
A Transfer-Learning-Based Deep Convolutional Neural Network for Predicting Leukemia-Related Phosphorylation Sites from Protein Primary Sequences


Abstract
:1. Introduction
2. Results and Discussion
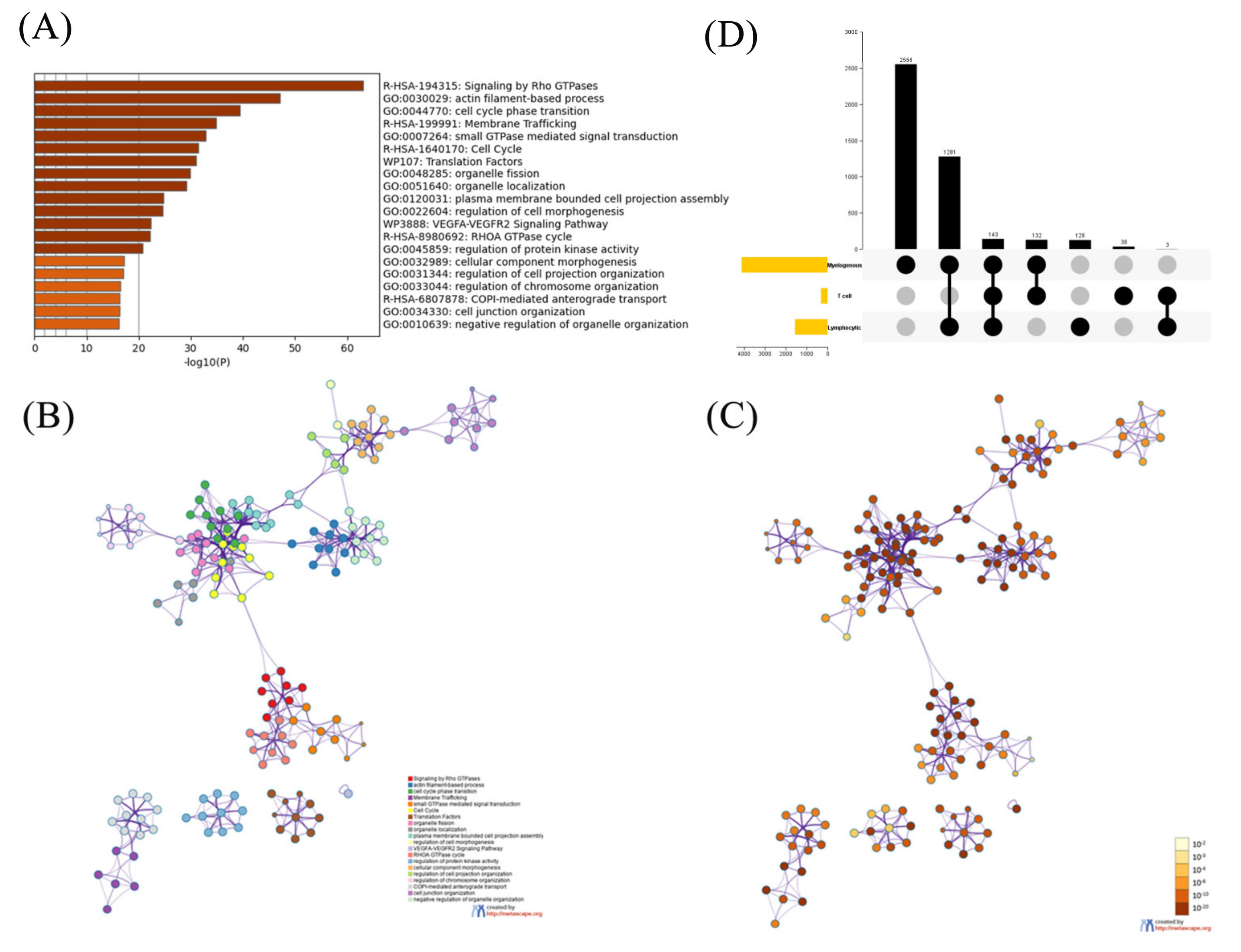
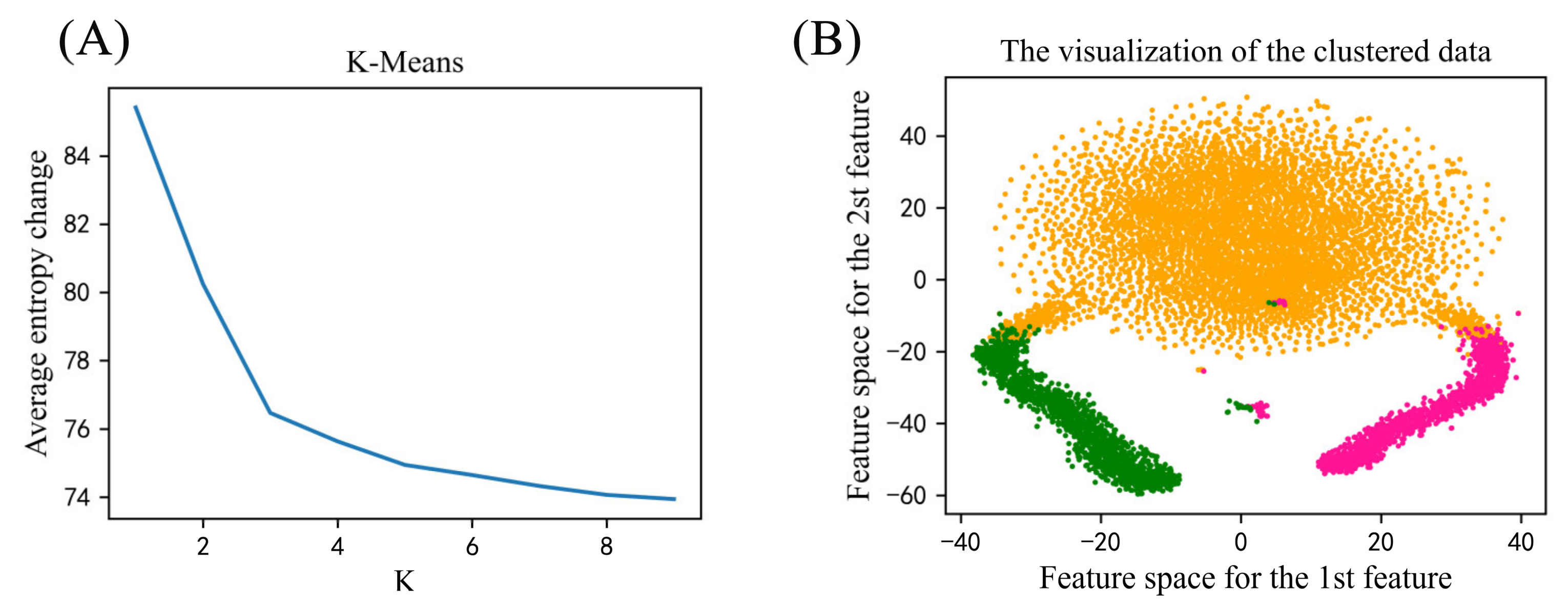
2.1. Functional Analysis on the Phosphorylated Proteins
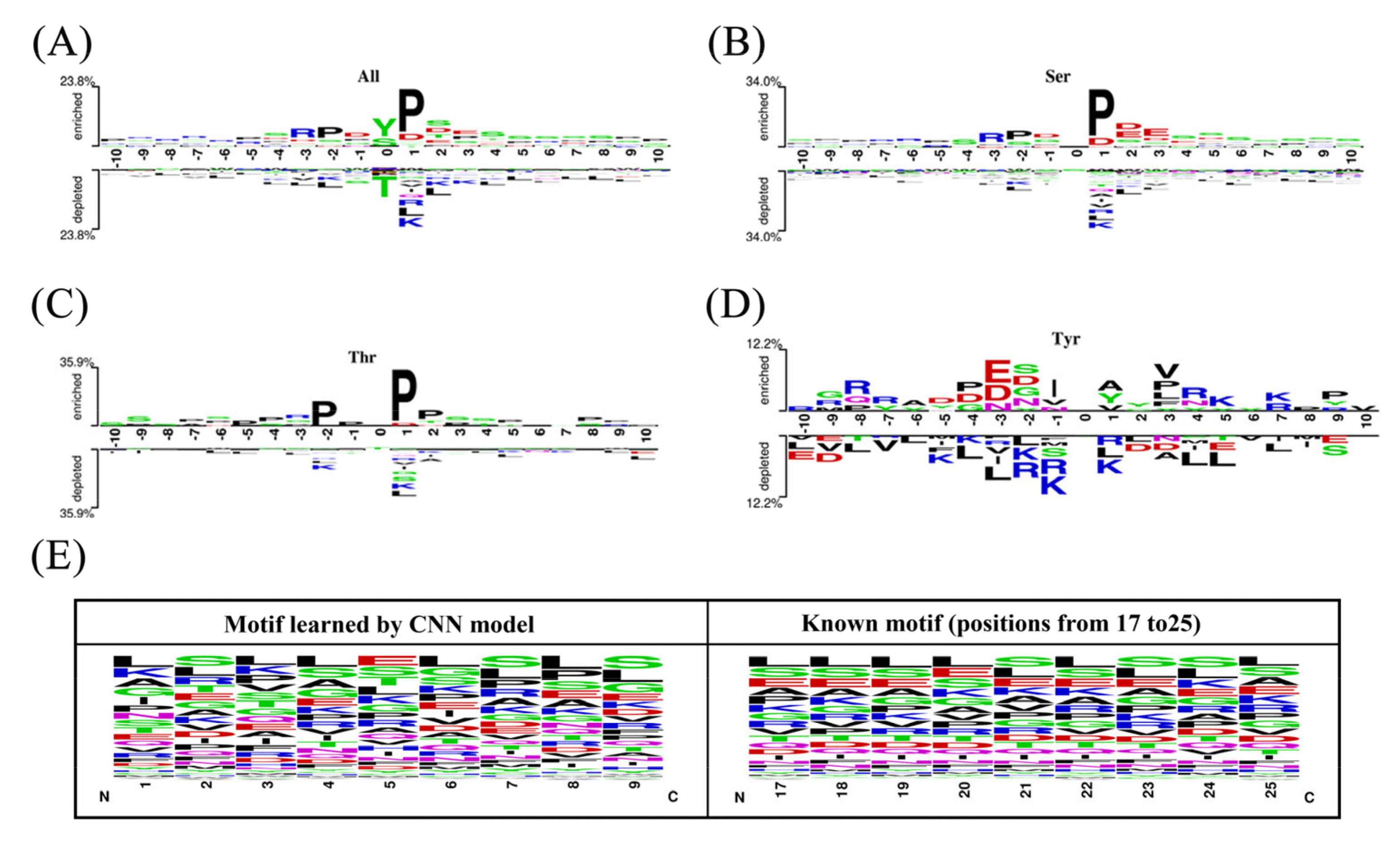
2.2. The Position-Specific Conversation Profiles of the Phosphorylation Peptides
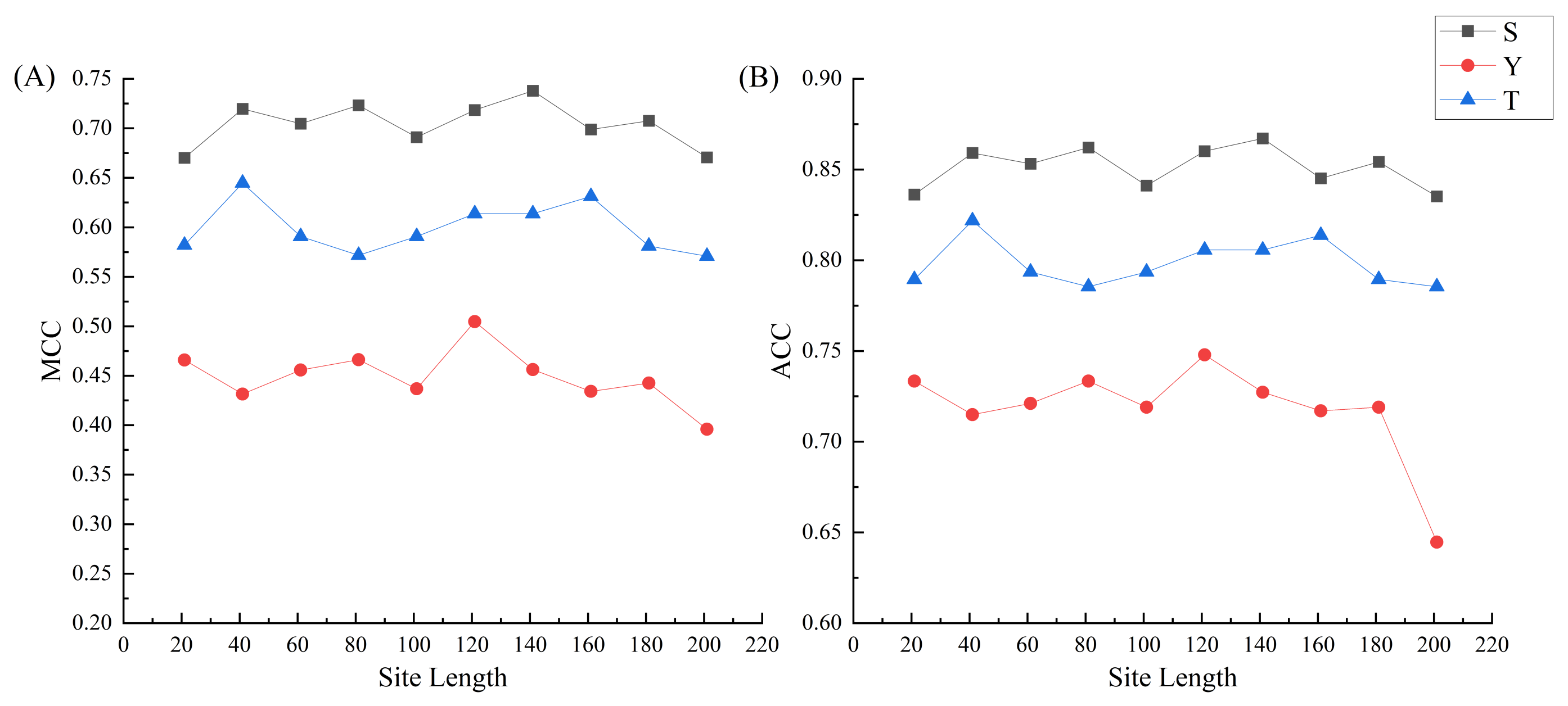
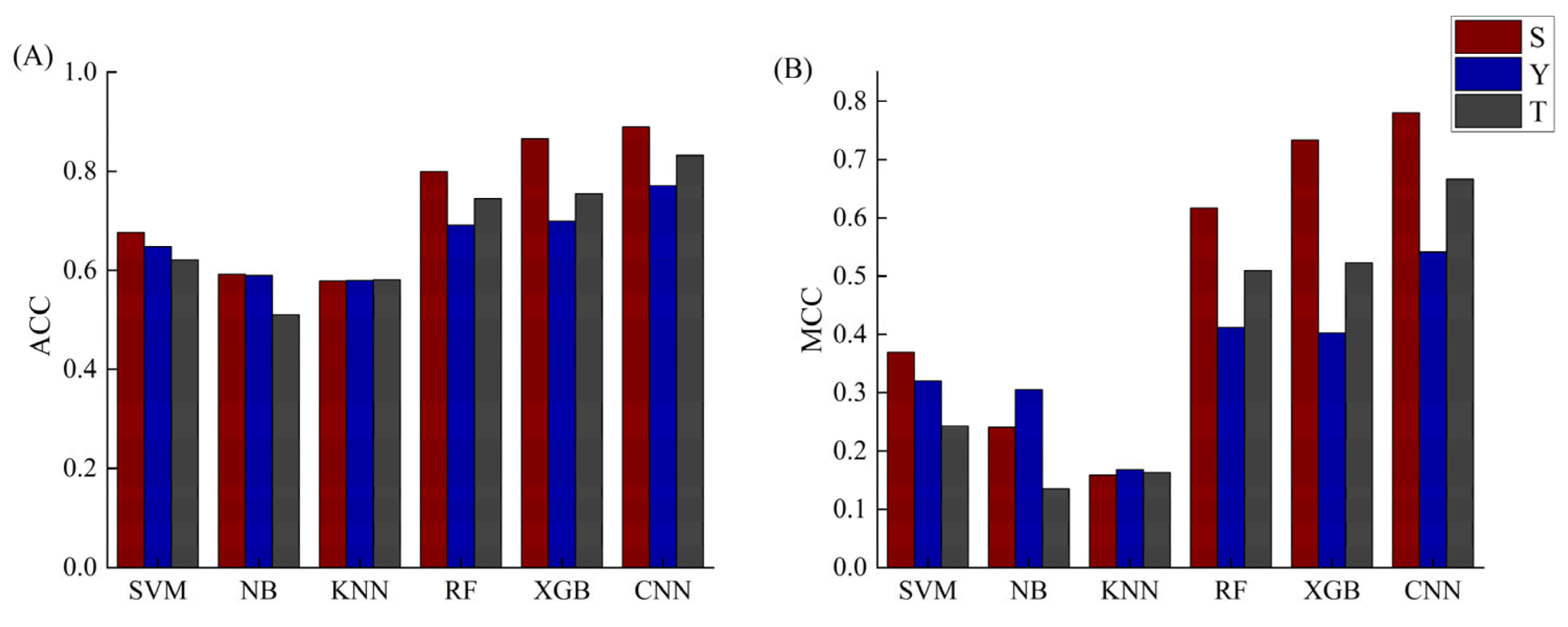
2.3. The Phosphorylation Peptide Length Optimization and Comparisons with Five Machine-Learning Methods
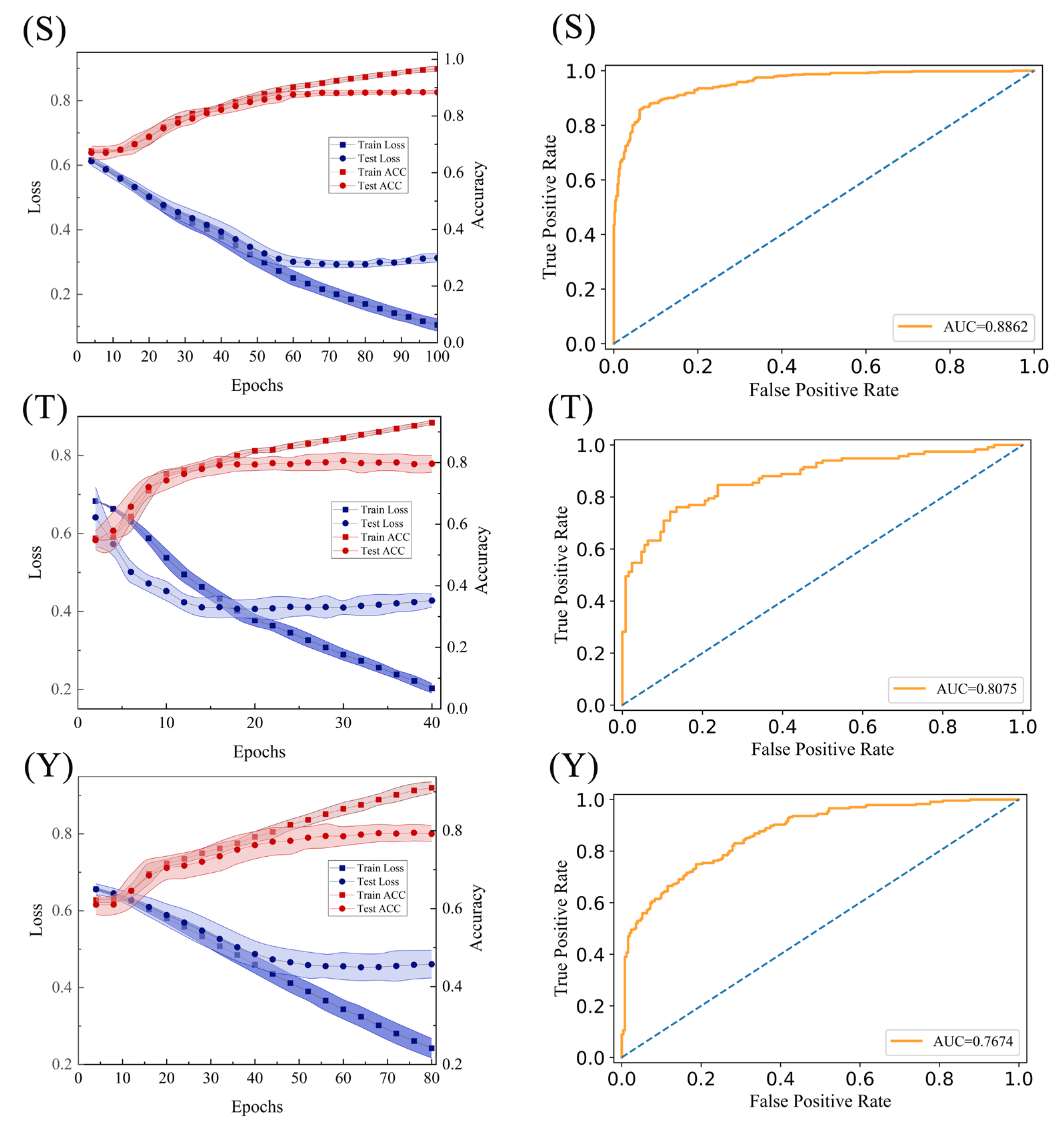
2.4. Construction of the Final Predict Models
2.5. Transfer-Learning on Two Other Small Size Datasets
3. Materials and Methods
3.1. Datasets
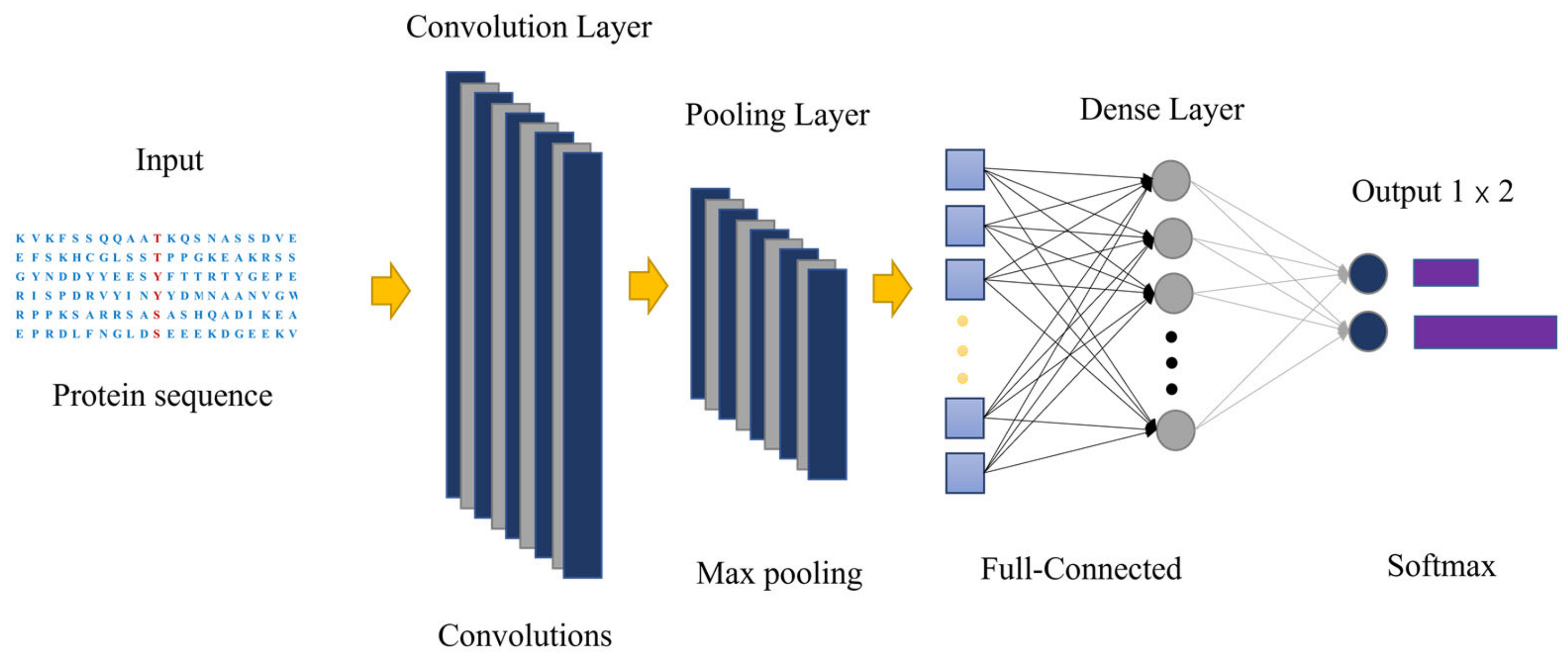
3.2. Convolutional Neural Network and Transfer-Learning
3.3. Traditional Machine-Learning Methods
3.4. Model Validation and Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Green, K.D.; Garneau-Tsodikova, S. Posttranslational Modification of Proteins. Compr. Nat. Prod. II 2010, 5, 433–468. [Google Scholar]
- Hunter, T. Signaling—2000 and Beyond. Cell 2000, 100, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Swaffer, M.P.; Jones, A.W.; Flynn, H.R.; Snijders, A.P.; Nurse, P. CDK Substrate Phosphorylation and Ordering the Cell Cycle. Cell 2016, 167, 1750–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Xu, J.; Wu, J.; Hu, Y.; Han, Y.; Gu, Y.; Zhao, K.; Zhang, Q.; Liu, X.; Liu, J. Phosphorylation-Mediated IFN-γR2 Membrane Translocation Is Required to Activate Macrophage Innate Response. Cell 2018, 184, 1393–1394. [Google Scholar] [CrossRef]
- Li, T.; Fei, L.; Zhang, X. Prediction of kinase-specific phosphorylation sites with sequence features by a log-odds ratio approach. Proteins Struct. Funct. Bioinform. 2010, 70, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.; Kusalik, A. Computational prediction of eukaryotic phosphorylation sites. Bioinformatics 2011, 27, 2927–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, X.; Jiangning, S.; Campbell, W.; Whisstock, J.C. PhosContext2vec: A distributed representation of residue-level sequence contexts and its application to general and kinase-specific phosphorylation site prediction. Sci. Rep. 2018, 8, 8240. [Google Scholar]
- Ismail, H.D.; Ahoi, J.; Kim, J.H.; Newman, R.H.; Kc, D.B. RF-Phos: A Novel General Phosphorylation Site Prediction Tool Based on Random Forest. BioMed Res. Int. 2016, 2016, e3281590. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xu, H.; Lin, S.; Deng, W.; Xue, Y. GPS 5.0: An Update on the Prediction of Kinase-specific Phosphorylation Sites in Proteins. Genom. Proteom. Bioinform. 2020, 18, 72–80. [Google Scholar] [CrossRef]
- Wang, D.; Zeng, S.; Xu, C.; Qiu, W.; Liang, Y.; Joshi, T.; Xu, D. MusiteDeep: A deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 2017, 33, 3909–3916. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Wang, M.; Liu, Y.; Zhao, X.M.; Li, A. DeepPhos: Prediction of protein phosphorylation sites with deep learning. Bioinformatics 2019, 35, 2766–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sa, A.; Mk, A.; Ma, A.; Zuk, B.; Djy, A. DeepPPSite: A deep learning-based model for analysis and prediction of phosphorylation sites using efficient sequence information. Anal. Biochem. 2021, 612, 113955. [Google Scholar]
- Shi, Y.; Zhang, Y.; Lin, S.; Wang, C.; Xue, Y. dbPSP 2.0, an updated database of protein phosphorylation sites in prokaryotes. Sci. Data 2020, 7, 164. [Google Scholar] [CrossRef] [PubMed]
- Bodenmiller, B.; Campbell, D.; Gerrits, B.; Lam, H.; Jovanovic, M.; Picotti, P.; Schlapbach, R.; Aebersold, R. PhosphoPep--a database of protein phosphorylation sites in model organisms. Nat. Biotechnol. 2008, 26, 1339–1340. [Google Scholar] [CrossRef] [Green Version]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.ELM: A database of phosphorylation sites--update 2011. Nucleic Acids Res. 2010, 39, 261–267. [Google Scholar] [CrossRef] [Green Version]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, 127–130. [Google Scholar] [CrossRef]
- Yu, K.; Zhang, Q.F.; Liu, Z.K.; Zhao, Q.; Zhang, X.L.; Wang, Y.; Wang, Z.X.; Jin, Y.; Li, X.X.; Liu, Z.X.; et al. qPhos: A database of protein phosphorylation dynamics in humans. Nucleic Acids Res. 2019, 47, 451–458. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.D.; Wang, Y.B.; Lin, S.F.; Deng, W.K.; Peng, D. PTMD: A Database of Human Disease-associated Post-translational Modifications. Genom. Proteom. Bioinform. 2018, 16, 244–251. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Zhang, B.; Beth, M.; Kornhauser, J.M.; Vaughan, L.; Elzbieta, S. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Randall, J.E.; Brian, C.K.; Nicole, F.L. Regulation of TDP-43 phosphorylation in aging and disease. GeroScience 2021, 43, 1605–1614. [Google Scholar]
- Zhang, T.; Chen, D.M.; Lee, T.H. Phosphorylation Signaling in APP Processing in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brustovetsky, N. Involvement of CRMP2 in Regulation of Mitochondrial Morphology and Motility in Hunti Kim ngton’s Disease. Cells 2021, 10, 3172. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Huart, A.S.; Temmerman, K.; Vahokoski, J.; Mertens, H.D.; Komadina, D.; Hoffmann, J.E.; Yumerefendi, H.; Svergun, D.I.; Kursula, P.; et al. Death-associated protein kinase activity is regulated by coupled calcium/calmodulin binding to two distinct sites. Structure 2016, 24, 851–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.W.; Brady, G.F.; Kwan, R.; Nesvizhskii, A.I.; Omary, M.B. Genotype-phenotype analysis of LMNA-related diseases predicts phenotype-selective alterations in lamin phosphorylation. FASEB J. 2020, 34, 9051–9073. [Google Scholar] [CrossRef]
- Xu, X.; Ao, L.; Wang, M. Prediction of human disease-specific phosphorylation sites with combined feature selection approach and support vector machine. IEEE Int. Conf. Bioinform. Biomed. 2015, 9, 155–163. [Google Scholar]
- Ri, B.; Ra, B.; Kobe, B. Structural basis and prediction of substrate specificity in protein serine/threonine kinases. Proc. Natl. Acad. Sci. USA 2003, 100, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Stewart, B.W.; Wild, C.P. World Cancer Report 2014; IARC Publications: Lyon, French, 2014. [Google Scholar]
- Du, Y.; Li, K.; Wang, X.; Kaushik, A.C.; Junaid, M.; Wei, D. Identification of chlorprothixene as a potential drug that induces apoptosis and autophagic cell death in acute myeloid leukemia cells. FEBS J. 2020, 287, 1645–1665. [Google Scholar] [CrossRef]
- Cecotka, A.; Polanska, J. Region-Specific Methylation Profiling in Acute Myeloid Leukemia. Interdiscip. Sci. Comput. Life Sci. 2018, 10, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Du, Y.; Wei, D.Q.; Zhang, F. CEBPE expression is an independent prognostic factor for acute myeloid leukemia. J. Transl. Med. 2020, 17, 188. [Google Scholar] [CrossRef]
- Labaj, W.; Papiez, A.; Polanski, A.; Polanska, J. Comprehensive Analysis of MILE Gene Expression Data Set Advances Discovery of Leukaemia Type and Subtype Biomarkers. Interdiscip. Sci. 2017, 9, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, M.L.; Li, B.Q.; Zhai, H.L.; Liu, J.J.; Li, S.Y. Prediction of phosphorylation sites based on Krawtchouk image moments. Proteins 2017, 85, 2231–2238. [Google Scholar] [CrossRef] [PubMed]
- Shashank, T.; Pohl, M.O.; Yingyao, Z.; Ariel, R.F.; Guojun, W.; Stein, D.A.; Hong, M.M.; Paul, D.J.; Jianwei, C.; Mulder, L. Meta-and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 2015, 18, 723–735. [Google Scholar]
- Brown, R.K.; Wilson, G.; Tucci, M.A.; Benghuzzi, H.A. The effects of thymoquinone and Doxorubicin on leukemia and cardiomyocyte cell lines. Biomed Sci. Instrum. 2014, 50, 391–396. [Google Scholar] [PubMed]
- Feng, Y.; Hu, J.; Xie, D.; Qin, J.; Gong, J. Subcellular localization of caspase-3 activation correlates with changes in apoptotic morphology in MOLT-4 leukemia cells exposed to X-ray irradiation. Int. J. Oncol. 2005, 27, 699–704. [Google Scholar]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef] [Green Version]
- Li, T.T.; Du, P.F.; Xu, N.F. Identifying Human Kinase-Specific Protein Phosphorylation Sites by Integrating Heterogeneous Information from Various Sources. PLoS ONE 2010, 5, e15411. [Google Scholar] [CrossRef]
- Nakariyakul, S.; Liu, Z.P.; Chen, L. A Sequence-based Computational Approach to Predicting PDZ Domain-Peptide Interactions. Biochim. Biophys. Acta 2014, 1844, 165–170. [Google Scholar] [CrossRef]
- Vacic, V.; Iakoucheva, L.M.; Radivojac, P. Two Sample Logo: A graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006, 22, 1536–1537. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Zhang, Y.Q. Review of Imbalanced Data Learning for Protein Methylation Prediction. In Algorithmic and Artificial Intelligence Methods for Protein Bioinformatics; Pan, Y., Wang, J.X., Li, M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 1, pp. 71–90. [Google Scholar]
- Wang, M.; Tai, C.; Weinan, E.; Wei, L. DeFine: Deep convolutional neural networks accurately quantify intensities of transcription factor-DNA binding and facilitate evaluation of functional non-coding variants. Nucleic Acids Res. 2018, 11, e69. [Google Scholar] [CrossRef] [Green Version]
- Crooks, G.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Daniel, V.; Uday, K.; Amarda, S. Deep Learning Improves Antimicrobial Peptide Recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar]
- Yu, L.; Liu, F.; Li, Y.; Luo, J.; Jing, R. DeepT3_4: A Hybrid Deep Neural Network Model for the Distinction Between Bacterial Type III and IV Secreted Effectors. Front. Microbiol. 2021, 12, 605782. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 years of PhosphoSitePlus: Integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2018, 8, 433–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomalka, A.G.; Stopford, C.M.; Lee, P.C.; Rietsch, A. A translocator-specific export signal establishes the translocator-effector secretion hierarchy that is important for type iii secretion system function. Mol. Microbiol. 2012, 86, 1464–1481. [Google Scholar] [CrossRef] [PubMed]
- Schesser, K.; Frithz-Lindsten, E.; Wolf-Watz, H.A. Delineation and mutational analysis of the Yersinia pseudotuberculosis YopE domains which mediate translocation across bacterial and eukaryotic cellular membranes. J. Bacteriol. 1996, 178, 7227–7233. [Google Scholar] [CrossRef] [Green Version]
- Karolchik, D.; Baertsch, R.; Diekhans, M.; Furey, T.S.; Hinrichs, A.; Lu, Y.T.; Roskin, K.M.; Schwartz, M.; Sugnet, C.W.; Thomas, D.J.; et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003, 1, 51–54. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Jurtz, V.I.; Rosenberg Johansen, A.; Nielsen, M.; Armenteros, J.A.; Nielsen, H.; Kaae Snderby, C.; Winther, O.; Kaae Snderby, S. An introduction to deep learning on biological sequence data: Examples and solutions. Bioinformatics 2017, 33, 3685–3690. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Jansen, R.A. Bayesian Networks Approach for Predicting Protein-Protein Interactions from Genomic Data. Science 2003, 302, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.L.; Zhou, Z.H. ML-KNN: A lazy learning approach to multi-label learning. Pattern Recognit. 2007, 40, 2038–2048. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Aledo, J.C.; Cantón, F.; Veredas, F.J. A machine learning approach for predicting methionine oxidation sites. BMC Bioinform. 2017, 18, 430. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.; Frank, E. Accelerating the XGBoost algorithm using GPU computing. PeerJ Comput. Sci. 2017, 3, e127. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phosphorylation | Length | Kernel_size | Filters | Hidden_dims | Epochs |
|---|---|---|---|---|---|
| Ser | 141 | 9 | 50 | 200 | 100 |
| Tyr | 121 | 9 | 100 | 50 | 80 |
| Thr | 41 | 7 | 200 | 100 | 40 |
| Phosphorylation | SE | SP | ACC | AUC | MCC |
|---|---|---|---|---|---|
| Ser | 0.8461 | 0.9318 | 0.8889 | 0.8862 | 0.7779 |
| Tyr | 0.6552 | 0.8927 | 0.7739 | 0.7674 | 0.5527 |
| Thr | 0.7695 | 0.8479 | 0.8087 | 0.8075 | 0.6195 |
| Class | SE | SP | ACC | MCC | |
|---|---|---|---|---|---|
| T-cell | Ser | 0.8857 | 0.9737 | 0.9305 | 0.8637 |
| Tyr | 0.9762 | 0.9524 | 0.9643 | 0.9288 | |
| Thr | 1.0000 | 0.8800 | 0.9400 | 0.8864 | |
| Lymphocytic | Ser | 0.9210 | 0.9508 | 0.9359 | 0.8719 |
| Tyr | 0.7801 | 0.9003 | 0.8289 | 0.6572 | |
| Thr | 1.0000 | 0.9919 | 0.9960 | 0.9920 | |
| Classes | Myelogenous | T-Cell Leukemia | Lymphocytic | |||
|---|---|---|---|---|---|---|
| Sites | Proteins | Sites | Proteins | Sites | Proteins | |
| Ser | 16,067 | 5520 | 385 | 275 | 645 | 349 |
| Tyr | 7518 | 3837 | 52 | 42s | 2002 | 1206 |
| Thr | 3821 | 2393 | 28 | 27 | 297 | 216 |
| Total | 27,406 | 8005 | 465 | 314 | 2944 | 1548 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; Wu, Y.; Pu, X.; Li, M.; Guo, Y. A Transfer-Learning-Based Deep Convolutional Neural Network for Predicting Leukemia-Related Phosphorylation Sites from Protein Primary Sequences. Int. J. Mol. Sci. 2022, 23, 1741. https://doi.org/10.3390/ijms23031741
He J, Wu Y, Pu X, Li M, Guo Y. A Transfer-Learning-Based Deep Convolutional Neural Network for Predicting Leukemia-Related Phosphorylation Sites from Protein Primary Sequences. International Journal of Molecular Sciences. 2022; 23(3):1741. https://doi.org/10.3390/ijms23031741
Chicago/Turabian StyleHe, Jian, Yanling Wu, Xuemei Pu, Menglong Li, and Yanzhi Guo. 2022. "A Transfer-Learning-Based Deep Convolutional Neural Network for Predicting Leukemia-Related Phosphorylation Sites from Protein Primary Sequences" International Journal of Molecular Sciences 23, no. 3: 1741. https://doi.org/10.3390/ijms23031741