. [Google Scholar] [CrossRef]
- Sonne, J.; Reddy, V.; Beato, M.R. Neuroanatomy, Substantia Nigra. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Brichta, L.; Greengard, P. Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: An update. Front. Neuroanat. 2014, 8, 152. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Palacino, J. A novel human embryonic stem cell-derived Huntington’s disease neuronal model exhibits mutant huntingtin (mHTT) aggregates and soluble mHTT-dependent neurodegeneration. FASEB J. 2013, 27, 1820–1829. [Google Scholar] [CrossRef] [Green Version]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Melland, H.; Carr, E.M.; Gordon, S.L. Disorders of synaptic vesicle fusion machinery. J. Neurochem. 2021, 157, 130–164. [Google Scholar] [CrossRef]
- Bradbury, A.; Bagel, J.; Sampson, M.; Farhat, N.; Ding, W.; Swain, G.; Prociuk, M.; O’Donnell, P.; Drobatz, K.; Gurda, B.; et al. Cerebrospinal Fluid Calbindin D Concentration as a Biomarker of Cerebellar Disease Progression in Niemann-Pick Type C1 Disease. J. Pharmacol. Exp. Ther. 2016, 358, 254–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, N.A.; Ghio, A.J.; Garrick, L.M.; Garrick, M.D.; Zhao, L.; Fenton, R.A.; Thevenod, F. Evidence for mitochondrial localization of divalent metal transporter 1 (DMT1). FASEB J. 2014, 28, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Xu, H.; Shi, L.; Jiang, Z.; Song, N.; Jiang, H.; Xie, J. Activation of ATP-sensitive potassium channels enhances DMT1-mediated iron uptake in SK-N-SH cells in vitro. Sci. Rep. 2016, 6, 33674. [Google Scholar] [CrossRef] [PubMed]
- Bazelon, M.; Fenichel, G.M.; Randall, J. Studies on neuromelanin. I. A melanin system in the human adult brainstem. Neurology 1967, 17, 512–519. [Google Scholar] [CrossRef]
- Carballo-Carbajal, I.; Laguna, A.; Romero-Gimenez, J.; Cuadros, T.; Bove, J.; Martinez-Vicente, M.; Parent, A.; Gonzalez-Sepulveda, M.; Penuelas, N.; Torra, A.; et al. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson’s disease pathogenesis. Nat. Commun. 2019, 10, 973. [Google Scholar] [CrossRef] [Green Version]
- Lautenschlager, J.; Stephens, A.D.; Fusco, G.; Strohl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [Green Version]
- Trexler, A.J.; Rhoades, E. N-Terminal acetylation is critical for forming alpha-helical oligomer of alpha-synuclein. Protein Sci. 2012, 21, 601–605. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.; Pan, B.; Gottlieb, L.; Petersson, E.J.; Marmorstein, R. Molecular basis for N-terminal alpha-synuclein acetylation by human NatB. Elife 2020, 9, e57491. [Google Scholar] [CrossRef]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein alpha-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.H.C.; Wislet-Gendebien, S.; Samuel, F.; Visanji, N.P.; Zhang, G.; Marsilio, D.; Langman, T.; Fraser, P.E.; Tandon, A. alpha-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J. Biol. Chem. 2013, 288, 7438–7449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci. Rep. 2016, 6, 24475. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Hong, W.; Lev, S. Tethering the assembly of SNARE complexes. Trends Cell Biol. 2014, 24, 35–43. [Google Scholar] [CrossRef]
- Hawk, B.J.D.; Khounlo, R.; Shin, Y.K. Alpha-Synuclein Continues to Enhance SNARE-Dependent Vesicle Docking at Exorbitant Concentrations. Front. Neurosci. 2019, 13, 216. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Wang, L.; Bao, H.; Premi, S.; Das, U.; Chapman, E.R.; Roy, S. Functional cooperation of alpha-synuclein and VAMP2 in synaptic vesicle recycling. Proc. Natl. Acad. Sci. USA 2019, 116, 11113–11115. [Google Scholar] [CrossRef] [Green Version]
- Schoch, S.; Deak, F.; Konigstorfer, A.; Mozhayeva, M.; Sara, Y.; Sudhof, T.C.; Kavalali, E.T. SNARE function analyzed in synaptobrevin/VAMP knockout mice. Science 2001, 294, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.J.; Markovic, S.; Puchkov, D.; Mahrenholz, C.C.; Beceren-Braun, F.; Maritzen, T.; Dernedde, J.; Volkmer, R.; Oschkinat, H.; Haucke, V. SNARE motif-mediated sorting of synaptobrevin by the endocytic adaptors clathrin assembly lymphoid myeloid leukemia (CALM) and AP180 at synapses. Proc. Natl. Acad. Sci. USA 2011, 108, 13540–13545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burre, J.; Sharma, M.; Sudhof, T.C. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgoyne, R.D.; Morgan, A. Cysteine string protein (CSP) and its role in preventing neurodegeneration. Semin. Cell Dev. Biol. 2015, 40, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Burre, J.; Sudhof, T.C. CSPalpha promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 2011, 13, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Burre, J.; Bronk, P.; Zhang, Y.; Xu, W.; Sudhof, T.C. CSPalpha knockout causes neurodegeneration by impairing SNAP-25 function. EMBO J. 2012, 31, 829–841. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Pang, Z.P.; Yang, X.; Zhang, Y.; Rosenmund, C.; Bacaj, T.; Sudhof, T.C. Syntaxin-1 N-peptide and Habc-domain perform distinct essential functions in synaptic vesicle fusion. EMBO J. 2013, 32, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, Y.; Gong, J.; Ye, S.; Yang, X.; Zhang, R.; Ma, C. Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nat. Commun. 2019, 10, 69. [Google Scholar] [CrossRef] [Green Version]
- Shao, K.; Li, F.; Yang, Y.; Wang, N.; Gao, X.D.; Nakanishi, H. Characteristics of SNARE proteins are defined by distinctive properties of SNARE motifs. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129658. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.J.; Sierecki, E.; Tomatis, V.M.; Gormal, R.S.; Giles, N.; Morrow, I.C.; Xia, D.; Gotz, J.; Parton, R.G.; Collins, B.M.; et al. Munc18-1 is a molecular chaperone for alpha-synuclein, controlling its self-replicating aggregation. J. Cell Biol. 2016, 214, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.K.; Huynh, C.; Proux-Gillardeaux, V.; Galli, T.; Andrews, N.W. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J. Biol. Chem. 2004, 279, 20471–20479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Yang, Y.; Su, Z.; Hyeon, C.; Lee, T.S.; Lee, H.W.; Kweon, D.H.; Shin, Y.K.; Yoon, T.Y. Dynamic Ca2+-dependent stimulation of vesicle fusion by membrane-anchored synaptotagmin 1. Science 2010, 328, 760–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Zhou, P.; Wang, A.L.; Wu, D.; Zhao, M.; Sudhof, T.C.; Brunger, A.T. The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 2017, 548, 420–425. [Google Scholar] [CrossRef] [Green Version]
- Abeliovich, A.; Schmitz, Y.; Farinas, I.; Choi-Lundberg, D.; Ho, W.H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.; Armanini, M.; Ryan, A.; et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef] [Green Version]
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine Transporter Activity Is Modulated by alpha-Synuclein. J. Biol. Chem. 2015, 290, 29542–29554. [Google Scholar] [CrossRef] [Green Version]
- Ishibashi, K.; Oda, K.; Ishiwata, K.; Ishii, K. Comparison of dopamine transporter decline in a patient with Parkinson’s disease and normal aging effect. J. Neurol. Sci. 2014, 339, 207–209. [Google Scholar] [CrossRef]
- Egana, L.A.; Cuevas, R.A.; Baust, T.B.; Parra, L.A.; Leak, R.K.; Hochendoner, S.; Pena, K.; Quiroz, M.; Hong, W.C.; Dorostkar, M.M.; et al. Physical and functional interaction between the dopamine transporter and the synaptic vesicle protein synaptogyrin-.-3. J. Neurosci. 2009, 29, 4592–4604. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.A. Synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baksi, S.; Singh, N. alpha-Synuclein impairs ferritinophagy in the retinal pigment epithelium: Implications for retinal iron dyshomeostasis in Parkinson’s disease. Sci. Rep. 2017, 7, 12843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leandrou, E.; Emmanouilidou, E.; Vekrellis, K. Voltage-Gated Calcium Channels and alpha-Synuclein: Implications in Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 237. [Google Scholar] [CrossRef] [PubMed]
- Llinas, R.; Sugimori, M.; Silver, R.B. Microdomains of high calcium concentration in a presynaptic terminal. Science 1992, 256, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Okita, Y.; Rcom-H’cheo-Gauthier, A.N.; Goulding, M.; Chung, R.S.; Faller, P.; Pountney, D.L. Metallothionein, copper and alpha-Synuclein in alpha-synucleinopathies. Front. Neurosci. 2017, 11, 114. [Google Scholar] [CrossRef]
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-synuclein is a cellular ferrireductase. PLoS ONE 2011, 6, e15814. [Google Scholar] [CrossRef]
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Interaction of alpha-synuclein with divalent metal ions reveals key differences: A link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [Google Scholar] [CrossRef]
- Chen, B.; Wen, X.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Interactions between iron and alpha-synuclein pathology in Parkinson’s disease. Free Radic. Biol. Med. 2019, 141, 253–260. [Google Scholar] [CrossRef]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G. The Interplay between Ca(2+) Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef] [Green Version]
- Serpell, L.C.; Berriman, J.; Jakes, R.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar] [CrossRef] [Green Version]
- Pountney, D.L.; Voelcker, N.H.; Gai, W.P. Annular alpha-synuclein oligomers are potentially toxic agents in alpha-synucleinopathy. Hypothesis. Neurotox. Res. 2005, 7, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Guiney, S.J.; Adlard, P.A.; Lei, P.; Mawal, C.H.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Fibrillar alpha-synuclein toxicity depends on functional lysosomes. J. Biol. Chem. 2020, 295, 17497–17513. [Google Scholar] [CrossRef] [PubMed]
- Hindeya Gebreyesus, H.; Gebrehiwot Gebremichael, T. The Potential Role of Astrocytes in Parkinson’s Disease (PD). Med. Sci. 2020, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Friedlich, A.L.; Tanzi, R.E.; Rogers, J.T. The 5’-untranslated region of Parkinson’s disease alpha-synuclein messengerRNA contains a predicted iron responsive element. Mol. Psychiatry 2007, 12, 222–223. [Google Scholar] [CrossRef]
- Ma, L.; Gholam Azad, M.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. 2021, 41, 101896. [Google Scholar] [CrossRef]
- Hare, D.J.; Double, K.L. Iron and dopamine: A toxic couple. Brain 2016, 139, 1026–1035. [Google Scholar] [CrossRef]
- Zucca, F.A.; Segura-Aguilar, J.; Ferrari, E.; Munoz, P.; Paris, I.; Sulzer, D.; Sarna, T.; Casella, L.; Zecca, L. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog. Neurobiol. 2017, 155, 96–119. [Google Scholar] [CrossRef]
- Shima, T.; Sarna, T.; Swartz, H.M.; Stroppolo, A.; Gerbasi, R.; Zecca, L. Binding of iron to neuromelanin of human substantia nigra and synthetic melanin: An electron paramagnetic resonance spectroscopy study. Free Radic. Biol. Med. 1997, 23, 110–119. [Google Scholar] [CrossRef]
- Ren, J.X.; Sun, X.; Yan, X.L.; Guo, Z.N.; Yang, Y. Ferroptosis in Neurological Diseases. Front. Cell Neurosci. 2020, 14, 218. [Google Scholar] [CrossRef]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef] [Green Version]
- Sulzer, D.; Cassidy, C.; Horga, G.; Kang, U.J.; Fahn, S.; Casella, L.; Pezzoli, G.; Langley, J.; Hu, X.P.; Zucca, F.A.; et al. Neuromelanin detection by magnetic resonance imaging (MRI) and its promise as a biomarker for Parkinson’s disease. NPJ Parkinsons Dis. 2018, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Laguna, A.; Carballo-Carbajal, I. Intracellular crowding by age-dependent neuromelanin accumulation disrupts neuronal proteostasis and triggers Parkinson disease pathology. Autophagy 2019, 15, 2028–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- del Toro, D.; Alberch, J.; Lazaro-Dieguez, F.; Martin-Ibanez, R.; Xifro, X.; Egea, G.; Canals, J.M. Mutant huntingtin impairs post-Golgi trafficking to lysosomes by delocalizing optineurin/Rab8 complex from the Golgi apparatus. Mol. Biol. Cell 2009, 20, 1478–1492. [Google Scholar] [CrossRef] [Green Version]
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: The case for isradipine. Biochim. Biophys. Acta 2011, 1812, 1584–1590. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.J.; Yun, S.M.; Jo, C.; Lee, D.H.; Choi, K.J.; Song, J.C.; Park, S.I.; Kim, Y.J.; Koh, Y.H. SUMO1 promotes Abeta production via the modulation of autophagy. Autophagy 2015, 11, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Tadokoro, K.; Yamashita, T.; Shang, J.; Ohta, Y.; Nomura, E.; Morihara, R.; Omote, Y.; Takemoto, M.; Abe, K. Switching the Proteolytic System from the Ubiquitin-Proteasome System to Autophagy in the Spinal Cord of an Amyotrophic Lateral Sclerosis Mouse Model. Neuroscience 2021, 466, 47–57. [Google Scholar] [CrossRef]
- Mayle, K.M.; Le, A.M.; Kamei, D.T. The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta 2012, 1820, 264–281. [Google Scholar] [CrossRef] [Green Version]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef]
- Wilson, R.L.; Wightman, R.M. Systemic and Nigral Application of Amphetamine Both Cause an Increase in Extracellular Concentration of Ascorbate in the Caudate-Nucleus of the Rat. Brain Res. 1985, 339, 219–226. [Google Scholar] [CrossRef]
- Schenk, J.O.; Miller, E.; Gaddis, R.; Adams, R.N. Homeostatic control of ascorbate concentration in CNS extracellular fluid. Brain Res. 1982, 253, 353–356. [Google Scholar] [CrossRef]
- Boag, M.K.; Ma, L.; Mellick, G.D.; Pountney, D.L.; Feng, Y.; Quinn, R.J.; Liew, A.W.-C.; Dharmasivam, M.; Azad, M.G.; Afroz, R.; et al. Calcium channels and iron metabolism: A redox catastrophe in Parkinson’s disease and an innovative path to novel therapies? Redox Biol. 2021, 47, 102136. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, A.B.; Johansen, T. Autophagy and endocytosis—interconnections and interdependencies. J. Cell Sci. 2020, 133, jcs228114. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Yang, L.; Ma, Y.; Li, X.; Yang, S.; Li, Y.; Wu, B.; Tang, S.; Zhang, F.; Zhang, B.; et al. IKKbeta activation promotes amphisome formation and extracellular vesicle secretion in tumor cells. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118857. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.E.; Colon-Ramos, D.A. The Journey of the Synaptic Autophagosome: A Cell Biological Perspective. Neuron 2020, 105, 961–973. [Google Scholar] [CrossRef] [Green Version]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef]
- Al-Refaei, M.A.; Makki, R.M.; Ali, H.M. Structure prediction of transferrin receptor protein 1 (TfR1) by homology modelling, docking, and molecular dynamics simulation studies. Heliyon 2020, 6, e03221. [Google Scholar] [CrossRef]
- Salazar, J.; Mena, N.; Hunot, S.; Prigent, A.; Alvarez-Fischer, D.; Arredondo, M.; Duyckaerts, C.; Sazdovitch, V.; Zhao, L.; Garrick, L.M.; et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 18578–18583. [Google Scholar] [CrossRef] [Green Version]
- Skjorringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Bi, M.; Du, X.; Jiao, Q.; Liu, Z.; Jiang, H. alpha-Synuclein Regulates Iron Homeostasis via Preventing Parkin-Mediated DMT1 Ubiquitylation in Parkinson’s Disease Models. ACS Chem. Neurosci. 2020, 11, 1682–1691. [Google Scholar] [CrossRef]
- Tchernitchko, D.; Bourgeois, M.; Martin, M.E.; Beaumont, C. Expression of the two mRNA isoforms of the iron transporter Nramp2/DMTI in mice and function of the iron responsive element. Biochem. J. 2002, 363, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Foot, N.J.; Dalton, H.E.; Shearwin-Whyatt, L.M.; Dorstyn, L.; Tan, S.S.; Yang, B.; Kumar, S. Regulation of the divalent metal ion transporter DMT1 and iron homeostasis by a ubiquitin-dependent mechanism involving Ndfips and WWP2. Blood 2008, 112, 4268–4275. [Google Scholar] [CrossRef] [PubMed]
- You, F.; Sun, H.; Zhou, X.; Sun, W.; Liang, S.; Zhai, Z.; Jiang, Z. PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat. Immunol. 2009, 10, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef] [Green Version]
- Frey, A.G.; Nandal, A.; Park, J.H.; Smith, P.M.; Yabe, T.; Ryu, M.S.; Ghosh, M.C.; Lee, J.; Rouault, T.A.; Park, M.H.; et al. Iron chaperones PCBP1 and PCBP2 mediate the metallation of the dinuclear iron enzyme deoxyhypusine hydroxylase. Proc. Natl. Acad. Sci. USA 2014, 111, 8031–8036. [Google Scholar] [CrossRef] [Green Version]
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. The new role of poly (rC)-binding proteins as iron transport chaperones: Proteins that could couple with inter-organelle interactions to safely traffic iron. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129685. [Google Scholar] [CrossRef]
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. The iron chaperone poly(rC)-binding protein 2 forms a metabolon with the heme oxygenase 1/cytochrome P450 reductase complex for heme catabolism and iron transfer. J. Biol. Chem. 2017, 292, 13205–13229. [Google Scholar] [CrossRef] [Green Version]
- Bae, D.H.; Lane, D.J.R.; Siafakas, A.R.; Sutak, R.; Paluncic, J.; Huang, M.L.H.; Jansson, P.J.; Rahmanto, Y.S.; Richardson, D.R. Acireductone dioxygenase 1 (ADI1) is regulated by cellular iron by a mechanism involving the iron chaperone, PCBP1, with PCBP2 acting as a potential co-chaperone. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165844. [Google Scholar] [CrossRef]
- Harrison, P.M. The structure and function of ferritin. Biochem. Educ. 1986, 14, 154–162. [Google Scholar] [CrossRef]
- Melman, A.; Bou-Abdallah, F. Iron mineralization and core dissociation in mammalian homopolymeric H-ferritin: Current understanding and future perspectives. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129700. [Google Scholar] [CrossRef]
- Arosio, P.; Elia, L.; Poli, M. Ferritin, cellular iron storage and regulation. IUBMB Life 2017, 69, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Campanella, A.; Rovelli, E.; Santambrogio, P.; Cozzi, A.; Taroni, F.; Levi, S. Mitochondrial ferritin limits oxidative damage regulating mitochondrial iron availability: Hypothesis for a protective role in Friedreich ataxia. Hum. Mol. Genet. 2009, 18, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, H.; Yang, H.; Yang, M.; Yanagisawa, D.; Bellier, J.P.; Mori, M.; Takahata, S.; Nonaka, T.; Zhao, S.; Tooyama, I. Mitochondrial ferritin protects SH-SY5Y cells against H2O2-induced oxidative stress and modulates alpha-synuclein expression. Exp. Neurol. 2017, 291, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fiskus, W.; Yong, B.; Atadja, P.; Takahashi, Y.; Pandita, T.K.; Wang, H.G.; Bhalla, K.N. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 6841–6846. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.; Huang, M.L.H.; Park, K.C.; Richardson, D.R. Antioxidant defense mechanisms and its dysfunctional regulation in the mitochondrial disease, Friedreich’s ataxia. Free Radic. Biol. Med. 2020, 159, 177–188. [Google Scholar] [CrossRef]
- Perfeito, R.; Lazaro, D.F.; Outeiro, T.F.; Rego, A.C. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell Neurosci. 2014, 62, 51–59. [Google Scholar] [CrossRef]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef] [Green Version]
- Valdinocci, D.; Simoes, R.F.; Kovarova, J.; Cunha-Oliveira, T.; Neuzil, J.; Pountney, D.L. Intracellular and Intercellular Mitochondrial Dynamics in Parkinson’s Disease. Front. Neurosci. 2019, 13, 930. [Google Scholar] [CrossRef] [Green Version]
- Valdinocci, D.; Kovarova, J.; Neuzil, J.; Pountney, D.L. Alpha-Synuclein Aggregates Associated with Mitochondria in Tunnelling Nanotubes. Neurotox. Res. 2021, 39, 429–443. [Google Scholar] [CrossRef]
- Mortimore, G.E.; Schworer, C.M. Induction of autophagy by amino-acid deprivation in perfused rat liver. Nature 1977, 270, 174–176. [Google Scholar] [CrossRef]
- Mortimore, G.E.; Poso, A.R. Lysosomal pathways in hepatic protein degradation: Regulatory role of amino acids. Fed. Proc. 1984, 43, 1289–1294. [Google Scholar] [PubMed]
- Nagy, M.; Fenton, W.A.; Li, D.; Furtak, K.; Horwich, A.L. Extended survival of misfolded G85R SOD1-linked ALS mice by transgenic expression of chaperone Hsp110. Proc. Natl. Acad. Sci. USA 2016, 113, 5424–5428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, D.M.; Balch, W.E. Q-bodies monitor the quinary state of the protein fold. Nat. Cell Biol. 2013, 15, 1137–1139. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.H.; Nichols, J.G.; Hsu, C.W.; Crooke, S.T. Hsc70 Facilitates Mannose-6-Phosphate Receptor-Mediated Intracellular Trafficking and Enhances Endosomal Release of Phosphorothioate-Modified Antisense Oligonucleotides. Nucleic Acid Ther. 2021, 31, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Mollapour, M.; Bourboulia, D.; Beebe, K.; Woodford, M.R.; Polier, S.; Hoang, A.; Chelluri, R.; Li, Y.; Guo, A.; Lee, M.J.; et al. Asymmetric Hsp90 N domain SUMOylation recruits Aha1 and ATP-competitive inhibitors. Mol. Cell 2014, 53, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Chen, Z.; Yu, F.; Chen, Q.; Tian, Y.; Ma, S.; Wang, T.; Liu, X. Hsp90 regulates autophagy and plays a role in cancer therapy. Tumour. Biol. 2016, 37, 1–6. [Google Scholar] [CrossRef]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. In Autophagosome and Phagosome; Deretic, V., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 77–88. [Google Scholar]
- Heras-Sandoval, D.; Perez-Rojas, J.M.; Hernandez-Damian, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.T.; Shahnazari, S.; Brech, A.; Lamark, T.; Johansen, T.; Brumell, J.H. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 2009, 183, 5909–5916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Zhuang, J.; Ji, X.; Zhu, Y.; Liu, W.; Sun, J.; Jiao, X.; Xu, X. Restriction of intracellular Salmonella typhimurium growth by the small-molecule autophagy inducer A77 1726 through the activation of the AMPK-ULK1 axis. Vet. Microbiol. 2021, 254, 108982. [Google Scholar] [CrossRef]
- Wang, C.; Wang, H.; Zhang, D.; Luo, W.; Liu, R.; Xu, D.; Diao, L.; Liao, L.; Liu, Z. Phosphorylation of ULK1 affects autophagosome fusion and links chaperone-mediated autophagy to macroautophagy. Nat. Commun. 2018, 9, 3492. [Google Scholar] [CrossRef]
- Moreau, K.; Ravikumar, B.; Renna, M.; Puri, C.; Rubinsztein, D.C. Autophagosome precursor maturation requires homotypic fusion. Cell 2011, 146, 303–317. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Lim, J.; Lachenmayer, M.L.; Wu, S.; Liu, W.; Kundu, M.; Wang, R.; Komatsu, M.; Oh, Y.J.; Zhao, Y.; Yue, Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015, 11, e1004987. [Google Scholar] [CrossRef]
- Menon, M.B.; Dhamija, S. Beclin 1 Phosphorylation—at the Center of Autophagy Regulation. Front. Cell Dev. Biol. 2018, 6, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popoff, V.; Mardones, G.A.; Bai, S.K.; Chambon, V.; Tenza, D.; Burgos, P.V.; Shi, A.; Benaroch, P.; Urbe, S.; Lamaze, C.; et al. Analysis of articulation between clathrin and retromer in retrograde sorting on early endosomes. Traffic 2009, 10, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Tabuchi, M.; Yanatori, I.; Kawai, Y.; Kishi, F. Retromer-mediated direct sorting is required for proper endosomal recycling of the mammalian iron transporter DMT1. J. Cell Sci. 2010, 123, 756–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilarino-Guell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Benet-Pages, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Gao, K.; Jankovic, J. The VPS35 gene and Parkinson’s disease. Mov. Disord. 2013, 28, 569–575. [Google Scholar] [CrossRef]
- MacLeod, D.A.; Rhinn, H.; Kuwahara, T.; Zolin, A.; Di Paolo, G.; McCabe, B.D.; Marder, K.S.; Honig, L.S.; Clark, L.N.; Small, S.A.; et al. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron 2013, 77, 425–439. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Orgaz, A.; Kvainickas, A.; Nagele, H.; Denner, J.; Eimer, S.; Dengjel, J.; Steinberg, F. Control of RAB7 activity and localization through the retromer-TBC1D5 complex enables RAB7-dependent mitophagy. EMBO J. 2018, 37, 235–254. [Google Scholar] [CrossRef]
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef]
- Ishiguro, M.; Li, Y.; Yoshino, H.; Daida, K.; Ishiguro, Y.; Oyama, G.; Saiki, S.; Funayama, M.; Hattori, N.; Nishioka, K. Clinical manifestations of Parkinson’s disease harboring VPS35 retromer complex component p.D620N with long-term follow-up. Parkinsonism Relat. Disord. 2021, 84, 139–143. [Google Scholar] [CrossRef]
- Toffoli, M.; Vieira, S.R.L.; Schapira, A.H.V. Genetic causes of PD: A pathway to disease modification. Neuropharmacology 2020, 170, 108022. [Google Scholar] [CrossRef] [PubMed]
- King, J.S.; Gueho, A.; Hagedorn, M.; Gopaldass, N.; Leuba, F.; Soldati, T.; Insall, R.H. WASH is required for lysosomal recycling and efficient autophagic and phagocytic digestion. Mol. Biol. Cell 2013, 24, 2714–2726. [Google Scholar] [CrossRef] [PubMed]
- Freeman, C.L.; Hesketh, G.; Seaman, M.N. RME-8 coordinates the activity of the WASH complex with the function of the retromer SNX dimer to control endosomal tubulation. J. Cell Sci. 2014, 127, 2053–2070. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Klionsky, D.J. Autophagic membrane delivery through ATG9. Cell Res. 2017, 27, 161–162. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, C.A.; Joachim, J.; Tooze, S.A. Chapter Two—Quantifying Autophagic Structures in Mammalian Cells Using Confocal Microscopy. In Methods in Enzymology; Galluzzi, L., Bravo-San Pedro, J.M., Kroemer, G., Eds.; Academic Press: New York, NY, USA, 2017; Volume 587, pp. 21–42. [Google Scholar]
- Kulkarni, V.V.; Maday, S. Compartment-specific dynamics and functions of autophagy in neurons. Dev. Neurobiol. 2018, 78, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Patoli, D.; Mignotte, F.; Deckert, V.; Dusuel, A.; Dumont, A.; Rieu, A.; Jalil, A.; Van Dongen, K.; Bourgeois, T.; Gautier, T.; et al. Inhibition of mitophagy drives macrophage activation and antibacterial defense during sepsis. J. Clin. Investig. 2020, 130, 5858–5874. [Google Scholar] [CrossRef]
- Mallard, F.; Tang, B.L.; Galli, T.; Tenza, D.; Saint-Pol, A.; Yue, X.; Antony, C.; Hong, W.; Goud, B.; Johannes, L. Early/recycling endosomes-to-TGN transport involves two SNARE complexes and a Rab6 isoform. J. Cell Biol. 2002, 156, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. alpha-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [Green Version]
- Haack, T.B.; Hogarth, P.H.; Kruer, M.C.; Gregory, A.; Wieland, T.; Schwarzmayr, T.; Graf, E.; Sanford, L.; Meyer, E.; Kara, E.; et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 2012, 91, 1144–1149. [Google Scholar] [CrossRef] [Green Version]
- Saitsu, H.; Nishimura, T.; Muramatsu, K.; Kodera, H.; Kumada, S.; Sugai, K.; Kasai-Yoshida, E.; Sawaura, N.; Nishida, H.; Hoshino, A.; et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013, 45, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.G.; Sun, L.; Miao, G.; Ji, C.; Zhao, H.; Sun, H.; Miao, L.; Yoshii, S.R.; Mizushima, N.; Wang, X.; et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy 2015, 11, 881–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, Y.; So, V.; Tijssen, M.A.J.; Verbeek, D.S.; Reggiori, F.; Mauthe, M. WDR45, one gene associated with multiple neurodevelopmental disorders. Autophagy 2021, 17, 3908–3923. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Zhao, H.; Li, D.; Sun, H.; Hao, J.; Chen, R.; Wang, X.; Zhang, H.; Zhao, Y.G. Role of Wdr45b in maintaining neural autophagy and cognitive function. Autophagy 2020, 16, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.I.; Dooley, H.C.; Tooze, S.A. WIPI2b and Atg16L1: Setting the stage for autophagosome formation. Biochem. Soc. Trans. 2014, 42, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Princely Abudu, Y.; Kumar, S.; Bissa, B.; Choi, S.W.; Jia, J.; Lazarou, M.; Eskelinen, E.L.; Johansen, T.; Deretic, V. Mammalian Atg8 proteins regulate lysosome and autolysosome biogenesis through SNAREs. EMBO J. 2019, 38, e101994. [Google Scholar] [CrossRef] [PubMed]
- Sora, V.; Kumar, M.; Maiani, E.; Lambrughi, M.; Tiberti, M.; Papaleo, E. Structure and Dynamics in the ATG8 Family From Experimental to Computational Techniques. Front. Cell Dev. Biol. 2020, 8, 420. [Google Scholar] [CrossRef]
- Maeda, S.; Otomo, C.; Otomo, T. The autophagic membrane tether ATG2A transfers lipids between membranes. Elife 2019, 8, e45777. [Google Scholar] [CrossRef]
- Dancourt, J.; Melia, T.J. Lipidation of the autophagy proteins LC3 and GABARAP is a membrane-curvature dependent process. Autophagy 2014, 10, 1470–1471. [Google Scholar] [CrossRef] [Green Version]
- Tanji, K.; Mori, F.; Wakabayashi, K. The Role of Atg8 Homologue in Lewy Body Disease. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2014; pp. 383–389. [Google Scholar]
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 2016, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Wang, X. Autophagy and p62 in cardiac protein quality control. Autophagy 2011, 7, 1382–1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nihira, K.; Miki, Y.; Ono, K.; Suzuki, T.; Sasano, H. An inhibition of p62/SQSTM1 caused autophagic cell death of several human carcinoma cells. Cancer Sci. 2014, 105, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.; Kim, J.H.; Shin, J. p62 forms a ternary complex with PKCzeta and PAR-4 and antagonizes PAR-4-induced PKCzeta inhibition. FEBS Lett. 2002, 510, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birgisdottir, A.B.; Lamark, T.; Johansen, T. The LIR motif—Crucial for selective autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef] [Green Version]
- Acharya, M.; Lingenfelter, D.J.; Huang, L.; Gage, P.J.; Walter, M.A. Human PRKC apoptosis WT1 regulator is a novel PITX2-interacting protein that regulates PITX2 transcriptional activity in ocular cells. J. Biol. Chem. 2009, 284, 34829–34838. [Google Scholar] [CrossRef] [Green Version]
- van der Zee, J.; Van Langenhove, T.; Kovacs, G.G.; Dillen, L.; Deschamps, W.; Engelborghs, S.; Matej, R.; Vandenbulcke, M.; Sieben, A.; Dermaut, B.; et al. Rare mutations in SQSTM1 modify susceptibility to frontotemporal lobar degeneration. Acta Neuropathol. 2014, 128, 397–410. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; Zhang, J.; Dong, M. Nrf2 as a potential target for Parkinson’s disease therapy. J. Mol. Med. 2021, 99, 917–931. [Google Scholar] [CrossRef]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [Green Version]
- Takats, S.; Pircs, K.; Nagy, P.; Varga, A.; Karpati, M.; Hegedus, K.; Kramer, H.; Kovacs, A.L.; Sass, M.; Juhasz, G. Interaction of the HOPS complex with Syntaxin 17 mediates autophagosome clearance in Drosophila. Mol. Biol. Cell 2014, 25, 1338–1354. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, K.; Itakura, M.; Fukutomi, T.; Nishiwaki, C.; Nakamichi, Y.; Torii, S.; Makiyama, T.; Harada, A.; Ohara-Imaizumi, M. VAMP7 Regulates Autophagosome Formation by Supporting Atg9a Functions in Pancreatic beta-Cells From Male Mice. Endocrinology 2018, 159, 3674–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.L. Syntaxin 16’s Newly Deciphered Roles in Autophagy. Cells 2019, 8, 1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatsuzawa, K.; Hirose, H.; Tani, K.; Yamamoto, A.; Scheller, R.H.; Tagaya, M. Syntaxin 18, a SNAP receptor that functions in the endoplasmic reticulum, intermediate compartment, and cis-Golgi vesicle trafficking. J. Biol. Chem. 2000, 275, 13713–13720. [Google Scholar] [CrossRef] [Green Version]
- Bennett, M.K.; Garcia-Arraras, J.E.; Elferink, L.A.; Peterson, K.; Fleming, A.M.; Hazuka, C.D.; Scheller, R.H. The syntaxin family of vesicular transport receptors. Cell 1993, 74, 863–873. [Google Scholar] [CrossRef]
- Nair, U.; Jotwani, A.; Geng, J.; Gammoh, N.; Richerson, D.; Yen, W.L.; Griffith, J.; Nag, S.; Wang, K.; Moss, T.; et al. SNARE proteins are required for macroautophagy. Cell 2011, 146, 290–302. [Google Scholar] [CrossRef] [Green Version]
- Moreau, K.; Renna, M.; Rubinsztein, D.C. Connections between SNAREs and autophagy. Trends Biochem. Sci. 2013, 38, 57–63. [Google Scholar] [CrossRef]
- Yoshida, S.; Hasegawa, T.; Suzuki, M.; Sugeno, N.; Kobayashi, J.; Ueyama, M.; Fukuda, M.; Ido-Fujibayashi, A.; Sekiguchi, K.; Ezura, M.; et al. Parkinson’s disease-linked DNAJC13 mutation aggravates alpha-synuclein-induced neurotoxicity through perturbation of endosomal trafficking. Hum. Mol. Genet. 2018, 27, 823–836. [Google Scholar] [CrossRef]
- Lee, H.-J.; Lee, K.-H.; Im, H.-N. Interaction of Human α-Synuclein with VTI1B May Modulate Vesicle Trafficking. Bull. Korean Chem. Soc. 2012, 33, 3071–3075. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Nayak, S.; Mindell, J.A.; Grabe, M. A model of lysosomal pH regulation. J. Gen. Physiol. 2013, 141, 705–720. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, I.; Chiba, M.; Ito, K.; Okamatsu, Y.; Suga, Y.; Kitahara, Y.; Nakahara, Y.; Endo, Y.; Takahashi, K.; Tagami, U.; et al. One-step construction of ferritin encapsulation drugs for cancer chemotherapy. Nanoscale 2021, 13, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Y.; Wan, W.P.; Zhao, S.; Ma, Z.G. L-type calcium channels are involved in iron-induced neurotoxicity in primary cultured ventral mesencephalon neurons of rats. Neurosci. Bull. 2020, 36, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Ravindranath, V. CaV1.3 L-Type Calcium Channels Increase the Vulnerability of Substantia Nigra Dopaminergic Neurons in MPTP Mouse Model of Parkinson’s Disease. Front. Aging Neurosci. 2019, 11, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal. Transduct Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M. NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef]
- Valdinocci, D.; Radford, R.A.W.; Goulding, M.; Hayashi, J.; Chung, R.S.; Pountney, D.L. Extracellular Interactions of Alpha-Synuclein in Multiple System Atrophy. Int. J. Mol. Sci. 2018, 19, 4129. [Google Scholar] [CrossRef] [Green Version]
- Jan, A.; Goncalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-Like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef]
- Ferreira, N.; Gonçalves, N.P.; Jan, A.; Jensen, N.M.; Van Der Laan, A.; Mohseni, S.; Vægter, C.B.; Jensen, P.H. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berge, N.; Ferreira, N.; Mikkelsen, T.W.; Alstrup, A.K.O.; Tamguney, G.; Karlsson, P.; Terkelsen, A.J.; Nyengaard, J.R.; Jensen, P.H.; Borghammer, P. Ageing promotes pathological alpha-synuclein propagation and autonomic dysfunction in wild-type rats. Brain 2021, 144, 1853–1868. [Google Scholar] [CrossRef] [PubMed]
- Ximerakis, M.; Pampalakis, G.; Roumeliotis, T.I.; Sykioti, V.S.; Garbis, S.D.; Stefanis, L.; Sotiropoulou, G.; Vekrellis, K. Resistance of naturally secreted alpha-synuclein to proteolysis. FASEB J. 2014, 28, 3146–3158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdinocci, D.; Grant, G.D.; Dickson, T.C.; Pountney, D.L. Epothilone D inhibits microglia-mediated spread of alpha-synuclein aggregates. Mol. Cell Neurosci. 2018, 89, 80–94. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Zhang, G.; Han, C.; Ma, K.; Guo, X.; Wan, F.; Kou, L.; Yin, S.; Liu, L.; Huang, J.; et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019, 10, 174. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimi, L.; Akbari, A.; Jabbari, N.; Mojarad, B.; Vahhabi, A.; Szafert, S.; Kalashani, S.A.; Soraya, H.; Nawaz, M.; Rezaie, J. Synergies in exosomes and autophagy pathways for cellular homeostasis and metastasis of tumor cells. Cell Biosci. 2020, 10, 64. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Y.; Song, N.; Wang, J.; Jiang, H.; Xie, J. New Progress on the Role of Glia in Iron Metabolism and Iron-Induced Degeneration of Dopamine Neurons in Parkinson’s Disease. Front. Mol. Neurosci. 2017, 10, 455. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Andersen, M.V.; Christoffersen, P.R.; Jensen, M.D.; Lichota, J.; Moos, T. Neurodegeneration with inflammation is accompanied by accumulation of iron and ferritin in microglia and neurons. Neurobiol. Dis. 2015, 81, 108–118. [Google Scholar] [CrossRef]
- Jellinger, K.; Paulus, W.; Grundke-Iqbal, I.; Riederer, P.; Youdim, M.B. Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J. Neural Transm. Park. Dis. Dement. Sect. 1990, 2, 327–340. [Google Scholar] [CrossRef]
- Shen, X.; Yang, H.; Zhang, D.; Jiang, H. Iron Concentration Does Not Differ in Blood but Tends to Decrease in Cerebrospinal Fluid in Parkinson’s Disease. Front. Neurosci 2019, 13, 939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, X.X.; Wang, S.F.; Tan, Y.; Song, J.X.; Zhu, Z.; Wang, Z.Y.; Wu, M.Y.; Cai, C.Z.; Huang, Z.J.; Tan, J.Q.; et al. Pharmacological enhancement of TFEB-mediated autophagy alleviated neuronal death in oxidative stress-induced Parkinson’s disease models. Cell Death Dis. 2020, 11, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Yang, C.; Iyaswamy, A.; Krishnamoorthi, S.; Sreenivasmurthy, S.G.; Liu, J.; Wang, Z.; Tong, B.C.; Song, J.; Lu, J.; et al. Balancing mTOR Signaling and Autophagy in the Treatment of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J.; Schumacker, P.T. Calcium, bioenergetics, and neuronal vulnerability in Parkinson’s disease. J. Biol. Chem. 2013, 288, 10736–10741. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Goodwin, J.; Engelborghs, Y.; Pountney, D.L. Raised calcium promotes alpha-synuclein aggregate formation. Mol. Cell Neurosci. 2011, 46, 516–526. [Google Scholar] [CrossRef]
- Wera, S.; Neyts, J. Calcineurin as a possible new target for treatment of Parkinson’s disease. Med. Hypotheses 1994, 43, 132–134. [Google Scholar] [CrossRef]
- Miller, A.J.; Levy, C.; Davis, I.J.; Razin, E.; Fisher, D.E. Sumoylation of MITF and its related family members TFE3 and TFEB. J. Biol. Chem. 2005, 280, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Henley, J.M.; Craig, T.J.; Wilkinson, K.A. Neuronal SUMOylation: Mechanisms, physiology, and roles in neuronal dysfunction. Physiol. Rev. 2014, 94, 1249–1285. [Google Scholar] [CrossRef]
- Yan, S.; Sun, X.; Xiang, B.; Cang, H.; Kang, X.; Chen, Y.; Li, H.; Shi, G.; Yeh, E.T.; Wang, B.; et al. Redox regulation of the stability of the SUMO protease SENP3 via interactions with CHIP and Hsp90. EMBO J. 2010, 29, 3773–3786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, I.; Ito, A.; Hirai, G.; Nishimura, S.; Kawasaki, H.; Saitoh, H.; Kimura, K.; Sodeoka, M.; Yoshida, M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009, 16, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Vijayakumaran, S.; Nakamura, Y.; Henley, J.M.; Pountney, D.L. Ginkgolic acid promotes autophagy-dependent clearance of intracellular alpha-synuclein aggregates. Mol. Cell Neurosci. 2019, 101, 103416. [Google Scholar] [CrossRef] [PubMed]
- Gerstmeier, J.; Seegers, J.; Witt, F.; Waltenberger, B.; Temml, V.; Rollinger, J.M.; Stuppner, H.; Koeberle, A.; Schuster, D.; Werz, O. Ginkgolic Acid is a Multi-Target Inhibitor of Key Enzymes in Pro-Inflammatory Lipid Mediator Biosynthesis. Front. Pharmacol. 2019, 10, 797. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yang, B.; Zhang, L.; Cong, X.; Liu, Z.; Hu, Y.; Zhang, J.; Hu, H. Ginkgolic acid induces interplay between apoptosis and autophagy regulated by ROS generation in colon cancer. Biochem. Biophys. Res. Commun. 2018, 498, 246–253. [Google Scholar] [CrossRef]
- Baek, S.H.; Ko, J.H.; Lee, J.H.; Kim, C.; Lee, H.; Nam, D.; Lee, J.; Lee, S.G.; Yang, W.M.; Um, J.Y.; et al. Ginkgolic Acid Inhibits Invasion and Migration and TGF-beta-Induced EMT of Lung Cancer Cells Through PI3K/Akt/mTOR Inactivation. J. Cell Physiol. 2017, 232, 346–354. [Google Scholar] [CrossRef]
- Yao, Q.Q.; Li, L.; Xu, M.C.; Hu, H.H.; Zhou, H.; Yu, L.S.; Zeng, S. The metabolism and hepatotoxicity of ginkgolic acid (17:1) in vitro. Chin. J. Nat. Med. 2018, 16, 829–837. [Google Scholar] [CrossRef]
- Brackett, C.M.; Garcia-Casas, A.; Castillo-Lluva, S.; Blagg, B.S.J. Synthesis and Evaluation of Ginkgolic Acid Derivatives as SUMOylation Inhibitors. ACS Med. Chem. Lett. 2020, 11, 2221–2226. [Google Scholar] [CrossRef]
- Chaari, A.; Hoarau-Vechot, J.; Ladjimi, M. Applying chaperones to protein-misfolding disorders: Molecular chaperones against alpha-synuclein in Parkinson’s disease. Int. J. Biol. Macromol. 2013, 60, 196–205. [Google Scholar] [CrossRef]
- Riedel, M.; Goldbaum, O.; Schwarz, L.; Schmitt, S.; Richter-Landsberg, C. 17-AAG induces cytoplasmic alpha-synuclein aggregate clearance by induction of autophagy. PLoS ONE 2010, 5, e8753. [Google Scholar] [CrossRef] [Green Version]
- Vijayakumaran, S.; Wong, M.B.; Antony, H.; Pountney, D.L. Direct and/or Indirect Roles for SUMO in Modulating Alpha-Synuclein Toxicity. Biomolecules 2015, 5, 1697–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Residue Location (Residue Number) | Flanking Residues | Motif | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Y-type | ||||||||||||||||
| FUND1 | 18–21 | S | D | D | D | S | Y-E-V-L | D | L | T | E | Y | Y | E | V | L |
| CRY1 | 273–276 | K | L | T | D | L | Y-K-K-V | K | K | N | S | S | Y | K | K | V |
| MAVS | 9–12 | A | E | D | K | T | Y-K-Y-I | C | R | N | F | S | Y | K | Y | I |
| MAPK15 | 340–343 | Y | R | S | R | V | Y-Q-M-I | L | E | C | G | G | Y | Q | M | I |
| NCOA4 | 71–74 | R | E | V | W | L | Y-E-Q-V | D | L | I | T | Q | Y | E | Q | V |
| NCOA4 | 428–431 | E | K | E | A | L | Y-K-W-L | L | K | K | E | G | Y | K | W | L |
| F-type | ||||||||||||||||
| ULK1 | 367–370 | C | D | T | D | D | F-V-M-V | P | A | Q | F | P | F | V | M | V |
| WDFY3 | 3346–3349 | D | E | K | D | G | F-I-F-V | N | Y | S | E | G | F | I | F | V |
| ATG2A | 1362–1365 | L | D | S | D | E | F-C-I-L | D | A | P | G | L | F | C | I | L |
| ATG13 | 444–447 | N | T | H | D | D | F-V-M-I | D | F | K | P | A | F | V | M | I |
| BECL1 | 97–100 | E | S | A | N | S | F-T-L-I | I | G | E | A | S | F | T | L | I |
| DISC1 | 210–213 | A | F | T | S | S | F-S-F-I | R | L | S | L | G | F | S | F | I |
| ATX3 | 74–77 | M | D | D | S | G | F-F-S-I | Q | V | I | S | N | F | F | S | I |
| NCOA4 | 99–102 | S | L | L | G | Q | F-N-C-L | T | H | Q | L | E | F | N | C | L |
| NCOA4 | 154–157 | Q | T | I | T | T | F-G-S-L | K | T | I | Q | I | F | G | S | L |
| NCOA4 | 328–331 | E | T | S | E | K | F-K-L-L | F | Q | S | Y | N | F | K | L | L |
| NCOA4 | 486–489 | R | I | A | D | S | F-Q-V-I | K | N | S | P | L | F | Q | V | I |
| W-type | ||||||||||||||||
| NEDD4 | 685–688 | E | S | S | E | N | W-E-I-I | R | E | D | E | A | W | E | I | I |
| p62 | 338–341 | G | G | D | D | D | W-T-H-L | S | S | K | E | V | W | T | H | L |
| Htt | 3035–3038 | S | M | V | R | D | W-V-M-L | S | L | S | N | F | W | V | M | L |
| Confirmed Motif | Positively Residues | Polar Residues | ||||||||||||||
| Speculated Motif | Negatively Charged | Non-Polar Residues | ||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boag, M.K.; Roberts, A.; Uversky, V.N.; Ma, L.; Richardson, D.R.; Pountney, D.L. Ferritinophagy and α-Synuclein: Pharmacological Targeting of Autophagy to Restore Iron Regulation in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 2378. https://doi.org/10.3390/ijms23042378
Boag MK, Roberts A, Uversky VN, Ma L, Richardson DR, Pountney DL. Ferritinophagy and α-Synuclein: Pharmacological Targeting of Autophagy to Restore Iron Regulation in Parkinson’s Disease. International Journal of Molecular Sciences. 2022; 23(4):2378. https://doi.org/10.3390/ijms23042378
Chicago/Turabian StyleBoag, Matthew K., Angus Roberts, Vladimir N. Uversky, Linlin Ma, Des R. Richardson, and Dean L. Pountney. 2022. "Ferritinophagy and α-Synuclein: Pharmacological Targeting of Autophagy to Restore Iron Regulation in Parkinson’s Disease" International Journal of Molecular Sciences 23, no. 4: 2378. https://doi.org/10.3390/ijms23042378