
Docking and Molecular Dynamic of Microalgae Compounds as Potential Inhibitors of Beta-Lactamase

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
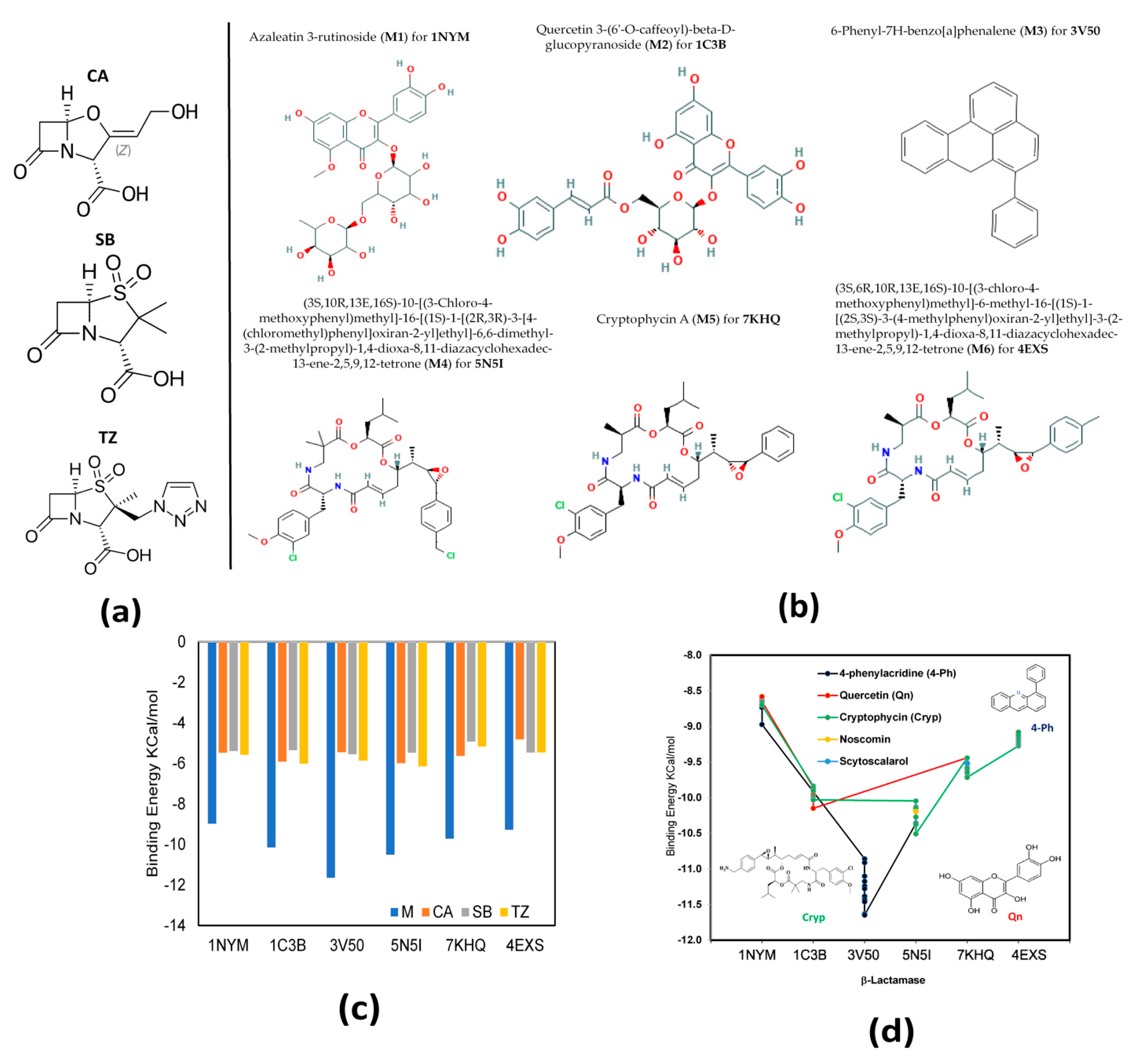
2.1. Molecular Docking
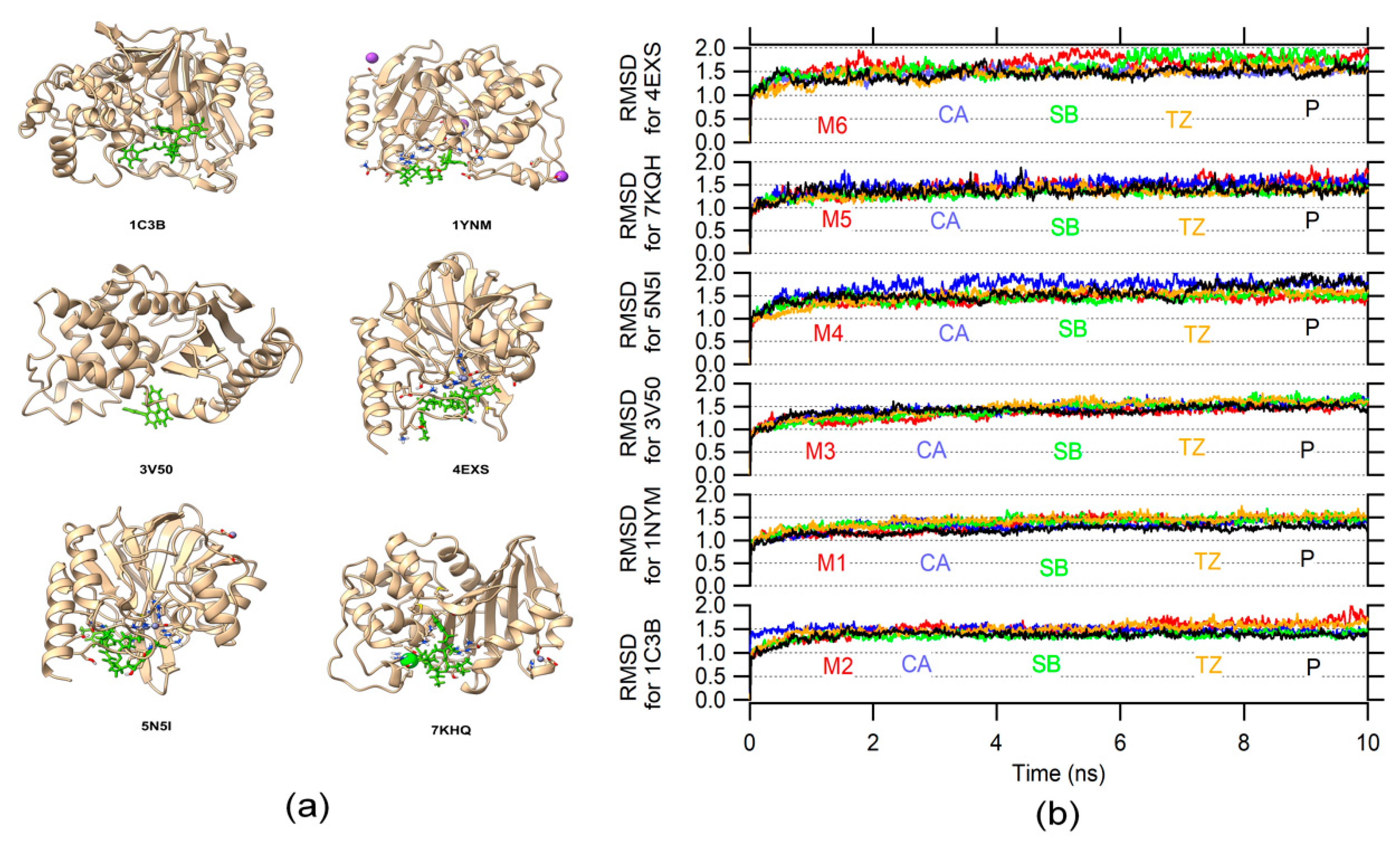
2.2. Molecular Dynamic
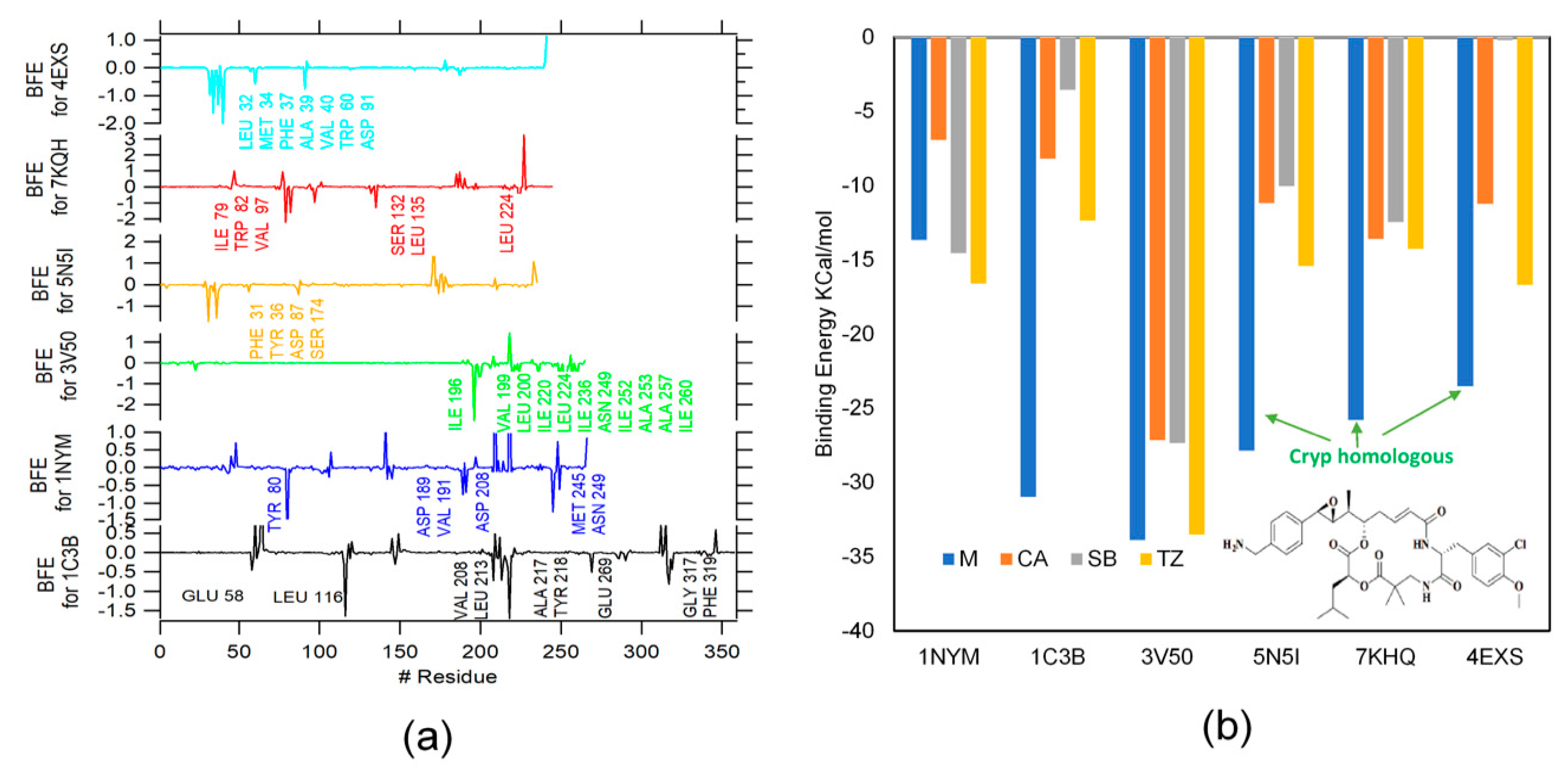
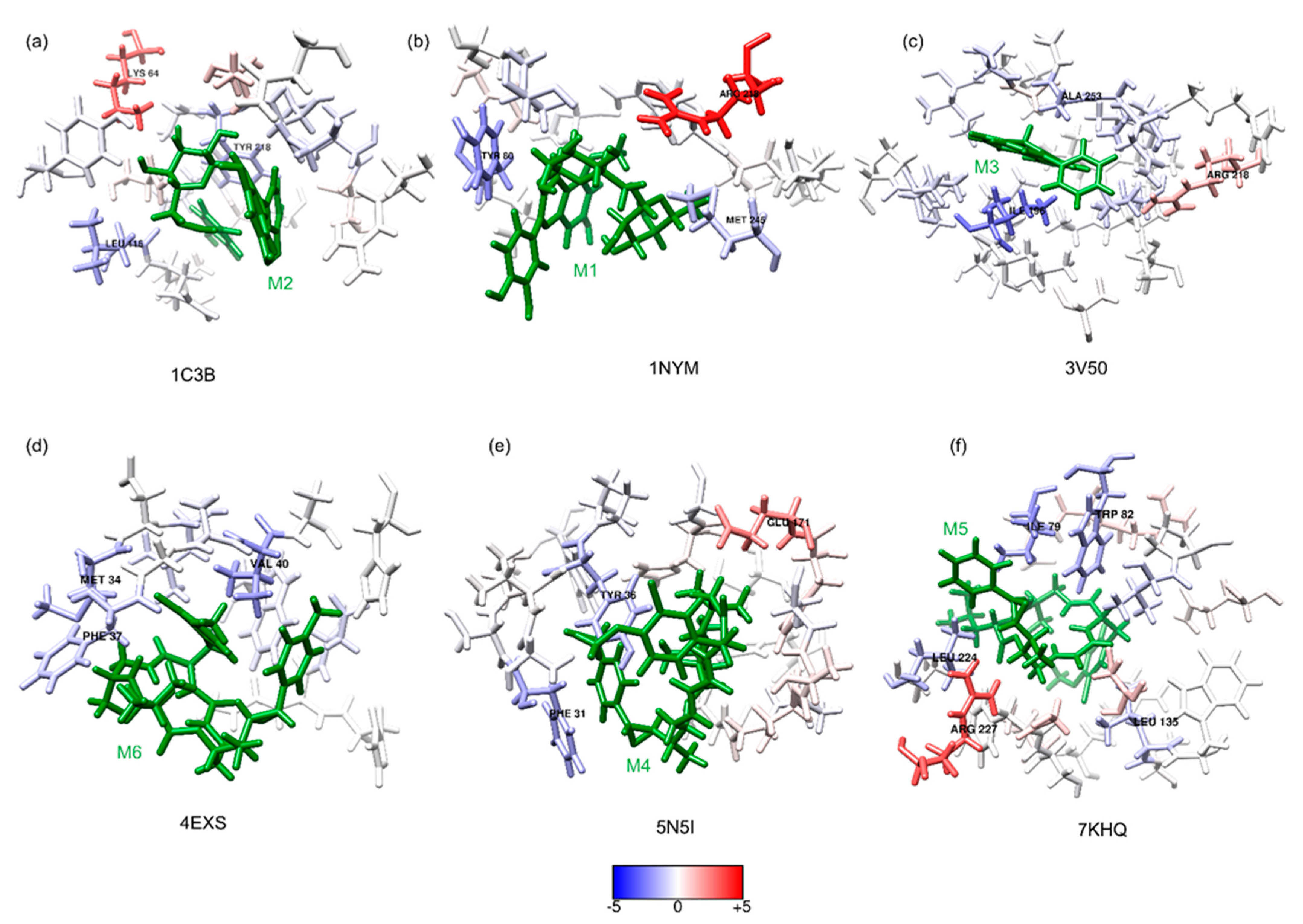
2.3. MMPBSA
3. Materials and Methods
3.1. Protein Selection
3.2. Ligands Selection
3.3. Molecular Docking
3.4. System Preparation for Molecular Dynamics Simulation
3.5. Molecular Dynamics (MD)
3.6. MMPBSA
3.7. RMSD Calculation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fleming, A. On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to Their Use in the Isolation of B. Influenzæ. Br. J. Exp. Pathol. 1929, 10, 226–233. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three Decades of β-Lactamase Inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [Green Version]
- Thakuria, B.; Lahon, K. The Beta Lactam Antibiotics as an Empirical Therapy in a Developing Country: An Update on Their Current Status and Recommendations to Counter the Resistance against Them. J. Clin. Diagn. Res. 2013, 7, 1207–1214. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Herrera, R.; Puc, L.E.C.; Sobrevilla, J.M.V.; Luque, D.; Cardona-Felix, C.S.; Aguilar-González, C.N.; Flores-Gallegos, A.C. Enzymes in the Pharmaceutical Industry for β-Lactam Antibiotic Production. In Enzymes in Food Biotechnology Production, Applications, and Future Prospects; Kuddus, M., Ed.; Elsevier: London, UK, 2019; pp. 627–643. [Google Scholar]
- Zeng, X.; Lin, J. Beta-Lactamase Induction and Cell Wall Metabolism in Gram-Negative Bacteria. Front. Microbiol. 2013, 4, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-Lactamases and β-Lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef]
- Fuda, C.C.S.; Fisher, J.F.; Mobashery, S. Beta-Lactam Resistance in Staphylococcus Aureus: The Adaptive Resistance of a Plastic Genome. Cell. Mol. Life Sci. CMLS 2005, 62, 2617–2633. [Google Scholar] [CrossRef]
- Choi, U.; Lee, C.-R. Distinct Roles of Outer Membrane Porins in Antibiotic Resistance and Membrane Integrity in Escherichia Coli. Front. Microbiol. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- King, D.T.; Sobhanifar, S.; Strynadka, N.C.J. The Mechanisms of Resistance to β-Lactam Antibiotics. In Handbook of Antimicrobial Resistance; Gotte, M., Berghuis, A., Matlashewski, G., Wainberg, M., Sheppard, D., Eds.; Springer: New York, NY, USA, 2017; pp. 177–201. [Google Scholar]
- Ceccarelli, D.; Kant, A.; van Essen-Zandbergen, A.; Dierikx, C.; Hordijk, J.; Wit, B.; Mevius, D.J.; Veldman, K.T. Diversity of Plasmids and Genes Encoding Resistance to Extended Spectrum Cephalosporins in Commensal Escherichia Coli From Dutch Livestock in 2007–2017. Front. Microbiol. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Jacoby, G.A.; Munoz-Price, L.S. The New β-Lactamases. N. Engl. J. Med. 2005, 352, 380–391. [Google Scholar] [CrossRef]
- Spyrakis, F.; Santucci, M.; Maso, L.; Cross, S.; Gianquinto, E.; Sannio, F.; Verdirosa, F.; De Luca, F.; Docquier, J.D.; Cendron, L.; et al. Virtual Screening Identifies Broad-Spectrum β-Lactamase Inhibitors with Activity on Clinically Relevant Serine- and Metallo-Carbapenemases. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef]
- Vázquez-Ucha, J.C.; Arca-Suárez, J.; Bou, G.; Beceiro, A. New Carbapenemase Inhibitors: Clearing the Way for the β-Lactams. Int. J. Mol. Sci. 2020, 21, 9308. [Google Scholar] [CrossRef]
- Hussein, U.A.-R. Assessment of the Antibacterial Activity of Macroalgae Cladophora Crispata Extract against Extended Spectrum Beta-Lactamase Producing Escherichia Coli Isolated from Diarrheic Children. Univ. Thi-Qar J. Sci. 2017, 6, 3–11. [Google Scholar]
- Ahmed, A.S.; Diab, H.M.; Alkahtani, M.A.; Alshehri, M.A.; Saber, H.; Badr, H.; Dandrawy, M.K.; El-Mansi, A.A.; Shati, A.A.; Ahmed, A.E. Molecular Epidemiology of Virulent E. Coli among Rural Small Scale Dairy Herds and Shops: Efficacy of Selected Marine Algal Extracts and Disinfectants. Int. J. Environ. Health Res. 2020, 32, 1–23. [Google Scholar] [CrossRef]
- Dhanasekaran, S.; Rajesh, A.; Mathimani, T.; Melvin Samuel, S.; Shanmuganathan, R.; Brindhadevi, K. Efficacy of Crude Extracts of Clitoria Ternatea for Antibacterial Activity against Gram Negative Bacterium (Proteus Mirabilis). Biocatal. Agric. Biotechnol. 2019, 21, 101328. [Google Scholar] [CrossRef]
- Ashford, W.A.; Golash, R.G.; Hemming, V.G. Penicillinase-Producing Neisseria Gonorrhoeae. Lancet 1976, 308, 657–658. [Google Scholar] [CrossRef]
- Gunn, B.A.; Woodall, J.B.; Jones, J.F.; Thornsberry, C. Letter: Ampicillin-Resistant Haemophilus Influenzae. Lancet 1974, 2, 845. [Google Scholar] [CrossRef]
- Cole, M. Biochemistry and Action of Clavulanic Acid. Scott. Med. J. 1982, 27, S10–S16. [Google Scholar] [CrossRef]
- Strynadka, N.C.J.; Jensen, S.E.; Johns, K.; Blanchard, H.; Page, M.; Matagne, A.; Frère, J.-M.; James, M.N.G. Structural and Kinetic Characterization of a β-Lactamase-Inhibitor Protein. Nature 1994, 368, 657–660. [Google Scholar] [CrossRef]
- Watkins, R.R.; Papp-Wallace, K.M.; Drawz, S.M.; Bonomo, R.A. Novel β-Lactamase Inhibitors: A Therapeutic Hope against the Scourge of Multidrug Resistance. Front. Microbiol. 2013, 4, 1–8. [Google Scholar] [CrossRef]
- Coleman, K. Diazabicyclooctanes (DBOs): A Potent New Class of Non-β-Lactam β-Lactamase Inhibitors. Curr. Opin. Microbiol. 2011, 14, 550–555. [Google Scholar] [CrossRef]
- Ehmann, D.E.; Jahic, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Kern, G.; Walkup, G.K.; Fisher, S.L. Avibactam Is a Covalent, Reversible, Non-Lactam-Lactamase Inhibitor. Proc. Natl. Acad. Sci. USA 2012, 109, 11663–11668. [Google Scholar] [CrossRef] [Green Version]
- Ehmann, D.E.; Jahić, H.; Ross, P.L.; Gu, R.-F.; Hu, J.; Durand-Réville, T.F.; Lahiri, S.; Thresher, J.; Livchak, S.; Gao, N.; et al. Kinetics of Avibactam Inhibition against Class A, C, and D β-Lactamases. J. Biol. Chem. 2013, 288, 27960–27971. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, J.; Wang, R.; Cai, Y. Resistance to Ceftazidime–Avibactam and Underlying Mechanisms. J. Glob. Antimicrob. Resist. 2020, 22, 18–27. [Google Scholar] [CrossRef]
- Rubio, A.M.; Kline, E.G.; Jones, C.E.; Chen, L.; Kreiswirth, B.N.; Nguyen, M.H.; Clancy, C.J.; Cooper, V.S.; Haidar, G.; Van Tyne, D.; et al. In Vitro Susceptibility of Multidrug-Resistant Pseudomonas Aeruginosa Following Treatment-Emergent Resistance to Ceftolozane-Tazobactam. Antimicrob. Agents Chemother. 2021, 65, e00084-21. [Google Scholar] [CrossRef]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (RPX7009) with Utility vs Class A Serine Carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef]
- Sun, D.; Rubio-Aparicio, D.; Nelson, K.; Dudley, M.N.; Lomovskaya, O. Meropenem-Vaborbactam Resistance Selection, Resistance Prevention, and Molecular Mechanisms in Mutants of KPC-Producing Klebsiella Pneumoniae. Antimicrob. Agents Chemother. 2017, 61, e01694-17. [Google Scholar] [CrossRef] [Green Version]
- Asempa, T.E.; Abdelraouf, K.; Nicolau, D.P. Metallo-β-Lactamase Resistance in Enterobacteriaceae Is an Artefact of Currently Utilized Antimicrobial Susceptibility Testing Methods. J. Antimicrob. Chemother. 2020, 75, 997–1005. [Google Scholar] [CrossRef]
- Salari-jazi, A.; Mahnam, K.; Sadeghi, P.; Damavandi, M.S.; Faghri, J. Discovery of Potential Inhibitors against New Delhi Metallo-β-Lactamase-1 from Natural Compounds: In Silico-Based Methods. Sci. Rep. 2021, 11, 2390. [Google Scholar] [CrossRef]
- Mondal, A.; Bose, S.; Banerjee, S.; Patra, J.K.; Malik, J.; Mandal, S.K.; Kilpatrick, K.L.; Das, G.; Kerry, R.G.; Fimognari, C.; et al. Marine Cyanobacteria and Microalgae Metabolites—A Rich Source of Potential Anticancer Drugs. Mar. Drugs 2020, 18, 476. [Google Scholar] [CrossRef]
- Kongkham, B.; Prabakaran, D.; Puttaswamy, H. Opportunities and Challenges in Managing Antibiotic Resistance in Bacteria Using Plant Secondary Metabolites. Fitoterapia 2020, 147, 104762. [Google Scholar] [CrossRef]
- Lauritano, C.; Ferrante, M.I.; Rogato, A. Marine Natural Products from Microalgae: An -Omics Overview. Mar. Drugs 2019, 17, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorokina, M.; Steinbeck, C. Review on Natural Products Databases: Where to Find Data in 2020. J. Cheminformatics 2020, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianquinto, E.; Tondi, D.; D’Arrigo, G.; Lazzarato, L.; Spyrakis, F. Can We Exploit β-Lactamases Intrinsic Dynamics for Designing More Effective Inhibitors? Antibiotics 2020, 9, 833. [Google Scholar] [CrossRef] [PubMed]
- Moreira, J.S.; Galvão, D.S.; Xavier, C.F.C.; Cunha, S.; da Rocha Pita, S.S.; Reis, J.N.; de Freitas, H.F. Phenotypic and in Silico Studies for a Series of Synthetic Thiosemicarbazones as New Delhi Metallo-Beta-Lactamase Carbapenemase Inhibitors. J. Biomol. Struct. Dyn. 2021, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ul Haq, F.; Abro, A.; Raza, S.; Liedl, K.R.; Azam, S.S. Molecular Dynamics Simulation Studies of Novel β-Lactamase Inhibitor. J. Mol. Graph. Model. 2017, 74, 143–152. [Google Scholar] [CrossRef]
- Gueto-Tettay, C.; Drosos, J.C.; Vivas-Reyes, R. Quantum Mechanics Study of the Hydroxyethylamines-BACE-1 Active Site Interaction Energies. J. Comput Aided Mol. Des. 2011, 25, 583–597. [Google Scholar] [CrossRef]
- Ahumedo Monterrosa, M.; Galindo, J.F.; Vergara Lorduy, J.; Alí-Torres, J.; Vivas-Reyes, R. The Role of LasR Active Site Amino Acids in the Interaction with the Acyl Homoserine Lactones (AHLs) Analogues: A Computational Study. J. Mol. Graph. Model. 2019, 86, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Gueto, C.; Torres, J.; Vivas-Reyes, R. CoMFA, LeapFrog and Blind Docking Studies on Sulfonanilide Derivatives Acting as Selective Aromatase Expression Regulators. Eur J. Med. Chem 2009, 44, 3445–3451. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clavulanic Acid—An Overview|ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/neuroscience/clavulanic-acid (accessed on 20 November 2021).
- Ho, K.; Su, S.; Lee, K. Molecular Docking and Simulation of the Interaction of Sulbactam with Acinetobacter Baumannii BaeSR and AdeSR. Biochem. Biophys. Res. Commun. 2021, 580, 81–86. [Google Scholar] [CrossRef]
- Pfaller, M.A.; Shortridge, D.; Harris, K.A.; Garrison, M.W.; DeRyke, C.A.; DePestel, D.D.; Moise, P.A.; Sader, H.S. Ceftolozane-Tazobactam Activity against Clinical Isolates of Pseudomonas Aeruginosa from ICU Patients with Pneumonia: United States, 2015–2018. Int. J. Infect. Dis. 2021, 112, 321–326. [Google Scholar] [CrossRef]
- Alshuniaber, M.A.; Krishnamoorthy, R.; AlQhtani, W.H. Antimicrobial Activity of Polyphenolic Compounds from Spirulina against Food-Borne Bacterial Pathogens. Saudi J. Biol. Sci. 2021, 28, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Al-Saif, S.S.A.; Abdel-Raouf, N.; El-Wazanani, H.A.; Aref, I.A. Antibacterial Substances from Marine Algae Isolated from Jeddah Coast of Red Sea, Saudi Arabia. Saudi J. Biol. Sci. 2014, 21, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggen, M.; Georg, G.I. The Cryptophycins: Their Synthesis and Anticancer Activity. Med. Res. Rev. 2002, 22, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.J.; Fortuna, M.; Media, J.; Wiegand, R.A.; Valeriote, F.A. Cryptophycin 1 Cellular Levels and Effects in Vitro Using L1210 Cells. Invest. New Drugs 1998, 16, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Trimurtulu, G.; Ohtani, I.; Patterson, G.M.L.; Moore, R.E.; Corbett, T.H.; Valeriote, F.A.; Demchik, L. Total Structures of Cryptophycins, Potent Antitumor Depsipeptides from the Blue-Green Alga Nostoc Sp. Strain GSV 224. J. Am. Chem. Soc. 1994, 116, 4729–4737. [Google Scholar] [CrossRef]
- Kyong-Hwa, K.; Se-Kwon, K. Beneficial Effect of Peptides from Microalgae on Anticancer. Curr. Protein Pept. Sci. 2013, 14, 212–217. [Google Scholar]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Hoffer, L.; Muller, C.; Roche, P.; Morelli, X. Chemistry-Driven Hit-to-Lead Optimization Guided by Structure-Based Approaches. Mol. Inform. 2018, 37, 1800059. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Raza, S.; Ranaghan, K.E.; van der Kamp, M.W.; Woods, C.J.; Mulholland, A.J.; Azam, S.S. Visualizing Protein-Ligand Binding with Chemical Energy-Wise Decomposition (CHEWD): Application to Ligand Binding in the Kallikrein-8 S1 Site. J. Comput. Aided Mol. Des. 2019, 33, 461–475. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.; Jacoby, G.A.; Medeiros, A.A. A Functional Classification Scheme for Beta-Lactamases and Its Correlation with Molecular Structure. Antimicrob Agents Chemother 1995, 39, 1211–1233. [Google Scholar] [CrossRef] [Green Version]
- Bush, K.; Jacoby, G.A. Updated Functional Classification of Beta-Lactamases. Antimicrob Agents Chemother 2010, 54, 969–976. [Google Scholar] [CrossRef] [Green Version]
- Powers, R.A.; Blázquez, J.; Weston, G.S.; Shoichet, B.K.; Morosini, M.-I.; Baquero, F. The Complexed Structure and Antimicrobial Activity of a Non-β-Lactam Inhibitor of AmpC β-Lactamase. Protein Sci. 1999, 8, 2330–2337. [Google Scholar] [CrossRef]
- Wang, X.; Minasov, G.; Blázquez, J.; Caselli, E.; Prati, F.; Shoichet, B.K. Recognition and Resistance in TEM β-Lactamase. Biochemistry 2003, 42, 8434–8444. [Google Scholar] [CrossRef]
- The Importance of the Trans-Enamine Intermediate as a Β-Lactamase Inhibition Strategy Probed in Inhibitor-Resistant SHV Β-Lactamase VariantsKe-2012-ChemMedChem—Wiley Online Library. Available online: https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cmdc.201200006 (accessed on 17 November 2021).
- Salimraj, R.; Hinchliffe, P.; Kosmopoulou, M.; Tyrrell, J.M.; Brem, J.; van Berkel, S.S.; Verma, A.; Owens, R.J.; McDonough, M.A.; Walsh, T.R.; et al. Crystal Structures of VIM-1 Complexes Explain Active Site Heterogeneity in VIM-Class Metallo-β-Lactamases. FEBS J. 2019, 286, 169–183. [Google Scholar] [CrossRef]
- Stojanoski, V.; Hu, L.; Sankaran, B.; Wang, F.; Tao, P.; Prasad, B.V.V.; Palzkill, T. Mechanistic Basis of OXA-48-like β-Lactamases’ Hydrolysis of Carbapenems. ACS Infect. Dis. 2021, 7, 445–460. [Google Scholar] [CrossRef]
- King, D.T.; Worrall, L.J.; Gruninger, R.; Strynadka, N.C.J. New Delhi Metallo-β-Lactamase: Structural Insights into β-Lactam Recognition and Inhibition. J. Am. Chem Soc. 2012, 134, 11362–11365. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended Tight-Binding Quantum Chemistry Methods. WIREs Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-XTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [Green Version]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham, T., III; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. AMBER 2018; University of California: San Francisco, CA, USA, 2018; Buscar Con Google (accessed on 22 November 2021). [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput Chem 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Tsui, V.; Case, D.A. Theory and Applications of the Generalized Born Solvation Model in Macromolecular Simulations. Biopolymers 2000, 56, 275–291. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring Protein Native States and Large-Scale Conformational Changes with a Modified Generalized Born Model. Proteins 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Su, P.-C.; Tsai, C.-C.; Mehboob, S.; Hevener, K.E.; Johnson, M.E. Comparison of Radii Sets, Entropy, QM Methods, and Sampling on MM-PBSA, MM-GBSA, and QM/MM-GBSA Ligand Binding Energies of F. Tularensis Enoyl-ACP Reductase (FabI). J. Comput. Chem. 2015, 36, 1859–1873. [Google Scholar] [CrossRef] [Green Version]
- Onufriev, A.V.; Izadi, S. Water Models for Biomolecular Simulations. WIREs Comput. Mol. Sci. 2018, 8, e1347. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Pestana-Nobles, R.; Leyva-Rojas, J.A.; Yosa, J. Searching Hit Potential Antimicrobials in Natural Compounds Space against Biofilm Formation. Molecules 2020, 25, 5334. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shalom, I.Y.; Pfeiffer-Marek, S.; Baringhaus, K.-H.; Gohlke, H. Efficient Approximation of Ligand Rotational and Translational Entropy Changes upon Binding for Use in MM-PBSA Calculations. J. Chem. Inf. Model. 2017, 57, 170–189. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. Comparison of the Efficiency of the LIE and MM/GBSA Methods to Calculate Ligand-Binding Energies. J. Chem. Theory Comput. 2011, 7, 3768–3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bush–Jacoby Group (2009) | Name | Bacteria | PDB Protein Data Bank |
|---|---|---|---|
| 1 | AMPC beta-lactamase | Escherichia coli | 1C3B [57] |
| 2b | M182T mutant of TEM-1 | Escherichia coli | 1NYM [58] |
| 2br | Complex of SHV S130G mutant beta-lactamase complexed to SA2-13 | Klebsiella pneumoniae | 3V50 [59] |
| 3a | VIM-1 metallo-beta-lactamase | Pseudomonas aeruginosa | 5N5I [60] |
| 2df | OXA-48 K73A | Klebsiella pneumoniae | 7KHQ [61] |
| 1 | NDM-1 | Klebsiella pneumoniae | 4EXS [62] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pestana-Nobles, R.; Aranguren-Díaz, Y.; Machado-Sierra, E.; Yosa, J.; Galan-Freyle, N.J.; Sepulveda-Montaño, L.X.; Kuroda, D.G.; Pacheco-Londoño, L.C. Docking and Molecular Dynamic of Microalgae Compounds as Potential Inhibitors of Beta-Lactamase. Int. J. Mol. Sci. 2022, 23, 1630. https://doi.org/10.3390/ijms23031630
Pestana-Nobles R, Aranguren-Díaz Y, Machado-Sierra E, Yosa J, Galan-Freyle NJ, Sepulveda-Montaño LX, Kuroda DG, Pacheco-Londoño LC. Docking and Molecular Dynamic of Microalgae Compounds as Potential Inhibitors of Beta-Lactamase. International Journal of Molecular Sciences. 2022; 23(3):1630. https://doi.org/10.3390/ijms23031630
Chicago/Turabian StylePestana-Nobles, Roberto, Yani Aranguren-Díaz, Elwi Machado-Sierra, Juvenal Yosa, Nataly J. Galan-Freyle, Laura X. Sepulveda-Montaño, Daniel G. Kuroda, and Leonardo C. Pacheco-Londoño. 2022. "Docking and Molecular Dynamic of Microalgae Compounds as Potential Inhibitors of Beta-Lactamase" International Journal of Molecular Sciences 23, no. 3: 1630. https://doi.org/10.3390/ijms23031630