
Pharmacogenomics of Anti-TNF Treatment Response Marks a New Era of Tailored Rheumatoid Arthritis Therapy


Abstract
:1. Introduction
2. The Pivotal Importance of TNF in RA
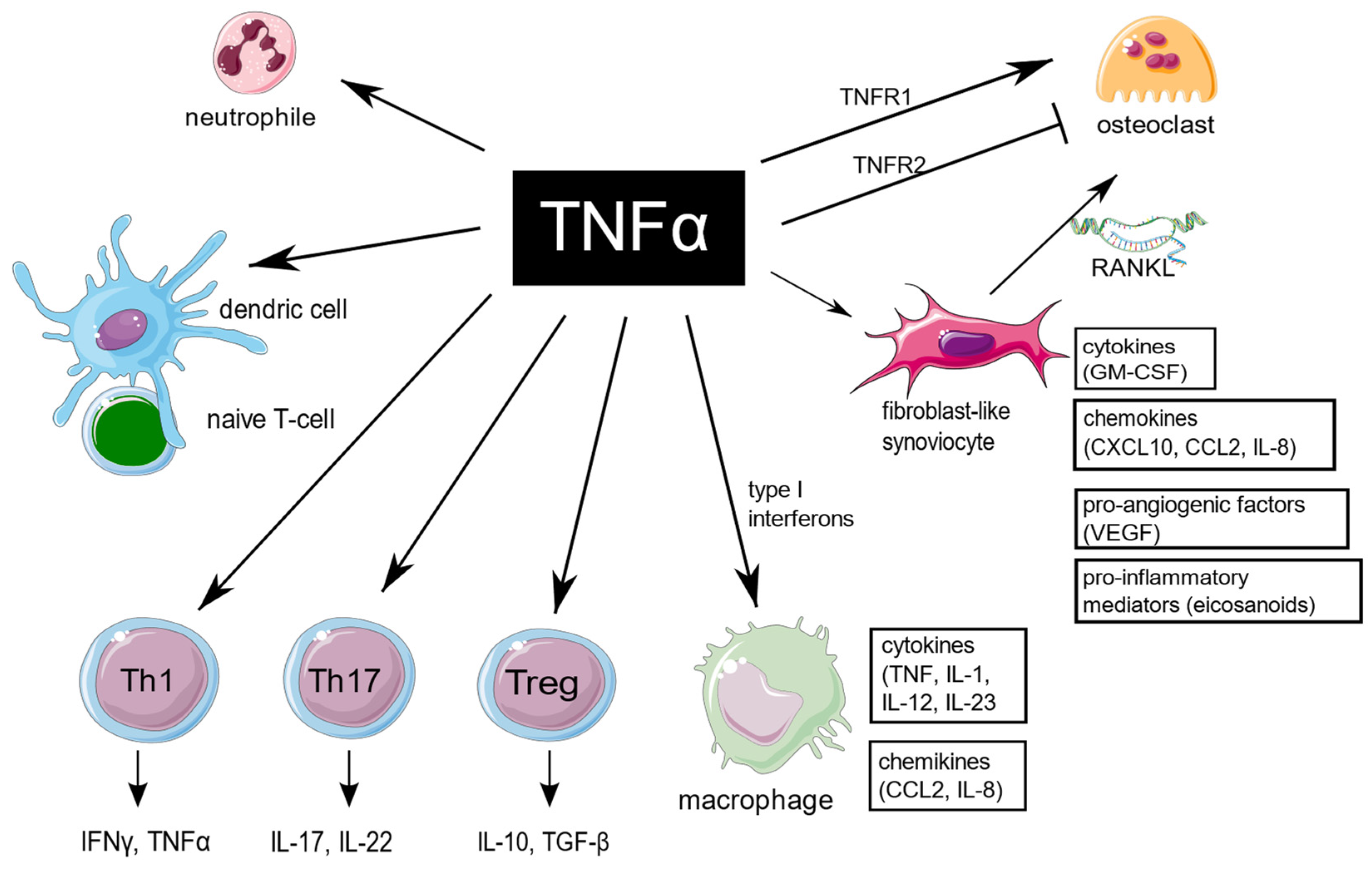
3. Diverse Influence of TNF on Target Cells in RA
3.1. Fibroblast-like Synoviocytes
3.2. Macrophages
3.3. Bone Marrow Stromal Cells and Osteoclastogenesis
3.4. Neutrophils
3.5. T Cells
3.6. Effects of TNF on Angiogenesis
3.7. Influence of TNF on Endothelial Dysfunction
4. TNF Inhibitors Characteristics
5. Primary and Secondary Non-Responsiveness to TNF Inhibitors
6. Anti-TNF Pharmacogenomics
6.1. Genetic Loci
6.2. Gene Expression
6.2.1. Pathotypes of RA Synovium and Synovial Tissue Gene Expression
6.2.2. Whole-Blood Transcriptome
6.2.3. Peripheral Blood Mononuclear Cells
6.2.4. Neutrophiles
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACPA | anti-citrullinated protein antibody |
| ADA | adalimumab |
| CZP | certolizumab pegol (CZP) |
| bDMARDs | biological disease-modifying antirheumatic drugs |
| csDMARDs | conventional synthetic disease-modifying antirheumatic drugs |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| ETA | etanercept |
| EULAR | European League Against Rheumatism |
| FcγR | Fc-gamma receptor |
| FISH | fluorescence in situ hybridization |
| FLS | fibroblast-like synoviocytes |
| GOL | golimumab |
| IFN-γ | interferon-γ |
| IHC | immunohistochemistry |
| IL-17 | interleukin-17 |
| IFX | infliximab |
| i.v. | intravenously |
| JNK | c-jun N-terminal kinase |
| MLS | macrophage-like synoviocytes |
| NGS | next generation sequencing |
| PEG | polyethylene glycol |
| PBMCs | peripheral blood mononuclear cells |
| RA | rheumatoid arthritis |
| RANKL | receptor activator of NF-κB ligand |
| ROS | reactive oxygen species |
| s.c. | subcutaneously |
| SNP | single-nucleotide polymorphism |
| TACE | TNF-alpha-converting enzyme |
| TLR | Toll-like receptor |
| sTNF | soluble Tumor Necrosis Factor |
| tmTNF | transmembrane Tumor Necrosis Factor |
| TNFR1/2 | Tumor Necrosis Factor receptor 1/2 |
| VEGF | vascular endothelial growth factor |
| WGS | whole genome sequencing |
| WES | whole exome sequencing |
References
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid arthritis. Lancet 2010, 376, 1094–1108. [Google Scholar] [CrossRef]
- Schettini, F.; Chic, N.; Braso-Maristany, F.; Pare, L.; Pascual, T.; Conte, B.; Martinez-Saez, O.; Adamo, B.; Vidal, M.; Barnadas, E.; et al. Clinical, pathological, and PAM50 gene expression features of HER2-low breast cancer. NPJ Breast Cancer 2021, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.N.; Lundy, S.K.; Fox, D.A. Synovial biology and T cells in rheumatoid arthritis. Pathophysiology Off. J. Int. Soc. Pathophysiol. 2005, 12, 183–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum. 2011, 63, 53–62. [Google Scholar] [CrossRef]
- Lu, M.C.; Lai, N.S.; Yin, W.Y.; Yu, H.C.; Huang, H.B.; Tung, C.H.; Huang, K.Y.; Yu, C.L. Anti-citrullinated protein antibodies activated ERK1/2 and JNK mitogen-activated protein kinases via binding to surface-expressed citrullinated GRP78 on mononuclear cells. J. Clin. Immunol. 2013, 33, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Lai, N.S.; Yu, H.C.; Yu, C.L.; Koo, M.; Huang, H.B.; Lu, M.C. Anti-citrullinated protein antibodies suppress let-7a expression in monocytes from patients with rheumatoid arthritis and facilitate the inflammatory responses in rheumatoid arthritis. Immunobiology 2015, 220, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Gregory, C.D.; Devitt, A. The macrophage and the apoptotic cell: An innate immune interaction viewed simplistically? Immunology 2004, 113, 1–14. [Google Scholar] [CrossRef]
- Holbrook, J.; Lara-Reyna, S.; Jarosz-Griffiths, H.; McDermott, M. Tumour necrosis factor signalling in health and disease. F1000Research 2019, 8, 111. [Google Scholar] [CrossRef]
- Smith, M.D. The normal synovium. Open Rheumatol. J. 2011, 5, 100–106. [Google Scholar] [CrossRef]
- Bromley, M.; Woolley, D.E. Histopathology of the rheumatoid lesion. Identification of cell types at sites of cartilage erosion. Arthritis Rheum. 1984, 27, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef]
- Armaka, M.; Apostolaki, M.; Jacques, P.; Kontoyiannis, D.L.; Elewaut, D.; Kollias, G. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J. Exp. Med. 2008, 205, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efimov, G.A.; Kruglov, A.A.; Khlopchatnikova, Z.V.; Rozov, F.N.; Mokhonov, V.V.; Rose-John, S.; Scheller, J.; Gordon, S.; Stacey, M.; Drutskaya, M.S.; et al. Cell-type-restricted anti-cytokine therapy: TNF inhibition from one pathogenic source. Proc. Natl. Acad. Sci. USA 2016, 113, 3006–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degboe, Y.; Rauwel, B.; Baron, M.; Boyer, J.F.; Ruyssen-Witrand, A.; Constantin, A.; Davignon, J.L. Polarization of Rheumatoid Macrophages by TNF Targeting Through an IL-10/STAT3 Mechanism. Front. Immunol. 2019, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Loh, C.; Park, S.H.; Lee, A.; Yuan, R.; Ivashkiv, L.B.; Kalliolias, G.D. TNF-induced inflammatory genes escape repression in fibroblast-like synoviocytes: Transcriptomic and epigenomic analysis. Ann. Rheum. Dis. 2019, 78, 1205–1214. [Google Scholar] [CrossRef]
- Park, S.H.; Kang, K.; Giannopoulou, E.; Qiao, Y.; Kang, K.; Kim, G.; Park-Min, K.H.; Ivashkiv, L.B. Type I interferons and the cytokine TNF cooperatively reprogram the macrophage epigenome to promote inflammatory activation. Nat. Immunol. 2017, 18, 1104–1116. [Google Scholar] [CrossRef] [Green Version]
- Muskardin, T.L.W.; Niewold, T.B. Type I interferon in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 214–228. [Google Scholar] [CrossRef]
- Abu-Amer, Y.; Erdmann, J.; Alexopoulou, L.; Kollias, G.; Ross, F.P.; Teitelbaum, S.L. Tumor necrosis factor receptors types 1 and 2 differentially regulate osteoclastogenesis. J. Biol. Chem. 2000, 275, 27307–27310. [Google Scholar] [CrossRef]
- Li, P.; Schwarz, E.M.; O’Keefe, R.J.; Ma, L.; Boyce, B.F.; Xing, L. RANK signaling is not required for TNFalpha-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J. Bone Miner. Res. Off. J. Am. Soc. Bone Mineral. Res. 2004, 19, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.C.; Rubin, J.; Nanes, M.S. The p55 TNF receptor mediates TNF inhibition of osteoblast differentiation independently of apoptosis. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E1011–E1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B. TNF and Bone Remodeling. Curr. Osteoporos. Rep. 2017, 15, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Heulsmann, A.; Tondravi, M.M.; Mukherjee, A.; Abu-Amer, Y. Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J. Biol. Chem. 2001, 276, 563–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiewchengchol, D.; Wright, H.L.; Thomas, H.B.; Lam, C.W.; Roberts, K.J.; Hirankarn, N.; Beresford, M.W.; Moots, R.J.; Edwards, S.W. Differential changes in gene expression in human neutrophils following TNF-alpha stimulation: Up-regulation of anti-apoptotic proteins and down-regulation of proteins involved in death receptor signaling. Immun. Inflamm. Dis. 2016, 4, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaczewska, J.; Abdulreda, M.H.; Yau, C.Y.; Schmitt, M.M.; Schubert, I.; Berggren, P.O.; Weber, C.; Koenen, R.R.; Moy, V.T.; Wojcikiewicz, E.P. TNF-alpha and IFN-gamma promote lymphocyte adhesion to endothelial junctional regions facilitating transendothelial migration. J. Leukoc. Biol. 2014, 95, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S.; Annunziato, F. Th17 and non-classic Th1 cells in chronic inflammatory disorders: Two sides of the same coin. Int. Arch. Allergy Immunol. 2014, 164, 171–177. [Google Scholar] [CrossRef]
- Dulic, S.; Vasarhelyi, Z.; Sava, F.; Berta, L.; Szalay, B.; Toldi, G.; Kovacs, L.; Balog, A. T cell Subsets in Rheumatoid Arthritis Patients on Long-Term Anti-TNF or IL-6 Receptor Blocker Therapy. Mediat. Inflamm. 2017, 2017, 6894374. [Google Scholar] [CrossRef] [Green Version]
- Aerts, N.E.; Ebo, D.G.; Bridts, C.H.; Stevens, W.J.; De Clerck, L.S. T cell signal transducer and activator of transcription (STAT) 4 and 6 are affected by adalimumab therapy in rheumatoid arthritis. Clin. Exp. Rheumatol. 2010, 28, 208–214. [Google Scholar]
- Hull, D.N.; Cooksley, H.; Chokshi, S.; Williams, R.O.; Abraham, S.; Taylor, P.C. Increase in circulating Th17 cells during anti-TNF therapy is associated with ultrasonographic improvement of synovitis in rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 303. [Google Scholar] [CrossRef] [Green Version]
- Talotta, R.; Berzi, A.; Atzeni, F.; Batticciotto, A.; Clerici, M.; Sarzi-Puttini, P.; Trabattoni, D. Paradoxical Expansion of Th1 and Th17 Lymphocytes in Rheumatoid Arthritis Following Infliximab Treatment: A Possible Explanation for a Lack of Clinical Response. J. Clin. Immunol. 2015, 35, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Kruglov, A.; Drutskaya, M.; Schlienz, D.; Gorshkova, E.; Kurz, K.; Morawietz, L.; Nedospasov, S. Contrasting contributions of TNF from distinct cellular sources in arthritis. Ann. Rheum. Dis. 2020, 79, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Valencia, X.; Stephens, G.; Goldbach-Mansky, R.; Wilson, M.; Shevach, E.M.; Lipsky, P.E. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood 2006, 108, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Khanniche, A.; Zhou, L.; Jiang, B.; Song, J.; Jin, Y.; Yin, J.; Wang, S.; Ji, P.; Shen, H.; Wang, Y.; et al. Restored and Enhanced Memory T Cell Immunity in Rheumatoid Arthritis After TNFalpha Blocker Treatment. Front. Immunol. 2019, 10, 887. [Google Scholar] [CrossRef] [PubMed]
- Pala, O.; Diaz, A.; Blomberg, B.B.; Frasca, D. B Lymphocytes in Rheumatoid Arthritis and the Effects of Anti-TNF-alpha Agents on B Lymphocytes: A Review of the Literature. Clin. Ther. 2018, 40, 1034–1045. [Google Scholar] [CrossRef]
- Clavel, G.; Bessis, N.; Boissier, M.C. Recent data on the role for angiogenesis in rheumatoid arthritis. Joint Bone Spine 2003, 70, 321–326. [Google Scholar] [CrossRef]
- Szekanecz, Z.; Koch, A.E. Targeting Angiogenesis in Rheumatoid Arthritis. Curr. Rheumatol. Rev. 2008, 4, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Petrache, I.; Birukova, A.; Ramirez, S.I.; Garcia, J.G.; Verin, A.D. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am. J. Respir. Cell Mol. Biol. 2003, 28, 574–581. [Google Scholar] [CrossRef] [Green Version]
- Chappell, D.; Hofmann-Kiefer, K.; Jacob, M.; Rehm, M.; Briegel, J.; Welsch, U.; Conzen, P.; Becker, B.F. TNF-alpha induced shedding of the endothelial glycocalyx is prevented by hydrocortisone and antithrombin. Basic Res. Cardiol. 2009, 104, 78–89. [Google Scholar] [CrossRef]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewe, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 79, 685–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Medicines Agency. Humira® Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/humira-epar-product-information_en.pdf (accessed on 18 February 2022).
- Levy, R.A.; Guzman, R.; Castaneda-Hernandez, G.; Martinez-Vazquez, M.; Damian, G.; Cara, C. Biology of anti-TNF agents in immune-mediated inflammatory diseases: Therapeutic implications. Immunotherapy 2016, 8, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. Simponi® Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/simponi-epar-product-information_en.pdf (accessed on 18 February 2022).
- European Medicines Agency. Remicade® Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/remicade-epar-product-information_en.pdf (accessed on 18 February 2022).
- European Medicines Agency. Cimzia® Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/cimzia-epar-product-information_en.pdf (accessed on 18 February 2022).
- European Medicines Agency. Enbrel® Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/enbrel-epar-product-information_en.pdf (accessed on 18 February 2022).
- Roda, G.; Jharap, B.; Neeraj, N.; Colombel, J.F. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin. Transl. Gastroenterol. 2016, 7, e135. [Google Scholar] [CrossRef] [PubMed]
- Buch, M.H.; Bingham, S.J.; Bryer, D.; Emery, P. Long-term infliximab treatment in rheumatoid arthritis: Subsequent outcome of initial responders. Rheumatology 2007, 46, 1153–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Knapp, K.; Wang, L.; Chen, C.I.; Craig, G.L.; Ferguson, K.; Schwartzman, S. Treatment Persistence and Clinical Outcomes of Tumor Necrosis Factor Inhibitor Cycling or Switching to a New Mechanism of Action Therapy: Real-world Observational Study of Rheumatoid Arthritis Patients in the United States with Prior Tumor Necrosis Factor Inhibitor Therapy. Advances Ther. 2017, 34, 1936–1952. [Google Scholar]
- Umicevic Mirkov, M.; Cui, J.; Vermeulen, S.H.; Stahl, E.A.; Toonen, E.J.; Makkinje, R.R.; Lee, A.T.; Huizinga, T.W.; Allaart, R.; Barton, A.; et al. Genome-wide association analysis of anti-TNF drug response in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 1375–1381. [Google Scholar] [CrossRef]
- Sánchez-Maldonado, J.M.; Cáliz Cáliz, R.; López-Nevot, M.Á.; Moñiz-Díez, A.; Cabrera-Serrano, A.J.; Canhão, H.; Ter Horst, R.; Escudero Contreras, A.; Sorensen, S.; Hetland, M.L.; et al. OP0017 Validation of GWAS-identified variants for anti-TNF drug response in rheumatoid arthritis: A meta-analysis of three large cohorts. Front. Immunol. 2021, 80, 9–10. [Google Scholar] [CrossRef]
- Julia, A.; Marsal, S. Pharmacogenomics of anti-TNF response in psoriasis, where are we? Pharmacogenomics 2016, 17, 323–326. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Stahl, E.A.; Saevarsdottir, S.; Miceli, C.; Diogo, D.; Trynka, G.; Raj, T.; Mirkov, M.U.; Canhao, H.; Ikari, K.; et al. Genome-wide association study and gene expression analysis identifies CD84 as a predictor of response to etanercept therapy in rheumatoid arthritis. PLoS Genet. 2013, 9, e1003394. [Google Scholar] [CrossRef] [Green Version]
- O’Rielly, D.D.; Roslin, N.M.; Beyene, J.; Pope, A.; Rahman, P. TNF-alpha-308 G/A polymorphism and responsiveness to TNF-alpha blockade therapy in moderate to severe rheumatoid arthritis: A systematic review and meta-analysis. Pharmacogenomics J. 2009, 9, 161–167. [Google Scholar] [CrossRef]
- Plant, D.; Bowes, J.; Potter, C.; Hyrich, K.L.; Morgan, A.W.; Wilson, A.G.; Isaacs, J.D.; Wellcome Trust Case Control Consortium; British Society for Rheumatology Biologics Register; Barton, A. Genome-wide association study of genetic predictors of anti-tumor necrosis factor treatment efficacy in rheumatoid arthritis identifies associations with polymorphisms at seven loci. Arthritis Rheum. 2011, 63, 645–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farragher, T.M.; Plant, D.; Flynn, E.; Eyre, S.; Bunn, D.; Thomson, W.; Symmons, D.; Barton, A. Association of a rheumatoid arthritis susceptibility variant at the CCL21 locus with premature mortality in inflammatory polyarthritis patients. Arthritis Care Res. 2010, 62, 676–682. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Ectopic germinal center formation in rheumatoid synovitis. Ann. N. Y. Acad. Sci. 2003, 987, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G., Jr.; Holweg, C.T.; Kummerfeld, S.K.; Choy, D.F.; Setiadi, A.F.; Hackney, J.A.; Haverty, P.M.; Gilbert, H.; Lin, W.Y.; Diehl, L.; et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res. Ther. 2014, 16, R90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, M.J.; Barnes, M.R.; Blighe, K.; Goldmann, K.; Rana, S.; Hackney, J.A.; Ramamoorthi, N.; John, C.R.; Watson, D.S.; Kummerfeld, S.K.; et al. Molecular Portraits of Early Rheumatoid Arthritis Identify Clinical and Treatment Response Phenotypes. Cell Rep. 2019, 28, 2455–2470.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lliso-Ribera, G.; Humby, F.; Lewis, M.; Nerviani, A.; Mauro, D.; Rivellese, F.; Kelly, S.; Hands, R.; Bene, F.; Ramamoorthi, N.; et al. Synovial tissue signatures enhance clinical classification and prognostic/treatment response algorithms in early inflammatory arthritis and predict requirement for subsequent biological therapy: Results from the pathobiology of early arthritis cohort (PEAC). Ann. Rheum. Dis. 2019, 78, 1642–1652. [Google Scholar]
- Humby, F.; Lewis, M.; Ramamoorthi, N.; Hackney, J.A.; Barnes, M.R.; Bombardieri, M.; Setiadi, A.F.; Kelly, S.; Bene, F.; DiCicco, M.; et al. Synovial cellular and molecular signatures stratify clinical response to csDMARD therapy and predict radiographic progression in early rheumatoid arthritis patients. Ann. Rheum. Dis. 2019, 78, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Nerviani, A.; Di Cicco, M.; Mahto, A.; Lliso-Ribera, G.; Rivellese, F.; Thorborn, G.; Hands, R.; Bellan, M.; Mauro, D.; Boutet, M.A.; et al. A Pauci-Immune Synovial Pathotype Predicts Inadequate Response to TNFalpha-Blockade in Rheumatoid Arthritis Patients. Front. Immunol. 2020, 11, 845. [Google Scholar] [CrossRef]
- Aterido, A.; Canete, J.D.; Tornero, J.; Blanco, F.; Fernandez-Gutierrez, B.; Perez, C.; Alperi-Lopez, M.; Olive, A.; Corominas, H.; Martinez-Taboada, V.; et al. A Combined Transcriptomic and Genomic Analysis Identifies a Gene Signature Associated With the Response to Anti-TNF Therapy in Rheumatoid Arthritis. Front. Immunol. 2019, 10, 1459. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nogales, A.; Martinez-Sobrido, L.; Topham, D.J. Interferon-Induced Protein 44 Interacts with Cellular FK506-Binding Protein 5, Negatively Regulates Host Antiviral Responses, and Supports Virus Replication. mBio 2019, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Herate, C.; Ramdani, G.; Grant, N.J.; Marion, S.; Gasman, S.; Niedergang, F.; Benichou, S.; Bouchet, J. Phospholipid Scramblase 1 Modulates FcR-Mediated Phagocytosis in Differentiated Macrophages. PLoS ONE 2016, 11, e0145617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poliska, S.; Besenyei, T.; Vegh, E.; Hamar, A.; Pusztai, A.; Vancsa, A.; Bodnar, N.; Szamosi, S.; Csumita, M.; Kerekes, G.; et al. Gene expression analysis of vascular pathophysiology related to anti-TNF treatment in rheumatoid arthritis. Arthritis Res. Ther. 2019, 21, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wampler Muskardin, T.L.; Fan, W.; Jin, Z.; Jensen, M.A.; Dorschner, J.M.; Ghodke-Puranik, Y.; Dicke, B.; Vsetecka, D.; Wright, K.; Mason, T.; et al. Distinct Single Cell Gene Expression in Peripheral Blood Monocytes Correlates With Tumor Necrosis Factor Inhibitor Treatment Response Groups Defined by Type I Interferon in Rheumatoid Arthritis. Front. Immunol. 2020, 11, 1384. [Google Scholar] [CrossRef]
- Tao, W.; Concepcion, A.N.; Vianen, M.; Marijnissen, A.C.A.; Lafeber, F.; Radstake, T.; Pandit, A. Multiomics and Machine Learning Accurately Predict Clinical Response to Adalimumab and Etanercept Therapy in Patients With Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 212–222. [Google Scholar] [CrossRef]
- Wright, H.L.; Cox, T.; Moots, R.J.; Edwards, S.W. Neutrophil biomarkers predict response to therapy with tumor necrosis factor inhibitors in rheumatoid arthritis. J. Leukoc. Biol. 2017, 101, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Wright, H.L.; Chikura, B.; Bucknall, R.C.; Moots, R.J.; Edwards, S.W. Changes in expression of membrane TNF, NF-{kappa}B activation and neutrophil apoptosis during active and resolved inflammation. Ann. Rheum. Dis. 2011, 70, 537–543. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| SNP | Gene | Function | Reference |
|---|---|---|---|
| rs12142623 | PLA2G4A | Phospholipase enzyme | Umicevic et al. |
| rs4651370 | PLA2G4A | Phospholipase enzyme | Umicevic et al. |
| rs2378945 | NUBPL | Assembly of the respiratory chain NADH dehydrogenase (complex I) | Umicevic et al. |
| rs1813443 | NCTN5 | Immunoglobulin superfamily | Umicevic et al. |
| rs4411591 | Unknown | Unclear | Umicevic et al. |
| rs7767069 | Unknown | Unclear | Umicevic et al. |
| rs1447722 | Unknown | Unclear | Umicevic et al. |
| rs1568885 | Unknown | Unclear | Umicevic et al. |
| rs7767069 | LINC02549 | Unclear | Sánchez-Maldonado et al. |
| rs717117G | LARRC55 | Unclear | Sánchez-Maldonado et al. |
| rs113878252 | MED15 | Polymerase II transcription | Julia et al. |
| rs6065221 | MAFB | Regulation of lineage-specific hematopoiesis | Julia et al. |
| rs6427528 | CD84, Non Coding Transcript Variant LOC105371468 | Self-ligand receptor of the signaling lymphocytic activation molecule | Cui et al. |
| rs1503860 | CD84: Non Coding Transcript Variant LOC105371468 | Self-ligand receptor of the signaling lymphocytic activation molecule | Cui et al. |
| rs1800629 | TNF | Tumor necrosis factor | O’Rielly et al. |
| rs17301249 | EYA4 | Transcription coactivator and phosphatase | O’Rielly et al. |
| rs1532269 | PDZD2 | Unclear | Plant et al. |
| rs2812378 | CCL21 | Chemokine ligand | Farragher et al. |
| Synovial Pathotype | Gene Expression Profile | Clinical Phenotype |
|---|---|---|
| lymphoid | B cell- and plasmablast lineage genes (including CD19, CD20, cd27, IGLL5, XBP1, immunoglobulin heavy and light chains, CD38 and CXCL13) | Good clinical response to TNF inhibitors Poor clinical response to IL-6R inhibitors |
| myeloid | NF-κB pathway genes (including TNF, IL-1β, IL-1RA, ICAM1, and MyD88), The inflammatory chemokines (CCL2, IL-8), Neutrophiles, macrophage and osteoclast—associated genes (S100A12, CD14, OSCAR, respectively) | Poor clinical response to TNF inhibitors Good clinical response to IL-6R inhibitors |
| fibroid | Fibroblast growth factor genes (FGF2, FGF9), Bone homeostasis—associated genes (BMP6, TNFRSF11b/ osteoprotogerin), Wnt and TGFβ pathways genes | Poor clinical response to TNF inhibitors |
| pauci-immune | SFRP3/FRZB gene | Poor clinical response to TNF inhibitors |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wysocki, T.; Paradowska-Gorycka, A. Pharmacogenomics of Anti-TNF Treatment Response Marks a New Era of Tailored Rheumatoid Arthritis Therapy. Int. J. Mol. Sci. 2022, 23, 2366. https://doi.org/10.3390/ijms23042366
Wysocki T, Paradowska-Gorycka A. Pharmacogenomics of Anti-TNF Treatment Response Marks a New Era of Tailored Rheumatoid Arthritis Therapy. International Journal of Molecular Sciences. 2022; 23(4):2366. https://doi.org/10.3390/ijms23042366
Chicago/Turabian StyleWysocki, Tomasz, and Agnieszka Paradowska-Gorycka. 2022. "Pharmacogenomics of Anti-TNF Treatment Response Marks a New Era of Tailored Rheumatoid Arthritis Therapy" International Journal of Molecular Sciences 23, no. 4: 2366. https://doi.org/10.3390/ijms23042366